
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, crystal structure and remote allosteric binding properties of cone thiacalix[4]pseudocrown receptors bearing anthraquinone function and different arms†
Xue-Xin Lv‡
,
Qian Wang‡,
Li-Jing Zhang,
Ling-Ling Liu,
Mei Zhao and
Dian-Shun Guo *
*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Shandong Normal University, Jinan 250014, P. R. China. E-mail: chdsguo@sdnu.edu.cn; Fax: +86 531 86928773; Tel: +86 531 86180743
First published on 17th July 2017
Abstract
Herein, three novel cone thiacalix[4]pseudocrown receptors 1–3, bearing anthraquinone and triazole functions as well as different side arms, were successfully synthesized via a double click reaction as the key step. Their recognition properties have been fully evaluated using optical, electrochemical, and theoretical methods. Receptor 1 features high selectivity for Pb2+, whereas receptor 2 can optically recognize Zn2+ in the presence of Cd2+. This difference of binding specificity to metal ions may be ascribed to the remote tuning effect from the side arms connected to the lower rim of thiacalix[4]arene that realizes the accommodation of binding specificity. Moreover, the plausible coordination modes have been presented.
1. Introduction
The design and synthesis of optical and redox-active receptors for the efficient detection of environmentally and biologically relevant ionic species is a challenging topic.1–5 This type of receptors combines signaling reporters with well-defined recognition centers to enable facile detection of ion binding through optical and electrochemical techniques. In particular, the selective detection of Zn2+ and Pb2+ ions is one of the most interesting projects as the former plays key roles in many biochemical processes6–10 and the latter is an ongoing danger to the human health, especially to children.11–14Thiacalix[4]arenes are a kind of versatile scaffolds for highly organized receptors because of their specific affinity and allosteric features in molecular recognition.15–21 In recent years, many optical receptors have been designed and synthesized based on the thiacalix[4]arene scaffold. Excellent examples include quinoline, rhodamine, naphthalene, pyrene, as well as coumarin-appended thiacalix[4]arene receptors.22–26 On the other hand, to date, less attention has been paid to developing the corresponding redox active receptors.27–29 In particular, there is a paucity of optical and redox-active receptors involving thiacalix[4]arene.27 Thiacalix[4]crown, with one or two crown ether(s) at the lower rim of the thiacalix[4]arene unit, possesses various donor atoms within its crown ring and can accommodate desired substrates.18,30,31 Thus, it is essential to develop optical and redox-active thiacalix[4]crowns as candidate receptors for the highly selective and sensitive detection of desired substrates as they provide facile access to multiple monitoring.
Anthraquinone can be used as a signal reporter in sensors because its optical and electrochemical properties can be significantly perturbed by chemical stimuli.32 The 9,10-anthraquinone probe is known to exhibit a pronounced color change and remarkable potential shift when it interacts with some metal ions.33,34 Recently, the click reaction has become a powerful strategy for molecular linking in synthetic chemistry.35–37 In view of these precedents, we combined the thiacalix[4]arene scaffold and 9,10-anthraquinone function through the double click reaction to construct new thiacalix[4]pseudocrown receptors, 1–3 (Scheme 1), in which the 9,10-anthraquinone group acted as an optical and electrochemical reporter, two 1,2,3-triazole rings acted as both linkers and potential binding sites, and various side arms could remotely regulate the binding specificity. Their ability of binding metal ions has been assessed by optical, electrochemical, and theoretical techniques.
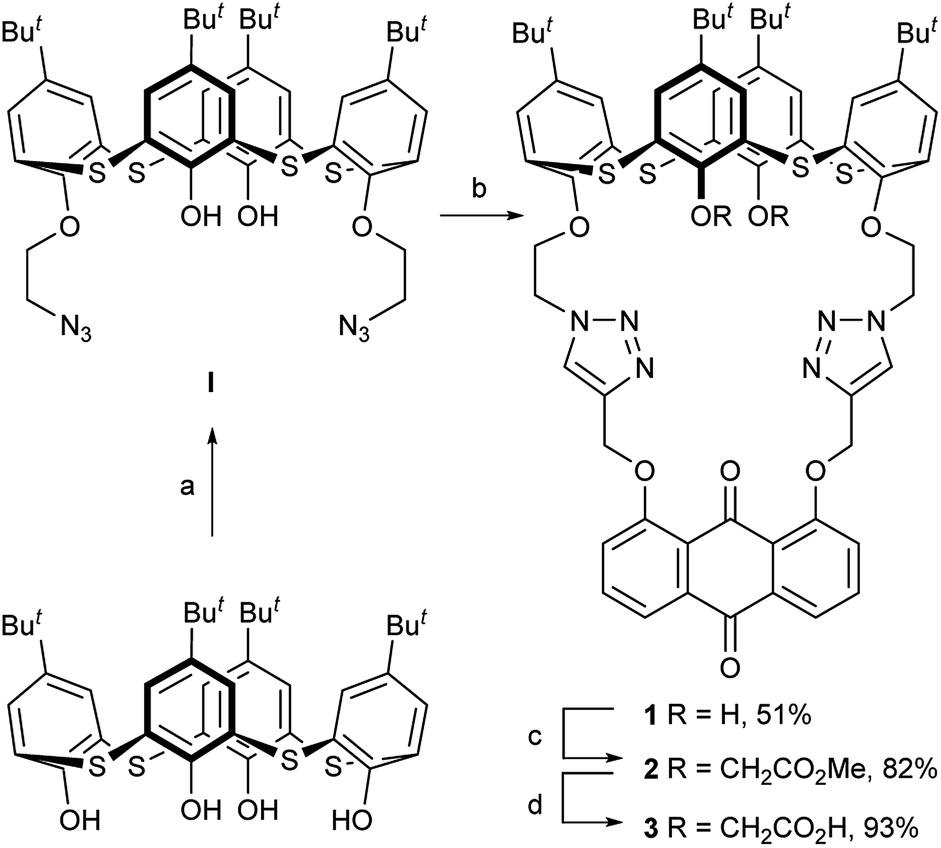 | ||
| Scheme 1 Reagents and conditions: (a) N3CH2CH2OTs, KI, K2CO3, acetone, and reflux. (b) 1,8-bis(2-propynyloxy)anthraquinone, Et3N, CuI, THF, H2O, and 60 °C. (c) BrCH2CO2Me, NaH, THF, and rt. (d) KOH, H2O, THF, and rt. | ||
2. Results and discussion
2.1 Synthesis and characterization of the receptors 1–3
The synthesis of the receptors 1–3 is shown in Scheme 1. Treatment of p-tert-butylthiacalix[4]arene with 2-azidoethyl-4-methylbenzene-sulfonate and KI in an acetone solution using anhydrous K2CO3 as a base furnished the intermediate I (a cone conformer) in 56% yield. Then, the double 1,3-dipolar cycloaddition ring-closure reaction of I with 1,8-bis(2-propynyloxy)anthraquinone catalyzed by CuI yielded the thiacalix[4]pseudocrown receptor 1 in 51% yield. Receptor 2 was obtained in good yield (82%) via treatment of 1 with methyl bromoacetate in the presence of NaH in a dry THF solution. Hydrolysis of the ester functions in 2 with KOH in THF provided the corresponding receptor 3 in 93% yield. All the new thiacalix[4]arene derivatives were fully characterized by 1H NMR, 13C NMR, IR, MS, and elemental analysis.The NMR spectral data indicate that the receptors 1–3 and intermediate I feature the cone conformation,21 showing two singlet peaks at 1.38–0.69 ppm in the 1H NMR and two doublet peaks at 31.5–30.7 ppm in the 13C NMR spectra. To further examine the 3D structure of thiacalix[4]pseudocrown receptors, the single crystal structure of 2 (Fig. 1) was determined by X-ray crystallography. Analysis of the data reveals that receptor 2 adopts a pinched cone conformation. The dihedral angles between the phenolic rings and the virtual plane defined by the four bridging sulfur atoms are 33.3(0), 56.0(1), 88.7(0), and 83.7(1)°. The average distance between two vicinal sulfur atoms is 5.523 Å, fitting the previously reported values.38 The two 1,2,3-triazole rings exhibit the same orientation, with dissimilar tilted angles. The dihedral angels between the triazole rings (N1–N2–N3–C14–C13) and (N4–N5–N6–C31–C32) and the anthraquinone ring (C23–C24–C26–C27–C28–C29) are 48.9(0) and 11.2(1)°, respectively. The anthraquinone unit in 2 shows a v-like shape, in which two outer phenyl rings make an interplanar angle of 21.1(1)°.39 Note that the intraannular carbonyl oxygen atom O3 is strongly bent out of the anthraquinone plane. This distortion may be ascribed to the intramolecular interactions between the carbonyl oxygen atom O3 and two ether bridges. The O3⋯O2 and O3⋯O5 distances are 2.665(6) and 2.674(7) Å, respectively, much shorter than the sum of the van der Waals radii of two oxygen atoms (3.04 Å).40 In the solid state, the molecule 2 possesses a well pre-organized array of oxygen atoms and nitrogen atoms of the triazole rings that are favorable for interaction with suitable guest species.
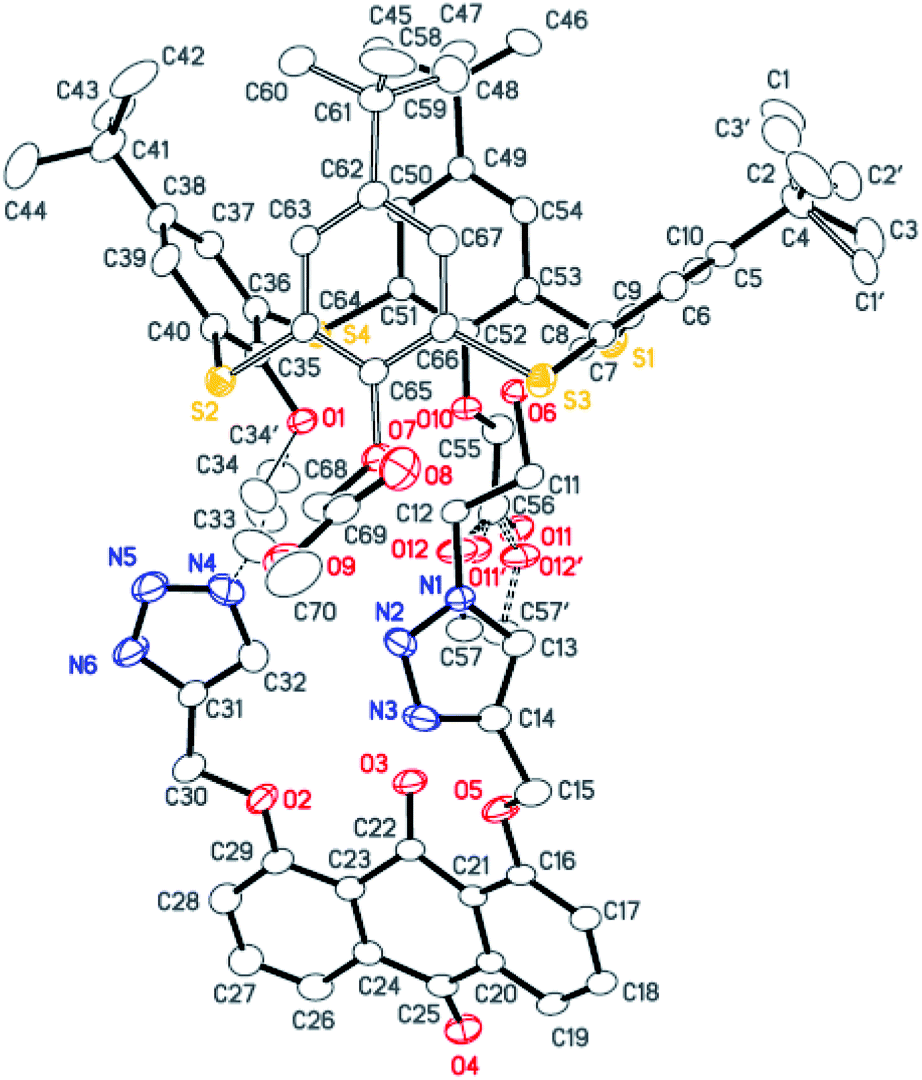 | ||
| Fig. 1 Crystal structure of 2, showing the atom-labelling scheme and the disordered atoms. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
2.2 UV-vis and fluorescence studies
The recognition behavior of the receptors towards various cations was first evaluated by UV-vis and fluorescence spectroscopy. UV-vis spectroscopy of 1 and 2 in CH2Cl2/MeCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution revealed absorption maxima at 372 and 376 nm (Fig. S1, ESI†), corresponding to the anthraquinone probe.34 The metal-binding properties of both receptors have been investigated by monitoring the absorption changes upon the addition of perchlorate salts of Li+, Na+, K+, Cs+, Mg2+, Ca2+, Co2+, Ni2+, Ag+, Cu2+, Hg2+, Zn2+, Cd2+, and Pb2+. As shown in Fig. 2, upon the addition of Zn2+ ions to the solution of 1, the absorption at 372 nm shifted to 399 nm with a significant absorbance enhancement. This change is probably due to the coordination of 1 with Zn2+. The fact is responsible for the turning of pale-yellow color into deep-yellow one that can be detected by the naked eye. The Job's plot for the binding of 1 with Zn2+ (Fig. S2, ESI†) provides a 1:1 stoichiometry. The association constant logKs obtained by fitting the titration data is 5.73 M−1. A similar behavior was observed for Cd2+ and Pb2+ ions, which exhibited a smaller shift to 385 and 387 nm (Fig. S3 and S4, ESI†), respectively. No obvious changes were observed in the UV-vis spectra upon the addition of other metal ions. Under the same conditions, receptor 2 exhibited similar but smaller changes in spectral absorption in the presence of Zn2+, Cd2+, and Pb2+ ions (Fig. S5–S7, ESI†). However, the association constants for 1 and 2 were nearly parallel (Table S1, ESI†). Similarly, the Job's plot for the binding of 2 with Pb2+ (Fig. S8, ESI†) also provided a 1:1 stoichiometry.
:1, v/v) solution revealed absorption maxima at 372 and 376 nm (Fig. S1, ESI†), corresponding to the anthraquinone probe.34 The metal-binding properties of both receptors have been investigated by monitoring the absorption changes upon the addition of perchlorate salts of Li+, Na+, K+, Cs+, Mg2+, Ca2+, Co2+, Ni2+, Ag+, Cu2+, Hg2+, Zn2+, Cd2+, and Pb2+. As shown in Fig. 2, upon the addition of Zn2+ ions to the solution of 1, the absorption at 372 nm shifted to 399 nm with a significant absorbance enhancement. This change is probably due to the coordination of 1 with Zn2+. The fact is responsible for the turning of pale-yellow color into deep-yellow one that can be detected by the naked eye. The Job's plot for the binding of 1 with Zn2+ (Fig. S2, ESI†) provides a 1:1 stoichiometry. The association constant logKs obtained by fitting the titration data is 5.73 M−1. A similar behavior was observed for Cd2+ and Pb2+ ions, which exhibited a smaller shift to 385 and 387 nm (Fig. S3 and S4, ESI†), respectively. No obvious changes were observed in the UV-vis spectra upon the addition of other metal ions. Under the same conditions, receptor 2 exhibited similar but smaller changes in spectral absorption in the presence of Zn2+, Cd2+, and Pb2+ ions (Fig. S5–S7, ESI†). However, the association constants for 1 and 2 were nearly parallel (Table S1, ESI†). Similarly, the Job's plot for the binding of 2 with Pb2+ (Fig. S8, ESI†) also provided a 1:1 stoichiometry.
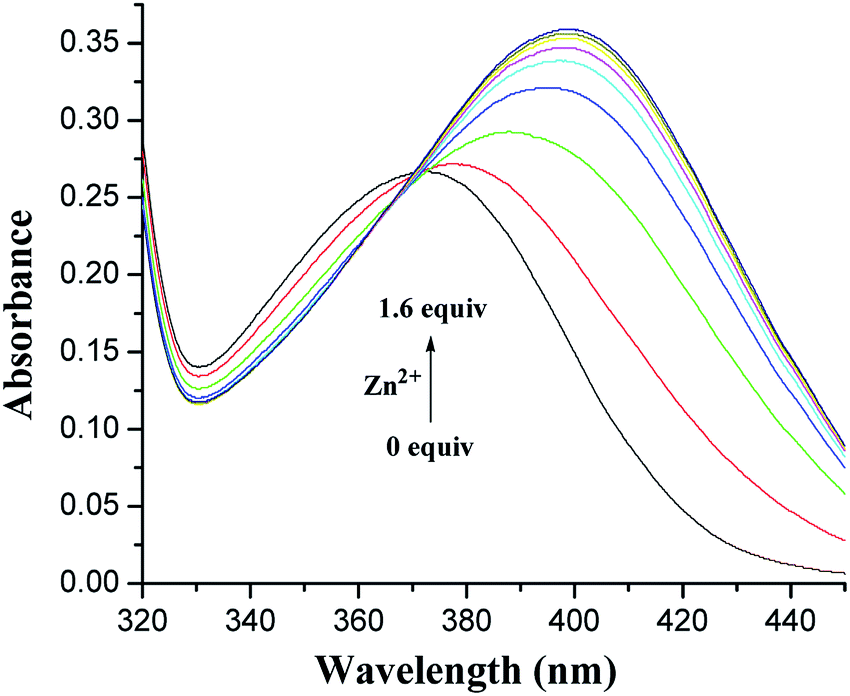 | ||
| Fig. 2 Changes in the absorption spectrum of 1 (5.0 × 10−5 M) in CH2Cl2/MeCN (1:1, v/v) upon the addition of increasing amounts of Zn2+. | ||
Assay of the ion affinity was also performed by monitoring the changes originating from the fluorescence intensity of 1–3 induced by metal ions. A systematic change in the fluorescence spectra associated with the addition of different metal ions is shown in Fig. 3.
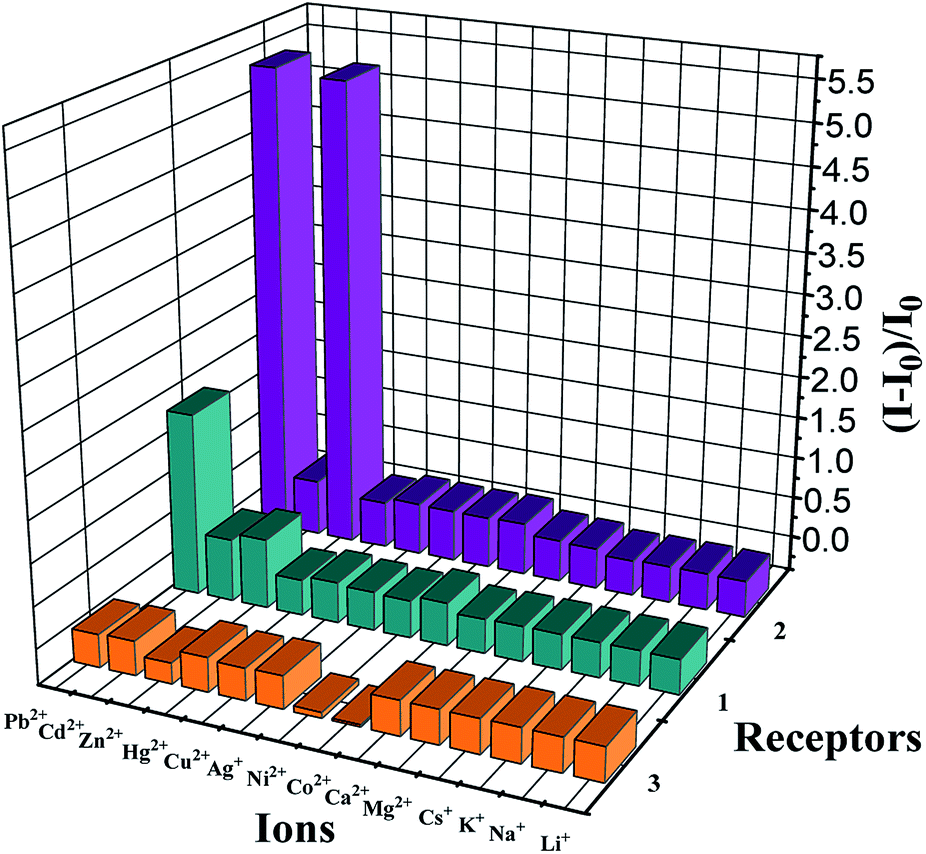 | ||
| Fig. 3 Changes in the fluorescence emission intensity of 1, 2 (5.0 × 10−5 M) in CH2Cl2/MeCN (1:1, v/v), and 3 (5.0 × 10−5 M) in THF/H2O (9:1, v/v) at pH 7.4 in HEPES after the addition of 1.0 equiv. metal ions. I0 and I refer to the fluorescence emission intensity of 1–3 and that after the addition of metal ions, respectively. | ||
In the fluorescence spectra, 1 exhibited a weak fluorescence with an emission band at 427 nm under excitation at 375 nm (Fig. S9, ESI†). A clear fluorescence enhancement (ca. 140%, Fig. 4) was observed upon the addition of Pb2+ at 465 nm, whereas only 42% and 33% enhancements were obtained in the cases of Zn2+ and Cd2+ at 428 nm, respectively. Under identical conditions, no increase in the fluorescent emission was detected upon the addition of other metal ions involving Li+, Na+, K+, Cs+, Mg2+, Ca2+, Co2+, Ni2+, Ag+, Cu2+, and Hg2+.
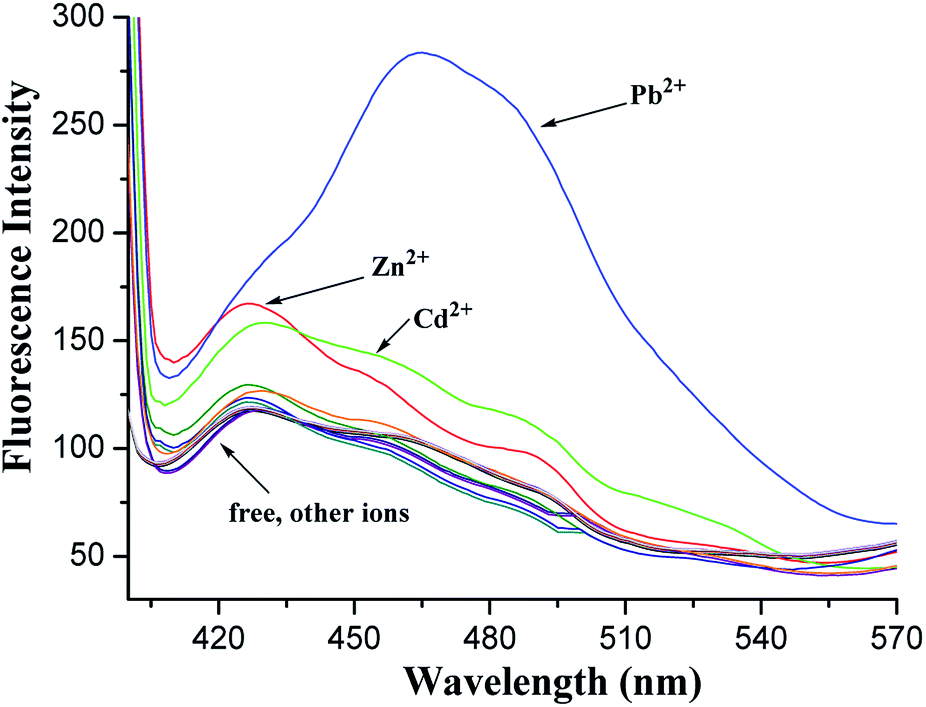 | ||
| Fig. 4 Fluorescence spectra of 1 (5.0 × 10−5 M) in CH2Cl2/MeCN (1:1, v/v) upon the addition of perchlorate salts of Li+, Na+, K+, Cs+, Mg2+, Ca2+, Co2+, Ni2+, Ag+, Cu2+, Hg2+, Zn2+, Cd2+, and Pb2+ under an excitation at 375 nm. | ||
The red shift in the emission band of 1 could be assigned to the ICT41,42 occurred via the coordination of 1 with Pb2+. When Pb2+ ion strongly interacts with the lone pair electrons of the intraannular carbonyl oxygen atom with the aid of two triazole rings (this has been ascertained by the electrochemical behavior of the [1·Pb2+] complex hereinafter), charge transfer from the 1,8-oxygen atoms to the electron-deficient carbonyl group becomes stronger. The enhancement of the fluorescence intensity is presumably ascribed to the metal ion binding with 1,2,3-triazole rings followed by PET suppression.43,44 This supposition has been initially confirmed by the following theoretical calculations.
Fluorescence titration of 1 with Pb2+ ions in CH2Cl2/MeCN (1:1, v/v) was also performed. Fig. 5 shows the changes in the fluorescence emission intensity of 1 upon the addition of increasing amounts of Pb2+ ions. Binding analysis using the method of continuous variations (Job's plot)45,46 established a 1:1 stoichiometry for the [1·Pb2+] complex (Fig. 5, inset). The association constant logKs was further determined to be 5.27 M−1 by fitting the titration result.47
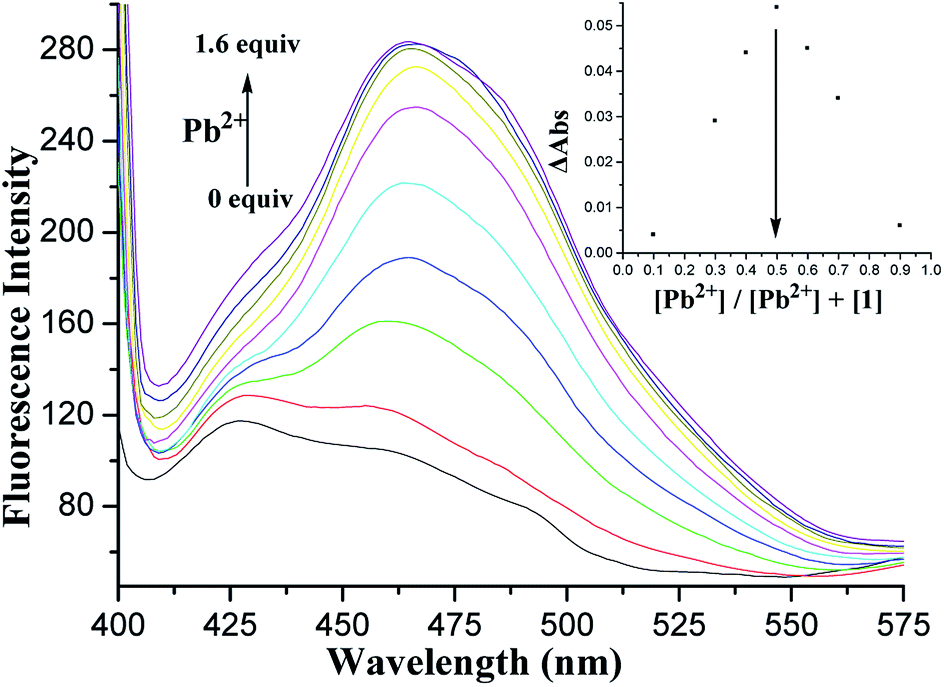 | ||
| Fig. 5 Changes in the fluorescence emission spectrum of 1 (5.0 × 10−5 M) in CH2Cl2/MeCN (1:1, v/v) upon the addition of Pb2+ ions under an excitation at 375 nm. The inset shows the Job's plot of 1 with Pb2+. | ||
The possible interferences by other metal ions were assessed through competitive experiments. The fluorescence changes of 1 in CH2Cl2/MeCN (1:1, v/v) solution were measured via treatment of 1.0 equiv. Pb2+ ions in the presence of 50 equiv. of other interfering metal ions involving Li+, Na+, K+, Cs+, Mg2+, Ca2+, Co2+, Ni2+, Ag+, Cu2+, Hg2+, Zn2+, and Cd2+. All the interfering metal ions showed little or no interference with the detection (Fig. 6, green bars), except Zn2+ and Cd2+ that quenched the fluorescence to some degree. This unique selectivity of 1 to Pb2+ may be ascribed to the result of the combination of the suitable coordination geometry of the chelating receptor, the radius of Pb2+, and the nitrogen-affinity character of Pb2+, indicating that receptor 1 can be potentially applied as a probe for Pb2+ ions.
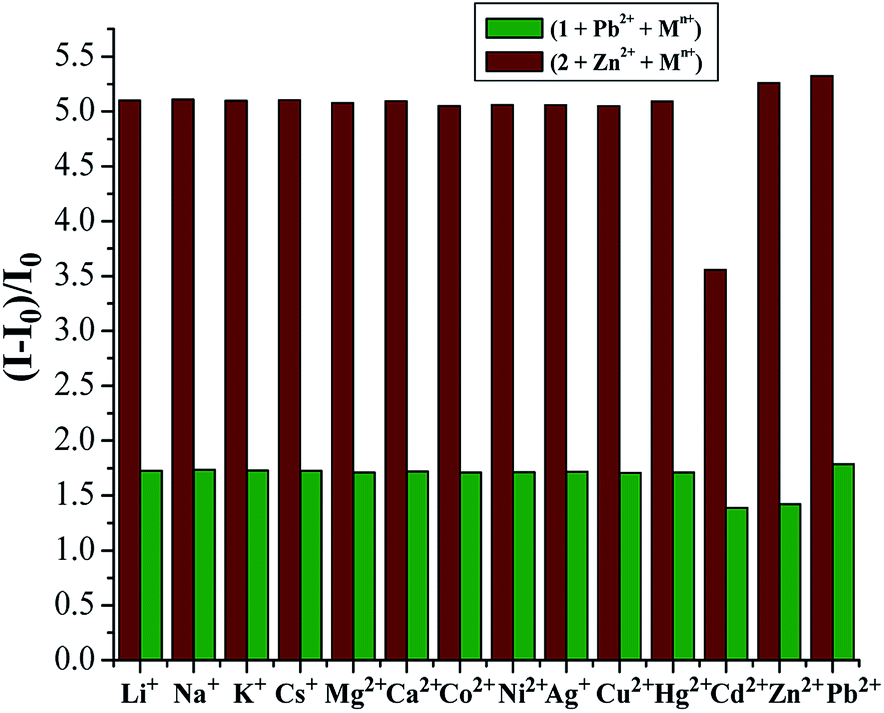 | ||
| Fig. 6 Competitive studies of [1·Pb2+] and [2·Zn2+] in CH2Cl2/MeCN (1:1, v/v) with different cations. Green bars: selectivity of 1 (5.0 × 10−5 M) towards Pb2+ upon the addition of different metal ions. Purplish red bars: selectivity of 2 (5.0 × 10−5 M) towards Zn2+ upon the addition of different metal ions. I0 and I refer to the fluorescence emission intensity of 1 and 2 and that after the addition of metal ions, respectively. | ||
Similar to 1, receptor 2, in which only two OH groups were transformed into OCH2CO2Me moieties via the O-alkylation reaction, showed a weak fluorescence with an emission band at 428 nm under excitation at 375 nm (Fig. S9, ESI†). A 4-fold fluorescence enhancement was obtained in the presence of Pb2+ ions at 465 nm, and the binding constant logKs was 5.82 M−1 (Fig. S10, ESI†), which was similar to the binding property of receptor 1 for Pb2+ ions. To our surprise, upon the addition of Zn2+ ions, 2 displayed a 3.75-fold fluorescence enhancement at 476 nm. Fig. 7 shows the detailed fluorescence changes of 2 upon the gradual addition of Zn2+ ions. The Job's plot (Fig. 7, inset) for the binding of 2 with Zn2+ shows a 1:1 stoichiometry, whereas the association constant logKs of the [2·Zn2+] complex has been calculated to be 5.56 M−1. This different selectivity of 2 may be attributed to the influence of two ester functions at the lower rim that can offer better flexibility of the thiacalix[4]arene unit to remotely regulate the size of its crown cavity as the ester functions cannot fix the structure via intramolecular hydrogen bonding interactions.21
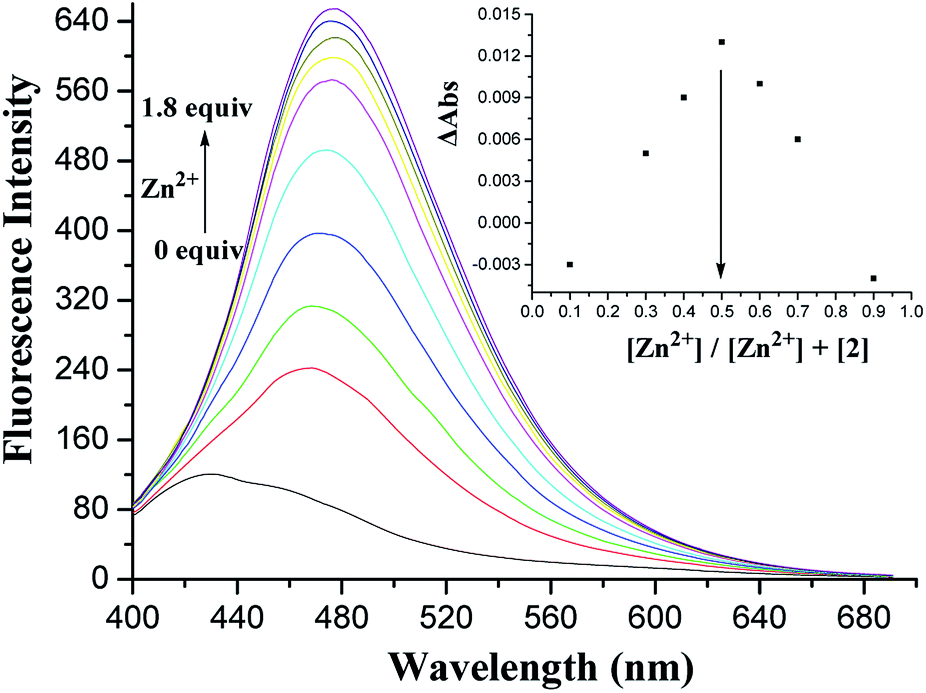 | ||
| Fig. 7 Changes in the fluorescence emission spectrum of 2 (5.0 × 10−5 M) in CH2Cl2/MeCN (1:1, v/v) upon the addition of Zn2+ ions under an excitation at 375 nm. The inset shows the Job's plot of 2 with Zn2+. | ||
In view of the better selectivity of 2, we tried to develop a chemosensor that could discriminate Zn2+ from Cd2+ to some extent, considering their similarity in chemical properties. Therefore, the competitive experiments of Zn2+ mixed with other metal ions were performed (Fig. 6, purplish red bars). As can be seen, among the metal ions tested herein, no significant spectral changes were observed upon the addition of alkali metal ions and some transition metal ions except for the case of Cd2+, where partial quenching was detected. The design of receptors that offer high Zn2+/Cd2+ selectivity is one of the current challenges in cation recognition chemistry.48–50 Thus, the ability to optically recognize Zn2+ in the presence of Cd2+ is an interesting feature of this thiacalix[4]pseudocrown receptor.
2.3 Electrochemistry studies
The recognition properties of 1 and 2 towards the abovementioned metal ions were also proven by monitoring the electrochemical behavior of 1 and 2 in the absence and presence of metal ions. Their electrochemical properties were initially measured using cyclic voltammetry (CV) in CH2Cl2/DMSO (3:1, v/v) containing 0.1 M n-Bu4NPF6 as the supporting electrolyte. The CV curves showed a quasi-reversible redox couple that became more and more irreversible upon the addition of metal ions. Alternatively, differential pulse voltammetry (DPV) was utilized. The cathodic peaks (Epc) at −0.95 and −0.98 V were observed for 1 and 2, respectively, which corresponded to the successive one-electron transfers to the anthraquinone moiety of the receptors. Negligible changes in the DPV voltammograms of both receptors were observed upon the addition of Li+, Na+, K+, Cs+, Mg2+, Ca2+, Co2+, Ni2+, Ag+, Cu2+, and Hg2+ metal ions, whereas significant modifications were found upon the addition of Zn2+, Cd2+, and Pb2+ ions (Fig. 8). On stepwise addition of Zn2+ ions to the receptor 1, a clear evolution of the reduction wave to Epc = −0.83 V (ΔEpc = 120 mV) with an increase in the intensity of the initial wave was observed (Fig. S11, ESI†), and the maximum shift of DPV was obtained with 1.0 equiv. Zn2+ ions. This result confirms the formation of the [1·Zn2+] complex. Receptor 1 also showed the shifts of the reduction waves in the presence of Cd2+ and Pb2+ ions, and new reduction waves at Epc = −0.87 V (ΔEpc = 80 mV) and Epc = −0.88 V (ΔEpc = 70 mV) positively shifted (Fig. S12, ESI†). In the presence of Zn2+, Cd2+, and Pb2+ ions, similar results were obtained for 2 (Fig. S13, ESI†). Based on the magnitude of these observed shifts, it can be speculated that complexation exerts a more powerful effect on the anthraquinone moiety and two triazole rings than on the thiacalix[4]arene cavity. The electrochemical selectivity of 1 and 2 may be attributed to the compatibility between the receptors and metal ions.
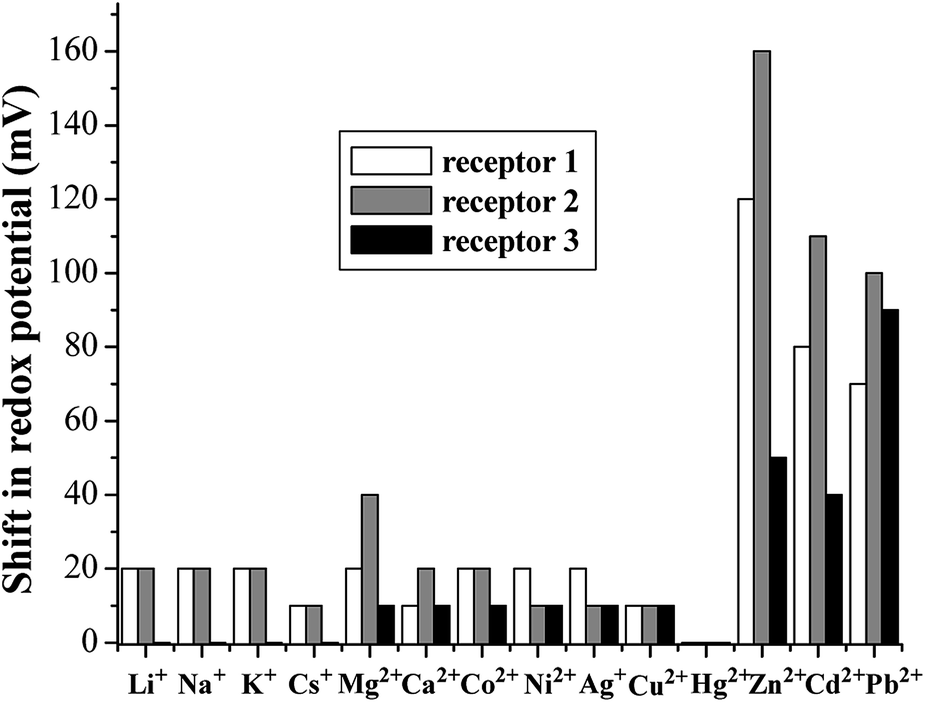 | ||
| Fig. 8 Shifts in the redox potential of 1–3 induced by the addition of 1.0 equiv. metal ions. The DPV of 1 and 2 were measured in CH2Cl2/DMSO (3:1, v/v), and that of 3 was measured in THF/H2O (9:1, v/v) at pH 7.4 in HEPES. | ||
2.4 1H NMR titrations
To explore the 1:1 stoichiometry and coordination modes, 1H NMR titration experiments were carried out for receptor 2 (9.0 × 10−3 M) in CDCl3 and Zn(ClO4)2 (3.0 × 10−1 M) in CD3CN at 25 °C (Fig. 9). The complexation process of 2 induced not only noticeable downfield changes in the chemical shifts of triazole-H (Δδ = 0.104 ppm) and OCH2 (Δδ = 0.278 ppm) linked to the anthraquinone unit but also relatively large upfield shifts in OCH2 (Δδ = 0.183 ppm) and CO2Me (Δδ = 0.130 ppm) of OCH2CO2Me. The downfield-shift spectral changes of the triazole ring protons and OCH2 linked to the anthraquinone unit can indicate that the two triazole rings and the anthraquinone unit are acting as effective binding sites in the complexation event; however, the upfield-shift changes show the adjustment of the thiacalix[4]arene conformation. The titration isotherm matches a 1:1 bonding model, which is in line with the results obtained by the UV-vis titrations and fluorescence titrations.
 | ||
| Fig. 9 Partial 1H NMR (300 MHz) spectra of 2 (9.0 × 10−3 M) in CDCl3 upon the addition of increasing amounts of Zn(ClO4)2 (3.0 × 10−1 M) in CD3CN. | ||
2.5 Theoretical studies
To understand the remotely regulated effect resulting from the different arms as well as the binding specificity of receptors 1 and 2, their structures were initially optimized via theoretical calculations at the AM1/3-21G level of theory.51,52 In the optimized structures of 1 and 2 (Fig. 10), two triazole rings exhibit the same orientation, and the anthraquinone unit has a v-like shape, which is similar to the crystal structure of 2. The main difference is the conformational shape of their thiacalix[4]arene core caused by the different arms at the lower rim, which can be exactly identified by the dihedral angle (88.7° for 1 and 116.2° for 2) between phenolic rings directly linked to the pseudocrown moiety. This confirms the feasibility of adjusting the pseudocrown cavity by changing the arms, which is the purpose of our design.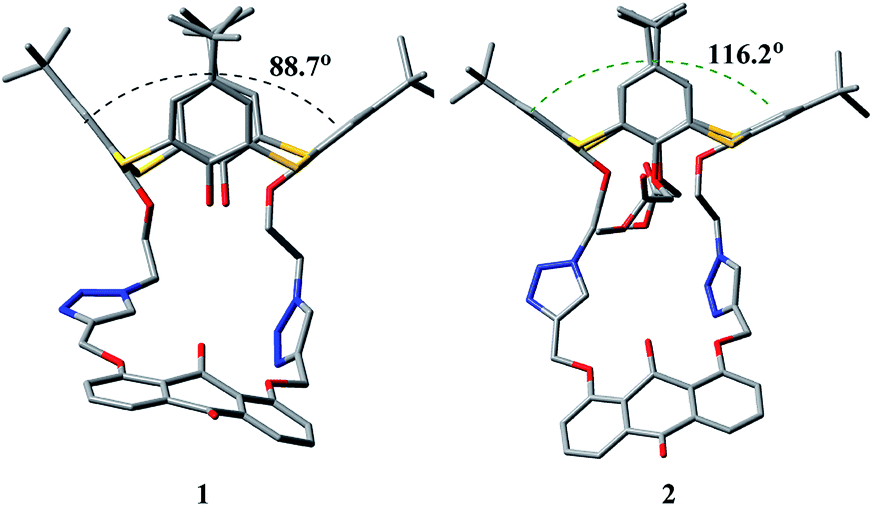 | ||
| Fig. 10 Optimized structures (AM1/3-21G) of 1 and 2, showing dihedral angles between opposite phenolic rings linked to the pseudocrown unit. Hydrogen atoms are omitted for clarity. | ||
Moreover, an optimized structure of [1·Pb2+] showed the binding modes between the receptor and Pb2+ (Fig. 11), where Pb2+ ion well occupied the pseudocrown cavity with the aid of the intraannular carbonyl oxygen atom of the anthraquinone group and nitrogen atoms of both 1,2,3-triazole rings. This confirmed the supposition about the binding sites. Remarkably, the most striking feature of [1·Pb2+] is that the v-like shape of the anthraquinone moiety was turning nearly into a plane. The relevant torsion angles O1–C1–C2–C3 and O1–C1–C4–C5 are 179.5 and 179.8° in [1·Pb2+], respectively, as compared to 153.3 and 155.9° in 1.
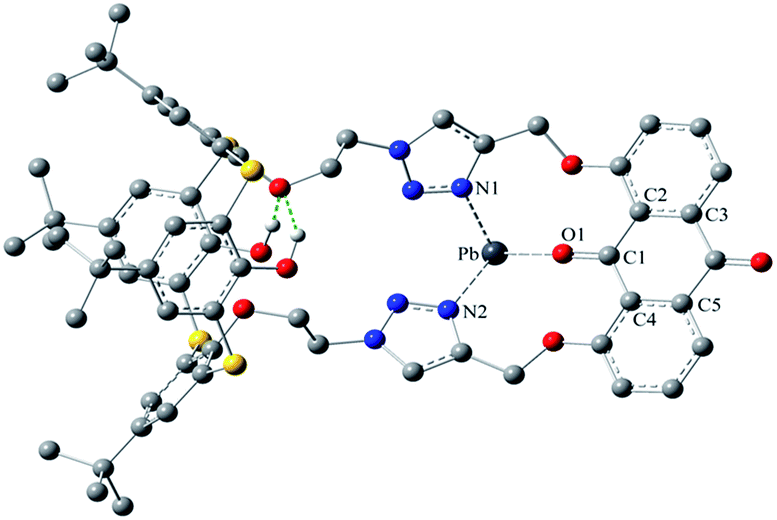 | ||
| Fig. 11 An optimized structure (AM1/3-21G) of [1·Pb2+], showing coordination modes. | ||
In the optimized structure of [1·Pb2+], there are intramolecular O–H⋯O hydrogen bonds, which can restrict the conformational transformation and fix the size of the pseudocrown cavity. This fact can explain the high specificity of 1 towards Pb2+. Similarly, this increase in the structural rigidity of [1·Pb2+] could rationalize the fluorescence enhancement, which was in agreement with the experimental phenomenon. However, in the case of [2·Zn2+] (Fig. S14, ESI†), no corresponding hydrogen bonds exist due to the replacement of OH groups with OCH2CO2Me arms; thus, the thiacalix[4]arene platform has enough flexibility to adjust the size of the pseudocrown cavity, fitting different cations. This may explain why 2 can recognize Pb2+ and Zn2+ ions at the same time.
The recognition properties of 3 towards different metal ions were also studied using similar methods. Its absorption band is centered at 386 nm. Changes in the absorption spectra of 3 upon the addition of Zn2+, Co2+, and Ni2+ were observed, and a new absorption band at 490 nm appeared (Fig. S15, ESI†). This result is attributable to the formation of [3·M2+] (M2+ = Zn2+, Co2+, and Ni2+). The fluorescence spectrum of 3 displays an emission peak at 484 nm under excitation at 375 nm. Compared with those of 1 and 2, the red-shifted fluorescence spectrum of 3 is ascribed to the dissociative proton of the carboxyl group binding to the carbonyl group of the anthraquinone function. Decreased emission bands were observed upon the addition of Zn2+, Co2+, and Ni2+ ions to 3 (Fig. S16, ESI†), which could be ascribed to a PET mechanism.44 Moreover, an obvious DPV response of 3 to Pb2+, Zn2+, and Cd2+ ions was found with a positive potential shift, especially in the case of Pb2+ ions. At present, the selectivity of 3 for metal ions is not desirable, but its solubility in aqueous media is improved. Therefore, we believe that better selective receptors can be obtained in the future by loading other water-soluble groups.
3. Experimental
3.1 Reagents and instruments
All reactions were carried out under a N2 atmosphere using standard Schlenk techniques. DMSO, MeCN, and CH2Cl2 were distilled under nitrogen over CaH2. K2CO3, TsCl, NaOH, KI, and 1,8-dihydroxy-9,10-anthraquinone were commercially obtained and used without further purification. 1H and 13C NMR spectra were obtained using a BRUKER ADVANCE 300 spectrometer. IR spectra were obtained using a BIO-RAD FTS-40 spectrometer. Mass spectrometric data were obtained using a HP1100 LC-API4000 TQ Mass Spectrometer. Elemental analyses were performed using a Perkin-Elmer 2400 analyzer. X-ray analysis was performed via a Bruker Smart Apex CCD diffractometer. Melting points were measured using a Yanaco MP-500 micro melting point apparatus and uncorrected. UV-vis absorption spectra were obtained using a Shimadzu UV-1700 absorption spectrometer. Fluorescence spectra were obtained using an FLS920 fluorescence spectrometer. Electrochemical experiments were performed using a CHI660 electrochemical analyzer.p-tert-Butylthiacalix[4]arene,53 2-azidoethyl-4-p-toluenesulfonate,54 and 1,8-bis(2-propynyloxy)anthraquinone55 were prepared according to literature methods.
3.2 Synthesis and characterization of I
A mixture of p-tert-butylthiacalix[4]arene (0.150 g, 0.21 mmol), 2-azidoethyl-4-methylbenzenesulfonate (1.000 g, 4.16 mmol), KI (0.697 g, 4.20 mmol), and K2CO3 (0.035 g, 0.25 mmol) in dry acetone (10 mL) was heated under reflux for 120 h and cooled down to room temperature. After the removal of the solvent under reduced pressure, the residue was dissolved in CH2Cl2 and washed with 5% HCl, saturated NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by flash column chromatography (silica gel, CH2Cl2/hexane = 6:1, Rf = 0.4) to obtain the intermediate I in a moderate 56% yield as a white solid, mp: 217–218 °C. IR (KBr, cm−1) νmax: 3127.12 (OH), 2101.00 (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+N−), 1245.29 (Ar–O–C), 1035.52 (C–O–C). 1H NMR (CDCl3, 300 Hz): 0.80 (s, 18H, t-Bu), 1.36 (s, 18H, t-Bu), 3.91 (t, 4H, J = 4.91 Hz, NCH2), 4.77 (t, 4H, J = 4.95 Hz, OCH2), 6.95 (s, 4H, Ar–H), 7.69 (s, 4H, Ar–H), 7.81 (s, 2H, OH). 13C NMR (CDCl3, 75 Hz): 30.73, 31.46, 34.01, 34.19, 51.33, 72.52, 122.04, 128.96, 132.75, 134.56, 142.88, 148.32, 155.45, 155.65. Anal. (%) calcd for C44H54N6O4S4: C, 61.51; H, 6.33; N, 9.78. Found: C, 61.26; H, 6.20; N, 9.71.
N+N−), 1245.29 (Ar–O–C), 1035.52 (C–O–C). 1H NMR (CDCl3, 300 Hz): 0.80 (s, 18H, t-Bu), 1.36 (s, 18H, t-Bu), 3.91 (t, 4H, J = 4.91 Hz, NCH2), 4.77 (t, 4H, J = 4.95 Hz, OCH2), 6.95 (s, 4H, Ar–H), 7.69 (s, 4H, Ar–H), 7.81 (s, 2H, OH). 13C NMR (CDCl3, 75 Hz): 30.73, 31.46, 34.01, 34.19, 51.33, 72.52, 122.04, 128.96, 132.75, 134.56, 142.88, 148.32, 155.45, 155.65. Anal. (%) calcd for C44H54N6O4S4: C, 61.51; H, 6.33; N, 9.78. Found: C, 61.26; H, 6.20; N, 9.71.
3.3 Synthesis and characterization of 1
To a stirred suspension of CuI (0.014 g, 0.08 mmol) and Et3N (0.18 mL, 3.02 mmol) in THF/H2O (12 mL, v/v = 3:1), a solution of 5 (0.130 g, 0.15 mmol) in dry THF (10 mL) and the other solution of 1,8-bis(2-propynyloxy)anthraquinone (0.048 g, 0.15 mmol) in dry THF (10 mL) were added at the same time. The resulting mixture was stirred for 4 h at 60 °C. After removal of the solvent under reduced pressure, the residue was dissolved in CH2Cl2 and washed with 5% HCl, saturated NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by flash column chromatography (silica gel, CH2Cl2/acetone = 2:1, Rf = 0.5) to obtain 1 in 51% yield as a yellow solid, mp: 247–249 °C. IR (KBr, cm−1) νmax: 3133.03 (OH), 1672.46 (CO), 1586.75 (NN), 1242.81 (Ar–O–C), 1040.64 (C–O–C). 1H NMR (DMSO-d6, 300 Hz): 0.69 (s, 18H, t-Bu), 1.30 (s, 18H, t-Bu), 4.90 (br s, 4H, NCH2), 5.06 (br s, 4H, OCH2), 5.49 (s, 4H, OCH2), 6.88 (s, 4H, Ar–H), 7.25 (s, 2H, OH), 7.47–7.68 (m, 10H, AQ–H, Ar–H, overlap), 8.48 (s, 2H, Tri–H). ESI-MS: m/z [M]+ calcd 1174.4, found: 1175.8 [M + 1]+, 1198.2 [M + Na]+. Anal. (%) calcd for C64H66N6O8S4: C, 65.39; H, 5.66; N, 7.15. Found: C, 65.10; H, 5.70; N, 7.18.
3.4 Synthesis and characterization of 2
A suspension of 1 (0.120 g, 0.10 mmol) and NaH (0.020 g, 0.82 mmol) in THF (6 mL) was stirred for 5 min, and then methyl bromoacetate (0.10 mL, 1.02 mmol) was added. The resulting mixture was stirred for 20 h at room temperature. After the removal of the solvent under reduced pressure, the residue was dissolved in CH2Cl2 and washed with 5% HCl, saturated NaHCO3, and brine. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by flash column chromatography (silica gel, EtOAc/hexane = 1:1, Rf = 0.4) to obtain compound 2 in 82% yield as a yellow solid, mp: 284–286 °C. IR (KBr, cm−1) νmax: 1759.02 (O–CO), 1673.31 (CO), 1587.68 (NN), 1241.83 (Ar–O–C), 1041.83 (C–O–C). 1H NMR (CDCl3, 300 Hz): 0.87 (s, 18H, t-Bu), 1.38 (s, 18H, t-Bu), 3.72 (s, 6H, CH3), 4.74 (s, 4H, OCH2), 4.83 (t, 4H, J = 7.97 Hz, NCH2), 5.34 (s, 4H, OCH2), 5.45 (t, 4H, J = 7.94 Hz, OCH2), 6.97 (s, 4H, Ar–H), 7.48 (d, 2H, J = 8.22 Hz, AQ–H), 7.71 (t, 2H, J = 7.91 Hz, AQ–H), 7.80 (s, 4H, Ar–H), 7.95 (d, 2H, J = 7.56 Hz, AQ–H), 8.43 (s, 2H, Tri–H). 13C NMR (CDCl3, 75 Hz): 30.80, 31.40, 33.97, 34.49, 49.26, 52.17, 65.24, 72.82, 72.87, 120.36, 121.42, 124.26, 125.27, 128.94, 129.54, 130.97, 133.10, 134.04, 134.93, 135.83, 143.55, 147.18, 147.58, 155.82, 158.01, 160.02, 169.41, 181.88, 183.78. ESI-MS: m/z [M]+ calcd 1318.4, found: 1320.1 [M + 2]+, 1341.8 [M + Na]+. Anal. (%) calcd for C70H74N6O12S4: C, 63.71; H, 5.65; N, 6.37. Found: C, 63.52; H, 5.38; N, 6.51.
3.5 Synthesis and characterization of 3
To a solution of 2 (0.200 g, 0.15 mmol) in THF (6 mL), KOH (0.021 g, 0.38 mmol) dissolved in water (1 mL) was added, and the mixture was stirred for 17 h at room temperature. After removal of the solvent under reduced pressure, the residue was dissolved in CH2Cl2 and washed with 5% HCl and brine. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by recrystallization to obtain compound 3 in 93% yield as a yellow solid, mp: 220–222 °C. IR (KBr, cm−1) νmax: 1746.04 (O–CO), 1671.30 (CO), 1587.62 (NN), 1242.54 (Ar–O–C), 1047.86 (C–O–C). 1H NMR (CDCl3, 300 Hz): 0.79 (s, 18H, t-Bu), 1.30 (s, 18H, t-Bu), 4.53 (br s, 4H, OCH2), 4.75 (s, 4H, NCH2), 4.90 (s, 4H, OCH2), 5.61 (br s, 4H, OCH2), 6.82 (s, 4H, Ar–H), 7.51 (s, 4H, Ar–H), 7.62 (br s, 4H, AQ–H), 7.92 (br s, 2H, AQ–H), 8.43 (s, 2H, Tri–H). 13C NMR (CDCl3, 75 Hz): 30.76, 31.30, 34.03, 34.42, 49.97, 62.30, 71.84, 75.60, 119.10, 119.93, 123.24, 126.46, 128.36, 129.03, 133.22, 133.86, 134.37, 136.47, 143.17, 148.02, 148.09, 155.30, 157.47, 157.80, 169.46, 182.34, 183.02. ESI-MS: m/z [M]+ calcd 1290.4, found: 1292.1 [M + 2]+, 1313.8 [M + Na]+. Anal. (%) calcd for C68H70N6O12S4: C, 63.23; H, 5.46; N, 6.51. Found: C, 62.98; H, 5.22; N, 6.33.
3.6 Crystallography
Crystal data for 2, C71H78N6O13S4, M = 1351.63, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 15.083(4) Å, b = 15.371(4) Å, c = 16.395(4) Å, α = 66.767(3)°, β = 81.148(4)°, γ = 80.089(4)°, V = 3425.3(14) Å3, μ = 0.206 mm−1, Z = 2, T = 173(2) K, crystal size 0.32 × 0.31 × 0.12 mm3, θ range 1.84–25.50°. Herein, 17948 reflections were obtained, of which 12489 reflections were unique (Rint = 0.0351). Final R indices [I > 2σ(I)]: R1 = 0.0652, wR2 = 0.1525, R indices (all data): R1 = 0.1098, wR2 = 0.1796, and goodness of fit on F2 = 1.022. CCDC: 863160.
, a = 15.083(4) Å, b = 15.371(4) Å, c = 16.395(4) Å, α = 66.767(3)°, β = 81.148(4)°, γ = 80.089(4)°, V = 3425.3(14) Å3, μ = 0.206 mm−1, Z = 2, T = 173(2) K, crystal size 0.32 × 0.31 × 0.12 mm3, θ range 1.84–25.50°. Herein, 17948 reflections were obtained, of which 12489 reflections were unique (Rint = 0.0351). Final R indices [I > 2σ(I)]: R1 = 0.0652, wR2 = 0.1525, R indices (all data): R1 = 0.1098, wR2 = 0.1796, and goodness of fit on F2 = 1.022. CCDC: 863160.
3.7 UV-vis/fluorescence tests
Stock solutions of the metal ions (5.0 × 10−3 M) were prepared in MeCN (for 1 and 2) and deionized water (for 3). A stock solution of 1 and 2 (5.0 × 10−5 M) was prepared in CH2Cl2/MeCN (1:1, v/v). A stock solution of 3 (5.0 × 10−5 M) was prepared in THF/H2O (9:1, v/v, pH = 7.4, HEPES). For fluorescence measurements, excitation was provided at 375 nm, and emission was obtained at 427 nm, 428 nm, and 484 nm, respectively, for 1, 2, and 3.
3.8 Electrochemistry tests
Electrochemical measurements were performed using a solution of 1, 2 (5.0 × 10−4 M) in CH2Cl2/DMSO (3:1, v/v), and 3 (5.0 × 10−4 M) in THF/H2O (9:1, v/v, pH = 7.4, HEPES) with 0.1 M n-Bu4NPF6 as a supporting electrolyte, Hg/Hg2Cl2 as the reference electrode, along with platinum working and auxiliary electrodes. Stock solutions of the metal ions (5.0 × 10−2 M) were prepared in DMSO (for 1 and 2) and deionized water (for 3). CV was scanned at 200 mVs−1. DPV was carried out with a 50 mV pulse amplitude at a scan rate of 5 mVs−1.
3.9 Theoretical calculations
The theoretical calculations were performed using the Gaussian 03 software package. The molecular structures of receptors 1 and 2 as well as their complexes were fully optimized using the AM1 (Austin Model vs.1) method with the tight convergence criteria in the gas-phase. The influence of counter-ions was not considered. This semi-empirical method is popularly used for the modeling of large molecular systems as it is a very convenient and time-saving technique.4. Conclusions
In summary, three novel cone thiacalix[4]pseudocrown receptors 1–3, bearing an anthraquinone function as an optical and electrochemical sensing probe and two 1,2,3-triazole rings as key binding sites, have been prepared. Through accommodating the crown cavity to alter the binding ability towards various ions, two cone thiacalix[4]pseudocrown receptors, 1 and 2, were obtained to monitor Pb2+ and Zn2+ ions, respectively, with high selectivity. This provides an insight into the regulation of the binding specificity of receptors by exchanging their side arms. Moreover, the better water-solubility of 3 is of importance for further development of this type of receptors.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant No. 21372147) and the Undergraduate Innovative Research Training Program of Shandong Normal University (grant No. 201610445067).Notes and references
- A. Caballero, R. Martínez, V. Lloveras, I. Ratera, J. Vidal-Gancedo, K. Wurst, A. Tárraga, P. Molina and J. Veciana, J. Am. Chem. Soc., 2005, 127, 15666–15667 CrossRef CAS PubMed.
- K. Huang, H. Yang, Z. Zhou, M. Yu, F. Li, X. Gao, T. Yi and C. Huang, Org. Lett., 2008, 10, 2557–2560 CrossRef CAS PubMed.
- F. Ge, H. Ye, J.-Z. Luo, S. Wang, Y.-J. Sun, B.-X. Zhao and J.-Y. Miao, Sens. Actuators, B, 2013, 181, 215–220 CrossRef CAS.
- C. Arivazhagan, R. Borthakur and S. Ghosh, Organometallics, 2015, 34, 1147–1155 CrossRef CAS.
- Y. Fang, Y. Zhou, Q. Rui and C. Yao, Organometallics, 2015, 34, 2962–2970 CrossRef CAS.
- R. J. P. Williams and J. J. R. F. da Silva, Coord. Chem. Rev., 2000, 200–202, 247–348 CrossRef CAS.
- J. J. R. Fraústo da Silva and R. J. P. Williams, Zinc: Lewis Acid Catalysis and Regulation, in The Biological Chemistry of Elements: The Inorganic Chemistry of Life, Oxford University Press, New York, 2nd edn, 2001 Search PubMed.
- P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205–229 CrossRef CAS.
- N. C. Lim, H. C. Freake and C. Brückner, Chem.–Eur. J., 2005, 11, 38–49 CrossRef PubMed.
- Z. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996–2006 RSC.
- A. R. Flegal and D. R. Smith, Environ. Res., 1992, 58, 125–133 CrossRef CAS PubMed.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- C.-Y. Li, Y. Zhou, Y.-F. Li, X.-F. Kong, C.-X. Zou and C. Weng, Anal. Chim. Acta, 2013, 774, 79–84 CrossRef CAS PubMed.
- T. Sun, Q. Niu, Z. Guo and T. Li, Tetrahedron Lett., 2017, 58, 252–256 CrossRef CAS.
- N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed.
- M. Kumar, N. Kumar and V. Bhalla, Dalton Trans., 2013, 42, 981–986 RSC.
- R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824–4870 RSC.
- H. Tomiyasu, J.-L. Zhao, X.-L. Ni, X. Zeng, M. R. J. Elsegood, B. Jones, C. Redshaw, S. J. Teat and T. Yamato, RSC Adv., 2015, 5, 14747–14755 RSC.
- M. Yamada, M. R. Gandhi, U. M. R. Kunda and F. Hamada, J. Inclusion Phenom. Macrocyclic Chem., 2016, 85, 1–18 CrossRef CAS.
- J.-L. Zhao, C. Wu, X. Zeng, S. Rahman, P. E. Georghiou, M. R. J. Elsegood Mr, T. G. Warwick, C. Redshaw, S. J. Teat and T. Yamato, ChemistrySelect, 2016, 1, 1541–1547 CrossRef CAS.
- M. Zhao, J. Lv and D.-S. Guo, RSC Adv., 2017, 7, 10021–10050 RSC.
- S. M. Darjee, D. R. Mishra, K. D. Bhatt, D. J. Vyas, K. M. Modi and V. K. Jain, Tetrahedron Lett., 2014, 55, 7094–7098 CrossRef CAS.
- M. Kumar, N. Kumar and V. Bhalla, Sens. Actuators, B, 2012, 161, 311–316 CrossRef CAS.
- F. Miao, J. Zhan, Z. Zou, D. Tian and H. Li, Tetrahedron, 2012, 68, 2409–2413 CrossRef CAS.
- J.-L. Zhao, H. Tomiyasu, C. Wu, H. Cong, X. Zeng, S. Rahman, P. E. Georghiou, D. L. Hughes, C. Redshaw and T. Yamato, Tetrahedron, 2015, 71, 8521–8527 CrossRef CAS.
- M.-Q. Ran, J.-Y. Yuana, Y.-H. Zhao, L. Mou, X. Zeng, C. Redshaw, J. L. Zhao and T. Yamato, Supramol. Chem., 2016, 28, 418–426 CrossRef CAS.
- D.-S. Guo, Z.-P. Liu, J.-P. Ma and R.-Q. Huang, Tetrahedron Lett., 2007, 48, 1221–1224 CrossRef CAS.
- B.-T. Zhao, Z. Zhou, Z.-N. Yan, E. Belhadj, F. L. Derf and M. Sallé, Tetrahedron Lett., 2010, 51, 5815–5818 CrossRef CAS.
- V. K. Gupta, B. Sethi, R. A. Sharma, S. Agarwal and A. Bharti, J. Mol. Liq., 2013, 177, 114–118 CrossRef CAS.
- V. Lamare, J.-F. Dozol, P. Thuéry, M. Nierlich, Z. Asfari and J. Vicens, J. Chem. Soc., Perkin Trans. 2, 2001, 1920–1926 CAS.
- A. Muravev, F. Galieva, O. Bazanova, D. Sharafutdinova, S. Solovieva, I. Antipin and A. Konovalov, Supramol. Chem., 2016, 28, 589–600 CrossRef CAS.
- A. Ghosh, D. A. Jose and R. Kaushik, Sens. Actuators, B, 2016, 229, 545–560 CrossRef CAS.
- D. Bethell, G. Dougherty and D. C. Cupertino, J. Chem. Soc., Chem. Commun., 1995, 675–676 RSC.
- D. Y. Han, J. M. Kim, J. Kim, H. S. Jung, Y. H. Lee, J. F. Zhang and J. S. Kim, Tetrahedron Lett., 2010, 51, 1947–1951 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. Wang, D. Ikhlef, S. Kahlal, J.-Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- F. W. B. van Leeuwen, H. Beijleveld, H. Kooijman, A. L. Spek, W. Verboom and D. N. Reinhoudt, J. Org. Chem., 2004, 69, 3928–3936 CrossRef CAS PubMed.
- S. G. Bott, S. J. Obrey, A. P. Marchand and K. A. Kumar, J. Chem. Crystallogr., 1996, 26, 677–681 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- S. Maruyama, K. Kikuchi, T. Hirano, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2002, 124, 10650–10651 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed.
- P. Job, Ann. Chim., 1928, 9, 113–203 CAS.
- V. M. S. Gil and N. C. Oliveira, J. Chem. Educ., 1990, 67, 473–478 CrossRef CAS.
- Y. Liu, B.-H. Han and Y.-T. Chen, J. Phys. Chem. B, 2002, 106, 4678–4687 CrossRef CAS.
- J. Jia, X. Tang, Y. He, M. Zhang and G. Xing, Chin. J. Org. Chem., 2012, 32, 1803–1811 CrossRef CAS.
- Y. Xu, L. Xiao, S. Sun, Z. Pei, Y. Peia and Y. Pang, Chem. Commun., 2014, 50, 7514–7516 RSC.
- A. Sil, A. Maity, D. Giri and S. K. Patra, Sens. Actuators, B, 2016, 226, 403–411 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision B.03, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healey and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971–3972 CrossRef CAS.
- B. Carboni, M. Vaultier and R. Carrié, Tetrahedron, 1987, 43, 1799–1810 CrossRef CAS.
- H. Sharghi, R. Khalifeh and M. M. Doroodmand, Adv. Synth. Catal., 2009, 351, 207–218 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral characterisation of compounds; protocols for UV-vis absorption, fluorescence emission spectra, 1H NMR and electrochemical titrations, as well as the calculated structure. CCDC 863160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06083a |
| ‡ Both authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |