
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine derivatives as PLK4 inhibitors for the treatment of breast cancer†
Zhihao Liu‡
,
Qian Lei‡,
Wei Wei,
Lu Xiong,
Yaojie Shi,
Guoyi Yan,
Chao Gao,
Tinghong Ye,
Ningyu Wang* and
Luoting Yu *
*
State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University and Collaborative Innovation Center, Chengdu 610041, China. E-mail: yuluot@scu.edu.cn; ningyuwang_sklb@scu.edu.cn; Fax: +86 28 8516 4060; Tel: +86 28 8516 4063
First published on 24th May 2017
Abstract
Polo-like kinase 4 (PLK4), a vital regulator of centriole duplication, is important for maintaining genome stability. Dysregulation of PLK4 has been found in several human cancers and is associated with a predisposition to tumorigenesis. Herein, we describe the discovery of (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine derivatives as potent PLK4 inhibitors with more concise structure using a scaffold hopping strategy. SAR exploration and preliminary assessment identified 14i as a new PLK4 inhibitor which displayed excellent potency in vitro. 14i could inhibit the activity of PLK4, perturb centriole replication, result in mitosis disorder and induce cell apoptosis in breast cancer cells. Moreover 14i demonstrated significant antitumor efficacy in the MDA-MB-468 and MDA-MB-231 xenograft models. This study suggested that this concise chemotype would represent a promising scaffold of PLK4 inhibitors for cancer therapy and 14i would be an attractive lead compound for further optimization and evaluation.
Introduction
The polo-like kinase family belongs to a class of highly conserved serine/threonine kinases which consists of five subtypes in mammals: PLK1, PLK2/Snk, PLK3/Fnk/Prk, PLK4/Sak and PLK5.1 They are involved in the regulation of cell cycle progression, including intersection of mitotic, spindle formation, cytokinesis, and are often deregulated in cancer.2 The N-terminus of PLKs has a highly homologous serine/threonine kinase catalytic domain except PLK5;3 and the C-terminus of PLKs contains one or two signature polo-box domains which are related to the location of PLKs into specific mitotic subcellular structures during mitosis.4,5One of the PLKs members, PLK4, shows obviously differences compared with other PLKs at the amino acid sequence level which contains only one polo-box domain instead of two.6,7 PLK4 is a main regulator of centriole duplication during mitosis and cell cycle.8–10 PLK4 is usually expressed in proliferative tissues with low abundance, but is aberrantly expressed in many tumor types, especially in breast cancer.11 High PLK4 levels in breast cancers are associated with poor prognosis and survival.12–14
Dysregulation of PLK4 often causes disorder of centrosome duplication and aneuploidy formation, thereby promoting genomic instability which is a characteristic observed in many types of cancers.15,16 Depletion of PLK4 by RNA interference in breast cancer cells and xenografts induces cell apoptosis and tumor growth suppression.16 Besides, PLK4 is crucial to PTEN-deficient cancer cells for survival suggesting that there is a synthetic lethal interaction between PLK4 and PTEN in breast cancer cells.17,18 Thus, PLK4 could be a potential target for breast cancer and draw our attention to discover PLK4 inhibitors as candidates against breast cancer.
Some typical PLK4 inhibitors are shown in Fig. 1. Multikinase inhibitor 1 (R1530) has been shown to be potent against PLK4 with an IC50 value of 6 nM. It could promote cancer cells genomic instability and facilitate cancer cells sensitive to chemotherapy.19 2 (Centrinone), a selective PLK4 inhibitor, could result in a p53-dependent cell cycle arrest in G1.20 And 3 (Axitinib), a well-known pan-VEGFR inhibitor, has been approved for renal cell carcinoma (RCC) by FDA and demonstrated collateral inhibitory activity against PLK4 with a Ki value of 4.2 nM.21 Recently, 4 (CFI-400437) with a scaffold of (E)-3-((1H-indazol-6-yl)methylene)indolin-2-ones was reported to be a PLK4 inhibitor with an IC50 value of 6 nM,22 and its spirooxindole counterparts 5, 6, and 7 exhibited excellent selectivity and potency.23–25 6 (CFI-400945) has demonstrated significant efficacy in animal models of breast cancer, colorectal cancer and pancreatic cancer, especially in PTEN-deficient cancer patient-derived xenografts.17,24,26
 | ||
| Fig. 1 Chemical structures of representative PLK4 inhibitors. | ||
To our knowledge, the reported scaffolds of PLK4 inhibitors are not diverse. What is more, although some bioactive molecules such as 1 (R1530) and 3 (Axitinib) demonstrated good inhibitory activity against PLK4, their main targets are not PLK4. Despite preferentially isolated as the E-isomer, the indazolylmethyleneindolinone compound 4 has the potential to isomerize in vivo.23 5, 6, and 7, as the spirooxindole counterparts of 4, own improved configurational stability and physicochemical properties, but their synthesis and purification are more complicated due to the spirooxindole motif with multiple chiral centers.27 Based on above status quo, we wished to explore a more concise scaffold of selective PLK4 inhibitors.
We firstly investigated the co-crystallization structure (PDB code 4JXF) to understand interactions between 5 and PLK4 kinase domain (Fig. 2). As a hinge binder, indazole ring of 5 formed two hydrogen bonds with the backbone carbonyl of Cys92 and NH of Glu90 in the hinge region. The arylvinyl group potentially fit into the solvent-exposed side of the adenine binding pocket, and the aryl fragment had a strong π-cation interaction with amino of Arg28. It is notable that all of 3, 4, 6 have the same motif of (E)-3-arylvinyl-1H-indazole as 5. Moreover, the carbonyl and NH of oxindole in 5 could form hydrogen bonds with side chain of Lys41 and the backbone carbonyl of Gln160, respectively. And 3, 4, 6 also have corresponding fragments to interact potentially with both these two amino acid residues (Fig. 3). Hence, we speculated that 3–6 might interact with PLK4 kinase domain in the same binding mode.
 | ||
| Fig. 2 X-ray crystal structure of 5 in the active site of PLK4 with interactions to the key residues (PDB code 4JXF). | ||
 | ||
| Fig. 3 Designed strategy of title molecules. | ||
Based on above analyses, to quest for a new and concise chemotype of PLK4 inhibitors, we kept (E)-3-arylvinyl-1H-indazole motif as hinge binder and focused on utilizing scaffold hopping strategy to simplify the skeletons at 6-position of indazole in 3–6 simultaneously retaining pharmacophores which could form H-bonds with Lys41 and Gln160 in our preconceived scaffold. After careful consideration, we cautiously incorporated 2-aminepyrimidin-4-yl at 6-position of indazole in order to facilitate interactions between compounds and PLK4 (Fig. 3). Herein we report the design, synthesis and biology evaluation of (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine derivatives as potent PLK4 inhibitors.
Results and discussion
Synthesis
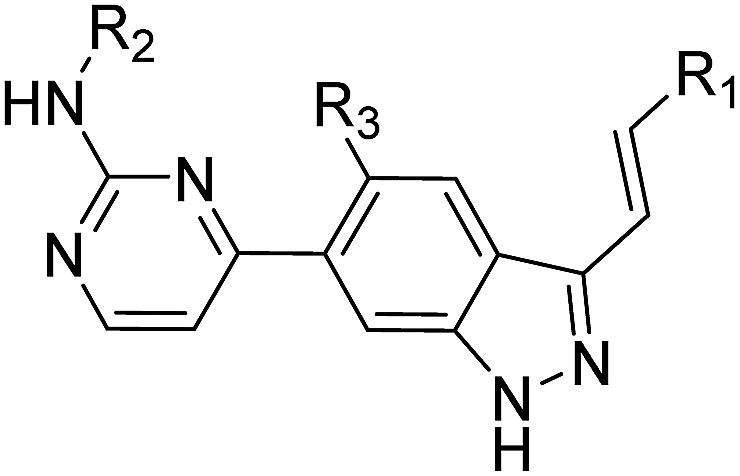
The synthesis of target compounds 14a–14y and 15a–15d is depicted in Scheme 1. Commercially available 6-bromo-1H-indazoles 8a–8b were reacted with bis(pinacolato)diboron using Pd(dppf)Cl2 and potassium acetate in dioxane at 90 °C respectively to afford corresponding boronic acid pinacol esters 9a–9b. Suzuki coupling of 9a–9b with 10a–10c respectively provided products 11a–11d. Then iodination of 11a–11d was carried out in DMF at 60 °C overnight and provided a good yield of intermediates 12a–12d. At last, Suzuki coupling of 12a–12d with trans-hetarylvinylboronates 13a–13y using Pd(dppf)Cl2 and cesium carbonate afforded target products 14a–14y and 15a–15d. | ||
Scheme 1 Synthetic route of title compounds 14a–14y, 15a–15d. Reagents and conditions: (a) bis(Pinacolato)diboron, AcOK, Pd(dppf)Cl2, dioxane, 90 °C; (b) Pd(dppf)Cl2, K2CO3, dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 4:1, 95 °C; (c) I2, K2CO3, DMF, 60 °C; (d) Pd(dppf)Cl2, Cs2CO3, dioxane:H2O = 4:1, 100 °C. :H2O = 4:1, 95 °C; (c) I2, K2CO3, DMF, 60 °C; (d) Pd(dppf)Cl2, Cs2CO3, dioxane:H2O = 4:1, 100 °C. | ||
Study on SAR
The title compounds were evaluated for their PLK4 potency as well as anti-proliferative potencies against MDA-MB-231 and MDA-MB-468 cell lines, and 3 (Axitinib) was used as a positive control. Axitinib demonstrated PLK4 potency with an IC50 value of 41.3 nM and moderate anti-proliferative activities against MDA-MB-231 and MDA-MB-468 cell lines. The SAR study began with the terminal aryl group of arylvinyl indazole motif. Compared with 3 (Axitinib), 14a containing a single phenyl displayed moderate PLK4 potency with an IC50 value of 74.9 nM, and it also exhibited anti-proliferative activities against MDA-MB-231 and MDA-MB-468 cell lines (IC50 = 6.2 and 8.7 μM, respectively), implying that our scaffold hopping strategy was feasible. Incorporation of a methoxy group at the 4-position of phenyl ring in 14a improved PLK4 potency but its cellular activity declined (14b), 3-methylamino substituted analogue 14c showed similar PLK4 potency with 14a. The phenyl ring in 14a replaced with indole (14d) resulted in a dramatic drop in PLK4 potency although its cellular activity improved slightly. Substituents on para-position of the phenyl ring with hydrophilic groups such as morpholino (14e) and N-methylpiperazine (14f) improved both PLK4 and cellular activities dramatically. The same tendency could also be observed in benzyl analogues 14g, 14h and 14i. However, meta-substituted phenyl analogues (14j, 14k) exhibited declined PLK4 and cellular activities, suggesting that the orientation of hydrophilic groups is very important for the interaction with PLK4.The pyridin-3(or 4)-yl analogues 14l and 14m were slightly better than 14a in activity. The H atom at 2-position of pyridine in 14m replaced with morpholino group (14n) or piperazinyl group (14o) improved the activity, but both PLK4 and cellular potencies declined with the increasing size of substituents, as exemplified by the N-methylpiperazinyl (14p) and N-Bocpiperazinyl (14q) analogues, suggesting a suitable size of group on this position was favorable. Among these compounds, 14i demonstrated excellent inhibitory activity against PLK4 (IC50 = 11.2 nM) and good potencies against MDA-MB-231 and MDA-MB-468 cell lines (IC50 = 0.09 and 0.06 μM, respectively). Then the influence of substituents on 2- and 3-position of the phenyl ring in 14i was investigated. Introduction of fluorine atom (14r & 14v), chlorine atom (14s & 14w), methyl group (14t & 14x) and methoxy (14u & 14y) on 2-position or 3-position impaired the enzymatic potency to the same extent, but a sharp loss of cellular potency was observed with respect to the 2-substituted groups (14r–14u). These results indicated it is the steric effect rather than the electric effect of substituents on the phenyl ring that was crucial to the PLK4 potency, because the hydrophobic pocket near the phenyl ring might be too narrow to accommodate any bulky substituents (Table 1).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R | PLK4 (IC50, nM) | Breast cancer cells IC50 (μM) | Entry | R | PLK4 (IC50, nM) | Breast cancer cells IC50 (μM) | ||
| MDA-MB-231 | MDA-MB-468 | MDA-MB-231 | MDA-MB-468 | ||||||
| a PLK4 IC50 values were determined from Kinase Profiler of Reaction Biology Corporation and all activity data represent the mean values of two independent experiments. | |||||||||
| 14a |  |
74.9 | 6.20 | 8.70 | 14n |  |
28.1 | 1.40 | 0.54 |
| 14b |  |
57.6 | >10 | >10 | 14o |  |
16.3 | 0.62 | 0.34 |
| 14c |  |
80.1 | 2.50 | 1.00 | 14p |  |
35.6 | 0.43 | 0.35 |
| 14d |  |
190 | 3.00 | 1.50 | 14q |  |
176 | 8.70 | 3.1 |
| 14e | 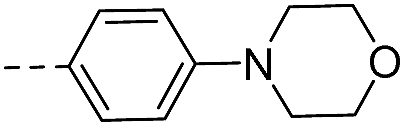 |
16.4 | 1.62 | 0.77 | 14r |  |
19.3 | 0.12 | 0.09 |
| 14f | 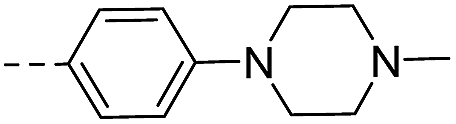 |
26.2 | 0.46 | 0.32 | 14s |  |
31.0 | 1.80 | 0.87 |
| 14g |  |
15.0 | 0.87 | 0.38 | 14t |  |
36.5 | 3.16 | 2.20 |
| 14h | 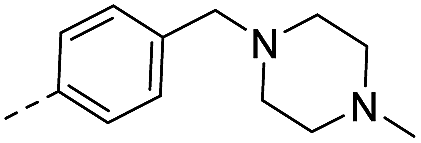 |
14.2 | 0.46 | 0.15 | 14u |  |
39.7 | 1.20 | 0.34 |
| 14i |  |
11.2 | 0.09 | 0.06 | 14v |  |
36.5 | 0.26 | 0.17 |
| 14j |  |
33.0 | 0.40 | 0.20 | 14w | 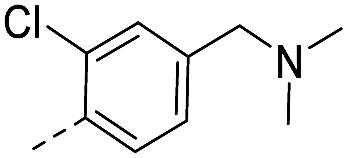 |
33.6 | 0.38 | 0.29 |
| 14k |  |
32.4 | 3.10 | 1.00 | 14x |  |
34.8 | 0.44 | 0.20 |
| 14l |  |
50.4 | 1.68 | 1.40 | 14y |  |
40.4 | 0.20 | 0.11 |
| 14m |  |
68.7 | 4.20 | 2.12 | Axitinib | 41.3 | >10 | 5.0 | |

Next, we investigated the influence of substituents on pyrimidin-2-amine and 5-position of indazole ring based on the structure of 14i (Table 2). The acetamino analogues of 14i or 14r (15a, 15b) and the methylamino analogue of 14i (15c) were slightly less potent than their corresponding counterparts, suggesting that substituents on this position might be tolerated. Introduction of methyl group at 5-position of indazole ring (15d) resulted in a dramatic drop in PLK4 potency (IC50 = 956 nM) and antiproliferative activities against MDA-MB-231 and MDA-MB-468 cell lines (IC50 = 8.60 and 5.43 μM, respectively). Interestingly, almost all of these compounds showed better anti-proliferative effects against MDA-MB-468 (PTEN-deficient cancer cells) than MDA-MB-231 (PTEN-wild type cancer cells), which implicated that PTEN deficient breast cancer cells could be more sensitive to PLK4 inhibitors like 14i because of the synthetic lethal interaction between PLK4 and PTEN.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | PLK4 (IC50, nM) | Breast cancer cells IC50 (μM) | |
| MDA-MB-231 | MDA-MB-468 | |||||
| a PLK4 IC50 values were determined from Kinase Profiler of Reaction Biology Corporation and all activity data represent the mean values of two independent experiments. | ||||||
| 15a |  |
 |
H– | 22.2 | 0.18 | 0.10 |
| 15b |  |
 |
H– | 54.5 | 0.30 | 0.14 |
| 15c |  |
CH3– | H– | 164 | 2.90 | 2.78 |
| 15d |  |
H– | CH3– | 956 | 8.60 | 5.43 |
| 14i |  |
H– | H– | 11.2 | 0.09 | 0.06 |
Solubility, Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P and LLE evaluation
P and LLE evaluation
Due to the preference of in vitro enzymatic and cellular potencies, compounds 14i, 14r, and 15a were selected to evaluate their solubility in water under different pH. And another two important parameters of drug-likeness, ClogP and LLE (ligand-lipophilicity efficiency), were also calculated28 (Table 3).
P, LLE and solubility of 14i, 14r and 15a
Both compounds 14i and 15a exhibited comparable ClogP and LLE, as well as solubility in H2O, which were much better than 14r. 14i was finally selected for further evaluation because of its excellent in vitro activity and molecular properties. Besides, 14i also exhibited good selectivity for PLK4 over other members of polo-like kinase family (Table 4).
| Entry | Inh% (100 nM) | |||
|---|---|---|---|---|
| PLK1 | PLK2 | PLK3 | PLK4 | |
| a Note: all compounds were tested at ATP concentration of 10 μM. The percent of inhibition is an average of two independent titrations. PLK5 does not have kinase domain, so it wasn't tested. | ||||
| 14i | 3.59 | 45.08 | 2.99 | 96.21 |
Molecular docking of 14i with PLK4
To understand the interaction of (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine derivatives with PLK4, we modelled the binding mode of 14i in the ATP pocket using a reported crystal structure of the PLK4 kinase domain (PDB ID: 4JXF). As shown in Fig. 4, 14i maintained H-bonding interactions with PLK4 at the backbone carbonyl of Cys92 and NH of Glu90 in the hinge region. Interestingly, unlike 5, the amino group of 14i formed two hydrogen bonds with the backbone carbonyls of Gln160 and Ala153, and the N atom of 1-pyrimidine in 14i had an interaction with crystalline water (HOH 424) instead of forming H-bond with Lys41. Furthermore, trans-arylvinyl group in 14i potentially extended to the solvent-exposed side. Overall, 14i could fit well into the ATP pocket of PLK4, which enable it to be a potent PLK4 inhibitor. | ||
| Fig. 4 Proposed binding mode of compound 14i to the PLK4 kinase domain. | ||
Inhibition of centriole duplication and mitosis by 14i
Because of the crucial role of PLK4 in centriole duplication, we examined the effect of 14i on centriole duplication in MDA-MB-468 and MDA-MB-231 cell lines by immunostaining DAPI, centrin2 and γ-tubulin to visualize cell nucleus (blue), centriole (green) and mitotic spindle (red) respectively. After 48 h exposure with 14i, cells with disordered centrosome replication increased in a dose-dependent manner (Fig. 5A), and 0.5 μM of 14i distinctly inhibited centrosome replication (Fig. 5B). | ||
| Fig. 5 (A) Cell numbers of abnormal centrosome replication after treated with 14i. (B) Effects of 14i on perturbing centrosome replication by immunofluorescence. MDA-MB-468 and MDA-MB-231 cells were treated with 14i or DMSO for 48 h. Cells were stained with centrin2, γ-tubulin, and DAPI. Insets show higher magnification views of the centrioles (boxed regions). | ||
By analysis of DNA content of MDA-MB-468 and MDA-MB-231 cells using flow cytometry, we found 14i induced polyploid formation in a dose-dependent manner (Fig. 6A), suggesting that 14i could result in mitotic disorder and promote genomic instability. Moreover, immunoblot analysis displayed the increase of activated cleaved-caspase-3 which represents the activation of apoptotic pathways and the reduction of P-H3 which indicates the unsmooth progression of mitosis in a does dependent manner after treated with 14i (Fig. 6B and C). Overall, 14i inhibited the activity of PLK4 thereby perturb the centriole replication and led to disorder of mitosis, which further induced cell apoptosis in MDA-MB-468 and MDA-MB-231 cells.
 | ||
| Fig. 6 (A) DNA content analysis by flow cytometry. MDA-MB-468 and MDA-MB-231 cells were treated with 14i or DMSO for 48 h prior to staining with propidium iodide. (B) and (C) MDA-MB-468 and MDA-MB-231 cells lysates were analyzed by immunoblot analysis with antibodies against P-H3 and CC3 (cleaved-caspase-3). | ||
In vivo antitumor activity of 14i
We finally evaluated the in vivo antitumor activity of compound 14i in MDA-MB-468 (PTEN-deficient breast cancer cells) and MDA-MB-231 (PTEN-wild type breast cancer cells) xenograft models. Compound 14i was administered orally at doses of 30 mg kg−1 or 90 mg kg−1 once daily for 18 consecutive days. The results showed that 14i significantly suppressed tumor growth in MDA-MB-468 and MDA-MB-231 xenograft models. Compared with MDA-MB-231 xenograft model, the tumor growth inhibition rate (TGI) of MDA-MB-468 xenograft was much higher, with TGI of 83.1% versus 67.6% and 68.3% versus 63.9% at the doses of 30 mg kg−1 and 90 mg kg−1 respectively (Fig. 7), which also confirmed that PTEN-deficient tumors were susceptible to PLK4 inhibitors in vivo. Moreover, no significant body weight loss was observed at both doses (Fig. 8). | ||
| Fig. 7 In vivo antitumor efficacy of 14i against MDA-MB-468 (A) and MDA-MB-231(B) tumor xenograft models (5 mice per group). Points indicate mean tumor volume (mm3); bars indicate SD. | ||
 | ||
| Fig. 8 Body weight measurements in vivo antitumor experiments. Note: results are expressed as the mean ± SEM (n = 5 for the inhibitor-treated group, n = 5 for the vehicle control group). | ||
Conclusion
In summary, using scaffold hopping strategy, we discovered (E)-4-(3-arylvinyl-1H-indazol-6-yl)pyrimidin-2-amine derivatives as a more concise chemotype of PLK4 inhibitors. SAR explorations and preliminary physicochemical properties assessment led to the identification of 14i as a new PLK4 inhibitor with potent PLK4 activity (IC50 = 11.2 nM) and significant antiproliferative effects (MDA-MB-231 IC50 = 0.09 and MDA-MB-468 IC50 = 0.06 μM, respectively). Besides, 14i exhibited good selectivity for PLK4 over other members of polo-like kinase family. Furthermore, 14i could perturb the centriole replication and induce disorder of mitosis as well as cancer cells apoptosis. Moreover, 14i demonstrated significant antitumor efficacy in the MDA-MB-468 and MDA-MB-231 xenograft models. All these data indicated that this chemotype would be a promising starting point for the development of PLK4 inhibitors as anticancer agents and 14i would be an attractive lead compound for further optimization and evaluation.Experimental
Synthesis of 9a–9b
To a solution of 8a–8b (40 mmol), bis(pinacolato)-diboron (15.2 g, 60 mmol) and KOAc (7.84 g, 80 mmol) in 1,4-dioxane (100 mL), Pd(dppf)Cl2 (2 mmol) was added and the mixture was degassed for 5 min. The mixture was heated to 90 °C for 8 h and cooled to r.t. The mixture was diluted with ethyl acetate and filtered through celite. The filtrate was washed successively with water and brine, dried over Na2SO4 and concentrated. The residue was purified by chromatography eluting with Pet:EtOAc = 2.5:1 to give products 9a–9b.
Synthesis of 11a–11d
To a solution of 9a–9b (15 mmol), 10a–10c (12.5 mmol) and KOAc (30 mmol) in 1,4-dioxane (100 mL), Pd(dppf)Cl2 (0.62 mmol) was added and the mixture was degassed for 5 min. The mixture was heated to 100 °C for 3 h and cooled to r.t. The mixture was diluted with ethyl acetate and filtered through celite. The filtrate was washed successively with water and brine, dried over Na2SO4 and concentrated. The residue was purified by chromatography eluting with DCM:CH3OH = 12:1 to afford products 11a–11d.
Synthesis of 12a–12d
11a–11d (8.0 mmol) and K2CO3 (16 mmol) were combined in DMF (15 mL), and I2 (16 mmol) dissolved in DMF (2 mL) was added dropwise to the mixture, then stirred 10 hours at 65 °C. The reaction was then poured into a solution of sodium dithionite (3.0 g) and K2CO3 (1.1 g) in water (50 mL). A white precipitate formed and was stirred at room temperature for 30 min. Product was isolated by filtration to give 12a–12d.Synthesis of 14a–14y, 15a–15d
To a solution of 12a–12d (0.36 mmol), trans-arylvinylboronates or trans-hetarylvinylboronates 13a–13y (0.43 mmol) and cesium carbonate (0.72 mmol) in 1,4-dioxane:H2O = 4:1 (15 mL), Pd(dppf)Cl2 (0.04 mmol) was added and the mixture was degassed for 5 min. The mixture was heated to 100 °C for 8 h and cooled to r.t. The mixture was diluted with ethyl acetate and filtered through celite. The filtrate was washed successively with water and brine, dried over Na2SO4 and concentrated. The residue was purified by chromatography eluting with DCM:CH3OH = 10:1 to afford the title compounds 14a–14y, 15a–15d.
Kinase inhibition assays
Kinase inhibition profiles were obtained using Kinase Profiler services provided by Reaction Biology Corporation, and ATP concentrations used are the ATP Km of corresponding kinases. Compounds were tested in a 10-dose IC50 mode with 3-fold serial dilution starting at 10 μM.Cell lines and cell culture conditions
All of cell lines used were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). All of them were cultured in RPIM 1640 or Iscove's Modified Dulbecco's Medium supplemented with 10% fetal bovine serum (FBS, Gibco, Auckland, N.Z.), penicillin-streptomycin (Life Technologies), and 4 mM L-glutamine. The cell lines were cultured in a humidified atmosphere with 5% CO2 at 37 °C.Cell viability assay
The cell viability treated with the compounds described above was measured by MTT assay. 100 μL of medium containing various concentrations of compound was added to 96-well micro-plates, then cells were seeded in each well at a density of 5–10 × 104 cells. After cultured for 72 h, the cells were added 20 μL MTT solution (5 mg mL−1) and incubated for another 2–4 h at 37 °C. When reached the appropriate time, the formazan crystal formed by living cells was dissolved with 50 μL of the 20% SDS solution overnight. Then, the optical density was measured using Spectra MAX M5 micro-plate spectrophotometer (Molecular Devices) at 570 nm and the IC50 values were carried out in Graphpad Prism 5 (GraphPad Software Inc.).Western blot analysis
The tumor cells MDA-MB-231 and MDA-MB-468 were incubated in medium containing different concentration of 14i for 48 h. Then harvested cells washed by PBS twice were lysed in RIPA buffer (Byotime, Beijing, China) for 20 min, then ultrasonicated and centrifugated. The protein concentrations were measured and equalized before loading. Protein was separated by SDS-PAGE gels. After electrophoresis, protein was transferred onto polyvinylidene fluoride membranes (Millipore), blocked in 5% non-fat milk for 2 h, washed in TBS-T and incubated with specific antibodies (Cell Signaling Technology) overnight at 4 °C. After incubated with the relevant secondary antibodies, the reactive bands were detected using an enhanced chemiluminescence kit (Millipore).Immunofluorescence analysis
For immunofluorescence staining, the treated cancer cells in 8 well chamber slides (Millipore) were washed twice in PBS and fixed in 4% paraformaldehyde for 15 min, then cells were blocked in 1% BSA for 1 h at room temperature. Primary antibody in block buffer was added to cells overnight at 4 °C and washed 3 times by PBS. Goat anti-rabbit and goat anti-mouse secondary antibodies conjugated to FITC and Alexa 594 were used to detect primary antibodies. After washed twice with PBS, the cells were counterstained with DAPI, and the images were captured using a Leica confocal microscope equipped with a Leica X63oil-immersion objective.Cell cycle analysis
For the cell cycle analysis, the 14i-treated cells were fixed and stained with PI staining solution for 5 min in the dark and then detected using flow cytometry (FCM). The data were analyzed using the NovoExpress software.Subcutaneous xenograft models
All animal experiments have been approved by the Institutional Animal Care and Treatment Committee of Sichuan University in China (Permit number: 20160188) and were carried out in accordance with the approved guidelines. Six-week-old female BALB/c nude mice were purchased from HFK Biotechnology Company (Beijing, China). Tumors were first prepared by subcutaneously implanting 1 × 107 cells/0.1 mL into the right flank region of mice. When the volume reached 200 mm3, tumors were dissected into fragments of approximated 10–20 mm3 and subsequently implanted into hind flank region of BALB/c nude mice. When the tumor reached 80–110 mm3, mice were selected and randomized into groups for further study (n = 5). Mice were given 14i and blank solvent once daily by oral gavage. Tumor size was monitored every 3 days using calipers and the volume was calculated using the following formula: tumor volume = length × width2 × 0.5.Solubility test29
The production of test solution: each compound was added in excess to 1000 μL solvent. The drug suspensions were placed in ultrasound for 15 min followed by filtration to get the saturated solution. Then the test solutions were obtained by diluting the saturated solution 100 times.The production of reference solution: adding accurate 2.000 mg (mr) compound into 1000 μL methanol. The solutions were placed in ultrasound for 15 min to dissolve completely. Then filtered and the reference solutions were obtained by diluting the filter liquor 100 times.
The concentration of each sample was measured with HPLC-UV-vis apparatus. The test condition is as follows:
Chromatographic column: Venusil XBP (C18) 5 μm 150 Å 4.6 × 250 mm.
Mobile phase: methanol (A) and pH = 6.8H2O (B)
Injection volumes: 40 μL per injection.
Column temperature: 30 °C.
Pressure: 120 Pa.
Flow rate: 1.00 mL min−1.
The calculation formula of solubility is as follows:
C = (mr × At)/(Ar × 1000)
At and Ar are respectively refer to the area of test solution's chromatographic peak and the area of reference solution's chromatographic peak.
Molecular modeling30
The binding mode of the compound 14i and PLK4 was conducted by structure based docking studies, which was carried out by using AutoDock 4.0. The structure of the receptor was taken from PDB (PDB ID: 4JXF). The binding site of the inhibitor was chosen as active site, grid dimensions were 40 × 40 × 40 Å3, which were defined by AutoDock tools.Acknowledgements
This work was supported by China Postdoctoral Science Foundation (No. 2015M570790, No. 2016T90860). We thank Lihua Zhou of State Key Laboratory of Biotherapy (Sichuan University) for NMR measurements.References
- G. D. Cárcer, G. Manning and M. Malumbres, Cell Cycle, 2011, 10, 2255–2262 CrossRef PubMed.
- F. A. Barr, H. H. Silljé and E. A. Nigg, Nat. Rev. Mol. Cell Biol., 2004, 5, 429–440 CrossRef CAS PubMed.
- C. G. De, B. Escobar, A. M. Higuero, L. García, A. Ansón, G. Pérez, M. Mollejo, G. Manning, B. Meléndez and J. Abad-Rodríguez, Mol. Cell. Biol., 2011, 31, 1225–1339 CrossRef PubMed.
- V. Archambault and D. M. Glover, Nat. Rev. Mol. Cell Biol., 2009, 10, 265–275 CrossRef CAS PubMed.
- S. M. Lens, E. E. Voest and R. H. Medema, Nat. Rev. Cancer, 2010, 10, 825–841 CrossRef CAS PubMed.
- G. C. Leung, J. W. Hudson, A. Kozarova, A. Davidson, J. W. Dennis and F. Sicheri, Nat. Struct. Biol., 2002, 9, 719–724 CrossRef CAS PubMed.
- J. E. Sillibourne and M. Bornens, Cell Div., 2010, 5, 25 CrossRef PubMed.
- M. Bettencourt-Dias, A. Rodrigues-Martins, L. Carpenter, M. Riparbelli, L. Lehmann, M. K. Gatt, N. Carmo, F. Balloux, G. Callaini and D. M. Glover, Curr. Biol., 2005, 15, 2199–2207 CrossRef CAS PubMed.
- J. Kleyleinsohn, J. Westendorf, C. M. Le, R. Habedanck, Y. D. Stierhof and E. A. Nigg, Dev. Cell, 2007, 13, 190–202 CrossRef CAS PubMed.
- R. Habedanck, Y. D. Stierhof, C. J. Wilkinson and E. A. Nigg, Nat. Cell Biol., 2005, 7, 1140–1146 CrossRef CAS PubMed.
- G. Salvatore, T. C. Nappi, P. Salerno, Y. Jiang, C. Garbi, C. Ugolini, P. Miccoli, F. Basolo, M. D. Castellone, A. M. Cirafici, R. M. Melillo, A. Fusco, M. L. Bittner and M. Santoro, Cancer Res., 2007, 67, 10148–10158 CrossRef CAS PubMed.
- V. D. V. Mj, Y. D. He, L. J. van't Veer, H. Dai, A. A. Hart, D. W. Voskuil, G. J. Schreiber, J. L. Peterse, C. Roberts and M. J. Marton, N. Engl. J. Med., 2002, 347, 1999–2009 CrossRef PubMed.
- Z. Hu, C. Fan, D. S. Oh, J. S. Marron, X. He, B. F. Qaqish, C. Livasy, L. A. Carey, E. Reynolds, L. Dressler, A. Nobel, J. Parker, M. G. Ewend, L. R. Sawyer, J. Wu, Y. Liu, R. Nanda, M. Tretiakova, A. Ruiz Orrico, D. Dreher, J. P. Palazzo, L. Perreard, E. Nelson, M. Mone, H. Hansen, M. Mullins, J. F. Quackenbush, M. J. Ellis, O. I. Olopade, P. S. Bernard and C. M. Perou, BMC Genomics, 2006, 7, 96 CrossRef PubMed.
- L. D. Miller, J. Smeds, J. George, V. B. Vega, L. Vergara, A. Ploner, Y. Pawitan, P. Hall, S. Klaar, E. T. Liu and J. Bergh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13550–13555 CrossRef CAS PubMed.
- R. Basto, K. Brunk, T. Vinadogrova, N. Peel, A. Franz, A. Khodjakov and J. W. Raff, Cell, 2008, 133, 1032–1042 CrossRef CAS PubMed.
- N. J. Ganem, S. A. Godinho and D. Pellman, Nature, 2009, 460, 278–282 CrossRef CAS PubMed.
- J. Mason, D. C. C. Lin, X. Wei, Y. Che, Y. Yao, R. Kiarash, D. Cescon, G. Fletcher, D. Awrey and M. Bray, Cancer Cell, 2014, 26, 163–176 CrossRef CAS PubMed.
- R. Brough, J. R. Frankum, D. Sims, A. Mackay, A. M. Mendespereira, I. Bajrami, S. Costacabral, R. Rafiq, A. S. Ahmad and M. A. Cerone, Cancer Discovery, 2011, 1, 260–273 CrossRef CAS PubMed.
- C. Tovar, B. Higgins, D. Deo, K. Kolinsky, J. Liu, D. C. Heimbrook and L. T. Vassilev, Cell Cycle, 2010, 9, 3364–3375 CrossRef CAS PubMed.
- L. W. Yao, J. V. Anzola, R. L. Davis, M. Yoon, A. Motamedi, A. Kroll, C. P. Seo, J. E. Hsia, K. K. Sun and J. W. Mitchell, Science, 2015, 348, 1155–1160 CrossRef PubMed.
- E. F. Johnson, K. D. Stewart, K. W. Woods, V. L. Giranda and Y. Luo, Biochemistry, 2007, 46, 9551–9563 CrossRef CAS PubMed.
- R. Laufer, B. Forrest, S. W. Li, Y. Liu, P. Sampson, L. Edwards, Y. Lang, D. E. Awrey, G. Mao and O. Plotnikova, J. Med. Chem., 2013, 56, 6069–6087 CrossRef CAS PubMed.
- P. B. Sampson, Y. Liu, N. K. Patel, M. Feher, B. Forrest, S. W. Li, L. Edwards, R. Laufer, Y. Lang and F. Ban, J. Med. Chem., 2015, 58, 130–146 CrossRef CAS PubMed.
- P. B. Sampson, Y. Liu, B. Forrest, G. Cumming, S. W. Li, N. K. Patel, L. Edwards, R. Laufer, M. Feher and F. Ban, J. Med. Chem., 2015, 58, 147–169 CrossRef CAS PubMed.
- S. W. Li, Y. Liu, P. B. Sampson, N. K. Patel, B. T. Forrest, L. Edwards, R. Laufer, M. Feher, F. Ban, D. E. Awrey, R. Hodgson, I. Beletskaya, G. Mao, J. M. Mason, X. Wei, X. Luo, R. Kiarash, E. Green, T. W. Mak, G. Pan and H. W. Pauls, Bioorg. Med. Chem. Lett., 2016, 26, 4625–4630 CrossRef CAS PubMed.
- I. Lohse, J. M. Mason, P. J. Cao, M. Pintilie, M. R. Bray and D. W. Hedley, Oncotarget, 2016, 8, 3064–3071 Search PubMed.
- B. Yu, Z. Yu, P. P. Qi, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 95, 35–40 CrossRef CAS PubMed.
- P. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
- G. Yao, Q. Yao, Z. Xia and Z. Li, J. Chem. Thermodyn., 2017, 179–186 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02518a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |