
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The synergetic effect of dual co-catalysts on the photocatalytic activity of square-like WO3 with different exposed facets
Huihua Gong,
Yue Cao,
Yifeng Zhang,
Yu Zhang,
Kewei Liu,
Hongmei Cao and
Hongjian Yan *
*
College of Chemistry, Sichuan University, P. R. China. E-mail: hjyan@scu.edu.cn; Tel: +86-1355-1341-892
First published on 30th March 2017
Abstract
Here we report the controlled selective deposition of Pt and PbOx dual-cocatalysts on the edged (200) and (020) facets and the main (002) facets of square-like WO3 nanoplates, respectively. The remarkably enhanced photocatalytic activities were observed for such assembled photocatalysts in photocatalytic water oxidation. The superior performance can be attributed to not only the light-induced preferential flow of photogenerated electrons and holes onto different facets of the square-like WO3, thus leading to the reduction and oxidation reactions taking place on the corresponding edged and major top facets, but also the selective deposition of suitable oxidation and reduction cocatalysts onto the needed facets of square-like WO3 to reduce the charge recombination and catalyze the redox reactions. These findings will be promising and intriguing for designing high efficiency WO3 system for visible light water splitting.
1. Introduction
Sustainable solar energy for the clean chemical energy (renewable energy sources) from H2 and O2 conversion using photocatalytic water splitting under visible light is one of the promising approaches to address the environmental issues and global energy shortage crisis.1–3 Tungsten oxide (WO3), an exceptionally important n-type visible-light semiconductor material with a narrow band gap of 2.4–2.8 eV, has attracted much attention in the field of photocatalysis due to its excellent electron transport properties and good resilience to photocorrosion effects in aqueous solution.2,4–7 However, pure WO3 is not an efficient photocatalyst due to the rapid recombination of photogenerated charge carriers and narrow photo-absorption range.8 Cocatalysts play a significant role because they can serve as the active sites for the water splitting process and trap the photoexcited electrons and holes to improve the charge-separation.9–15 It has been reported that selective deposition of suitable dual-cocatalysts (oxidation cocatalysts and reduction cocatalysts) on corresponding sites of semiconductor-based crystals (e.g., BiVO4, TiO2, SrTiO3, PbTiO3) resulted in a remarkable enhanced photocatalytic performance due to the spatial separation of the photogenerated charges between different facets and the synergetic effect of dual-cocatalysts deposited onto the needed facets.13,16–22 Our recent research works revealed that the square-like WO3 nanoplates with Pt loaded mainly on dominant facets shows higher photocatalytic activity than that with Pt loaded on the edged facets due to light-induced charge separation between different facets of square-like WO3.23 However, the activity difference is not very big owe to that platinum particles were not effective oxidation reaction promoters for promoting O2 evolution on the dominant (002) facets of square-like WO3. Therefore, the selective deposition of suitable cocatalysts on the needed facets of square-like WO3, particularly loading suitable oxygen-generation promoters on the dominant (002) facets to stress the surface recombination of charge carriers and thus improve photocatalytic performance is feasible and urgent based on the charge separation between different facets.In this work, we have successfully prepared Pt/PbOx/WO3 by using a two-step photodeposition method. PbOx nanoparticles as water oxidation cocatalyst were selectively deposited on the dominant (002) facets of square-like WO3, while Pt as reduction cocatalyst were preferentially deposited on the minor (020) and (200) facets of square-like WO3. The prepared Pt/PbOx/WO3 photocatalyst exhibits a significantly increased photocatalytic O2 evolution rate in the presence of Ag+ as sacrifice agent, exceeding that of WO3 deposited with single PbOx or Pt cocatalyst. The superior performance can be attributed to the intrinsic nature of charge separation between the different facets of WO3, together with the co-loading of both Pt and PbOx on reduction facets and oxidation facets of WO3 to capture photogenerated electrons and holes and then reduce the charge recombination.
2. Experimental
2.1 Preparation of catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:1 v/v) was added into the solution drop by drop and magnetically stirred for 30 minutes. The obtained yellow precipitate was washed thoroughly with distilled water for removing Cl−, and then transferred into a 30 mL of Teflon-lined stainless steel autoclave for hydrothermal treatment at 180 °C for 24 h. As a result, the yellow precipitates were collected by centrifugation and washed with deionized water for several times and dried at 60 °C for overnight.
:H2O = 1:1 v/v) was added into the solution drop by drop and magnetically stirred for 30 minutes. The obtained yellow precipitate was washed thoroughly with distilled water for removing Cl−, and then transferred into a 30 mL of Teflon-lined stainless steel autoclave for hydrothermal treatment at 180 °C for 24 h. As a result, the yellow precipitates were collected by centrifugation and washed with deionized water for several times and dried at 60 °C for overnight.The photo-deposition of the PbOx was achieved with Pb(NO3)2 as the precursor. For PbOx/WO3 preparation, 0.500 g of WO3 powder and appropriate amount of Pb(NO3)2 were suspended in 200 mL H2O solution with different pH values by the addition of nitric acid. After photoirradiation for 1 h, the resulting suspension was centrifuged, washed with deionized water and ethanol for several times, and finally dried at 60 °C for overnight. Pt/PbOx/WO3 was prepared using the similar method by loading PbOx nanoparticles on the as-prepared platinized WO3 powder (Pt/WO3).
The photo-deposition of MnOx or CoOx was achieved with MnSO4 or Co(NO3)2 as the precursor, and NaIO3 was employed as the electron acceptor. Briefly, 0.500 g of WO3 powder was suspended in 200 mL 0.01 M NaIO3 aqueous solution containing a certain amount of MnSO4 or Co(NO3)2. Then the mixture was illuminated with a Xe lamp for an hour under stirring after removing the dissolved oxygen completely. The product was collected, washed with deionized water and ethanol for several times, and finally dried at 60 °C for overnight.
RuO2 was loaded by an impregnation method by using Ru3(CO)12 as the precursor according to the method presented previously.24 Typically, WO3 powder was immersed in a tetrahydrofuran (THF) solution containing appropriate amount of dissolved Ru3(CO)12 and stirred at 333 K for 6 h for 4–5 h. The samples were finally evaporated to dryness and calcined in air at 623 K for 5 h to convert the Ru species to RuO2. The amount of RuO2 loaded was 0.1 wt%.
2.2 Photocatalytic reactions
The photocatalytic water splitting O2 evolution reactions were performed in a top-irradiation reactor vessel connected to a glass closed gas circulation system. The evolved gases were analyzed by an online gas chromatograph equipped with a thermal conductivity detector (SPSIC, GC-112AT, argon carrier). Typically, 0.300 g of the as-prepared photocatalyst was dispersed in 200 mL of 0.01 M aqueous AgNO3 solution (AgNO3 acts as the sacrificial reagent). Before irradiated by a 300 W Xe lamp without cutoff filter, the reaction mixture was evacuated for 1 h to completely remove air and establish the adsorption–desorption equilibrium between the solution and photocatalyst with a constant magnetic stirring. The temperature of the reaction solution was maintained at 283 K by a flow of cooling water during the reaction. The gas generated was analyzed every 1 h.2.3 Characterization
Scanning electron microscope (SEM) images of the samples was performed with JSM-5900LV Scanning Electron Microscopy (SEM, JEOL, Japan). Transmission electron microscopy images (TEM), high-resolution (HR) TEM images of the samples were obtained on transmission electron microscope (TEM; Tecnai G2 F20 S-TWIN). Powder X-ray diffraction (XRD) patterns were obtained using a X-Pert Pro diffractometer with Cu Kα radiation (λ = 1.5406 Å) at a scanning speed of 4° min−1. The UV-vis diffuse reflectance absorption spectra (DRS) were recorded on a UV-vis spectrophotometer (UV3600, Shimadzu) by using BaSO4 as a reference. The photoluminescence (PL) spectra of the samples was carried out with a Hitachi F-7000 photoluminescence spectrophotometer (Hitachi F-7000) and the excitation wavelength was 230 nm. X-ray photoelectron spectroscopy (XPS) spectra were measured on a V4105 instrument (Thermo Electron, USA) with a Mn Kα radiation source.3. Result and discussion
Fig. 1 shows the SEM image of square-like WO3 nanoplates synthesized by hydrothermal method. The size of the WO3 nanoplates was between several hundred nanometres and micrometres in length with a thickness of approximately 15–20 nm. The rectangular square-like WO3 nanoplate with predominant facets of (002) and side facets of (020) and (200) based on our recent works.23 It is known that Pt was used as the powerful reduction cocatalysts for high activity WO3.5,25 The choice of suitable oxidation cocatalysts is another important factor for photocatalytic water oxidation reaction. Fig. 2 shows the photocatalytic water oxidation activities of WO3 loaded with different oxidation cocatalysts when platinum is fixed as the reduction cocatalyst. Four metal oxides, MnOx, RuO2, CoOx, and PbOx, known as water oxidation cocatalysts, can catalyze the water oxidation reactions to enhance the photocatalytic oxygen evolution of WO3.26–28 MnOx, CoOx, PbOx, and Pt were deposited by photo-deposition method except that RuO2 was deposited by the impregnation method. The photocatalytic O2 evolution rate over WO3 loaded with MnOx, RuO2, CoOx or PbOx is 446.8, 490.6, 636.7, 929.7 μmol h−1 g−1, respectively. The activity order is MnOx < RuO2 < CoOx < PbOx. Compared to O2 evolution rate of Pt/WO3 (479.1 μmol h−1 g−1), the photocatalytic activity of Pt–MnOx/WO3 was lower than Pt/WO3, indicating that the MnOx have less contribution to the O2 evolution of WO3. Almost very little improvement of photocatalytic activity was observed when RuO2 was deposited. CoOx show more effect on the enhanced photocatalytic activity than RuO2. The water oxidation activity was increased much more obvious when PbOx is used as the oxidation cocatalyst, suggesting that the loading of PbOx and Pt as cocatalysts together shows a synergistic effect in photocatalysis. | ||
| Fig. 1 SEM image of as-prepared square-like WO3. | ||
 | ||
| Fig. 2 Photocatalytic water oxidation activities of WO3 loaded with different oxidation cocatalysts and Pt as the reduction cocatalyst. The amount of Pt and metal oxide nanoparticles cocatalysts were all set to be 0.5 wt% and 0.1 wt% respectively. | ||
The band edge position for WO3 and some metal oxides are shown in Fig. 3. The electronic structures of PbO and Pb3O4 have been relatively well understood, while the band structure and the nature of metallic behaviour of PbO2 are still controversial in recent years.29 Therefore the band gap of PbO2 is not listed in Fig. 3. The photogenerated holes in the valence band (VB) of WO3 can be transfer to metal oxides, since the VB potential of WO3 is more positive than that of metal oxides. The different activities of MOx/Pt/WO3 (M = Co, Ru, Pb, Mn) may be ascribed to the different matching degrees of energy band level and electronic structures between WO3 and metal oxides.22,30,31 It is known that the cocatalyst and semiconductor should have compatible lattice and electronic structures with suitable Fermi-levels or band levels, thus the migration of photogenerated carriers can be promoted by the internal field at the interface.22,30,31 Furthermore, the processing condition and method of preparation metal oxides catalyst also play major roles in the stability, crystallinity, and the size of metal oxides particles, which are greatly affect the activities of catalysts.32 Possible differences in roughness factors also can contribute to difference in performance of as-prepared samples. For example, stability for RuO2, the size of CoOx particles affects photocatalytic activity of photocatalyst.33,34
 | ||
| Fig. 3 Band structures of WO3 and related transition metal oxides. Reference: WO3,2 MnO2,35,36 Co3O4,31 Pb3O4,37,38 RuO2.39–41 | ||
Photo-oxidation deposition of PbOx on WO3 was prepared by using Pb(NO3)2 as the precursor in solution with different acidity. Fig. 4 shows the effect of the different acidity condition of loading PbOx on photocatalytic activity over Pt/PbOx/WO3. The pH values were 1.8, 2.4, 3.77, 4.8, and 7 in the order from small to large. It clearly shows that the highest activity of Pt/PbOx/WO3 was observed at approximately pH in the range of 2 to 4. In order to clarify the influence of acidity on the photocatalytic activity, PbOx/WO3 (0.5 wt%, Pb) samples prepared by photooxidative deposition of PbOx at three different pH values of 1.8, 2.4 and 4.8 were selected to characterize by X-ray photoelectron spectroscopy (XPS). The survey XPS spectrum (Fig. 5a) shows that the main elements on the surface of the composite were W, O, Pb and C. The C 1s peak observed in the survey scan is due to the carbon contamination which was used to calibrate the binding energy. For PbOx/WO3 samples prepared at pH values of 1.8 and 2.4, the peaks locates at around 35.50 and 37.61 eV (see Fig. 5b), related to W 4f7/2 and W 4f5/2 binding energies of W(VI) oxidation state in the tungsten oxide sample.8,26 The O 1s peak (Fig. 5c) at 530.30 eV is associated with the lattice oxygen in WO3, and the other peak at 531.97 eV corresponds with surface adsorbed hydroxide and water.8,26,42,43
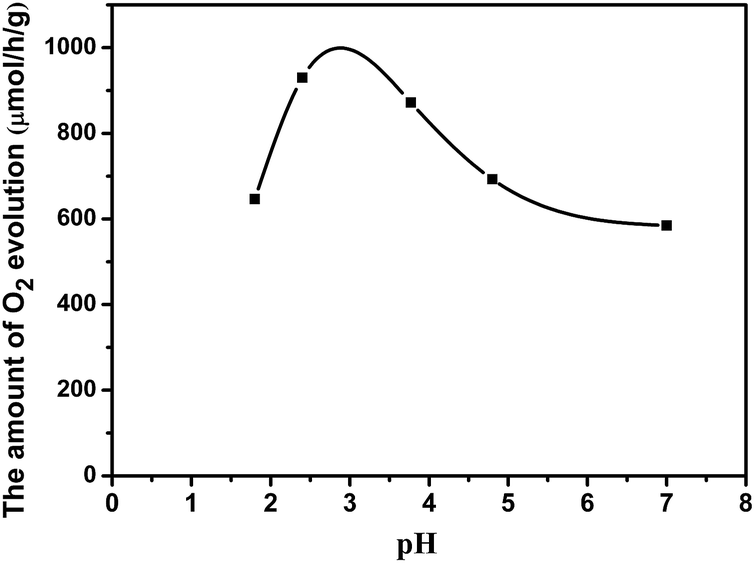 | ||
| Fig. 4 Photocatalytic water oxidation activities over Pt/PbOx/WO3, where the PbOx was loaded in solution with different acidity condition. The amount of Pt and PbOx nanoparticles cocatalysts were set to be 0.5 wt% and 0.1 wt% respectively. | ||
 | ||
| Fig. 5 The XPS survey scan (a), and the corresponding high-resolution XPS spectra of W 4f (b), O 1s (c), and Pb 4f (d) of PbOx/WO3 samples prepared by photo-deposition at different acidity. | ||
The Pb 4f spectrum for PbOx/WO3 sample prepared at pH value of 2.4 exhibited two binding energies at 138.80 and 143.64 eV, corresponding to Pb 4f7/2 and Pb 4f5/2 for Pb4+, respectively. This result indicated that PbO2 was formed when the PbOx was loaded at pH value of 2.4.26,42–46 For PbO2/WO3 sample prepared at pH values of 4.8 (near neutral pH), the Pb 4f spectrum exhibited two peaks at 138.95 and 143.85 eV, corresponding to Pb 4f7/2 and Pb 4f5/2 in the form of Pb3O4,42,47,48 respectively, which indicated both states of Pb(II) and Pb(IV) exist in this photocatalyst. With the increase in acidity, nitric acid will gradually dissolve lead oxide to produce Pb2+, then the Pb2+ was oxidized by photo-generated holes (Pb2+ + 2H2O + 2h+ = PbO2 + 4H+). The increased peak intensity of Pb observed in the XPS of sample with pH value of 2.4 suggests that solution pH not only affect the chemical states of Pb in PbOx but also affect the loading amount of PbOx. Therefore, the different photocatalytic performances (as shown in Fig. 4) of Pt/PbOx/WO3 prepared by pH values of 2.4 and 1.8 may be caused by the loading amount of PbOx, and the different performances of Pt/PbOx/WO3 prepared at pH = 2.4 to 7 may be caused by the slow change of valence states of lead in loaded oxides of lead. Compared to the samples prepared at pH values of 2.4 and 1.8, the W 4f and O 1s peaks of PbOx/WO3 sample prepared at near neutral pH (pH 4.8) show a small shift about 0.2 eV toward high binding energy, which could be due to the effect of states in the loaded PbOx.
The XRD patterns of the prepared catalysts are shown in Fig. 6. The patterns clearly demonstrated that the pure WO3 exhibits a monoclinic phase structure with obvious characteristic diffraction peaks at 2θ values of 23.15, 23.61, 24.37, 33.30, and 34.19°, corresponding to the (002), (020), (200), (022) and (202) planes, respectively (JCPDS no. 72-1465, a = 7.300 Å, b = 7.530 Å, and c = 7.680 Å). All samples have monoclinic WO3 structure and the cocatalysts loading does not influence the crystal structures of WO3. In the XRD patterns of Pt/WO3, and Pt/PbOx/WO3 samples, no related peaks of Pt or PbOx except for monoclinic WO3 are observed because of the tiny amount of Pt(0.5 wt%), PbOx(0.1 wt%), small size and high dispersion in the samples.
 | ||
| Fig. 6 The X-ray diffraction (XRD) patterns of as-synthesized WO3, Pt/WO3, PbOx/WO3 and Pt/PbOx/WO3 samples. PbOx in the samples were loaded at pH = 3.77. | ||
The morphology of square-like WO3 deposited with Pt cocatalyst (0.5 wt%) was observed, and the SEM and TEM image was shown in Fig. 7a and b, respectively. Obviously, Pt nanoparticles with a primary size of 2–3 nm were mainly deposited on the side (020) and (200) facets of the square-like WO3 nanoplates and seriously aggregated. The phenomenon of preferred deposition of Pt is due to intrinsic differences in surface charge of square-like WO3 nanoplates leading sorption-induced deposition of Pt on square-like WO3.49 Prior to photoreduction, since the edges appear positively charged and then preferentially adsorb the negatively charged [PtCl6]2− ions, thus leading to the observed sorption-determined preferred deposition of platinum on the edged facets of square-like WO3.49 Fig. 7c shows the SEM image of WO3 deposited with PbOx (Pb, 0.1 wt%) by photo-oxidation deposition method. As shown in Fig. 7c, PbOx with size of more than 5 nm were mainly loaded on the dominant (002) facets of square-like WO3, while almost no PbOx particles were observed on the side (020) and (200) facets.
 | ||
| Fig. 7 SEM image (a) and TEM image (b) of as-prepared Pt/WO3, and SEM image of PbOx/WO3 prepared at pH = 2.4 (c). | ||
The TEM and SEM images of the square-like WO3 deposited with Pt and PbOx dual cocatalysts by in situ photo-reduction and photo-oxidation deposition methods were also obtained and are shown in Fig. 8. The HRTEM image (Fig. 8d) shows that the lattice fringes of nanoparticles on side (020) and (200) facets is 0.227 nm, which can be indexed to the (111) plane of Pt (JCPDS, no. 70-2431), and it is also hard to find out the PbOx on the side (020) and (200) facets of square-like WO3. As shown in Fig. 8a, it is clearly observed that the surface of tungsten trioxide including the small and dominant facets are covered with cocatalysts. PbOx and Pt can be easily discerned through the particle sizes. Combining both the TEM and SEM images shown in Fig. 7 and 8, we can clearly confirm that the PbOx nanoparticles with a diameter of around 5–10 nm are selectively deposited on the dominant (002) facets of square-like WO3, while Pt nanoparticles with smaller particle size were deposited on the side (020) and (200) facets of square-like WO3. Considering that the basal (002) planes indicates a weak negative surface charge,49 dark sorption of the positively charged Pb2+ ions prior to photoreduction is probably preferred at the dominant (002) facets rather than positively charged edges. When illumination, light-induced spatial separation of the photogenerated electrons and holes toward the edged facets and the dominant crystal facets of square-like WO3 nanoplates, respectively.23 The photogenerated holes that directional migration to (002) facets will oxidize the divalent lead ions to PbOx, thus resulting in preferred loading of PbOx on the dominant planes of WO3.
 | ||
| Fig. 8 SEM image (a), TEM image (b), and HRTEM (d) images of Pt/PbOx/WO3, on which PbOx was loaded at pH = 2.4. The enlargement TEM image of circle part in (b) is shown in (c). | ||
As shown in Fig. 9, the photocatalytic oxygen evolution activity is strongly dependent on the cocatalysts photo-deposited on the surface of the square-like WO3. The highest water oxidation activity is achieved for the Pt/PbOx/WO3 sample with metallic platinum and PbOx particles selectively photo-deposited on the edged and dominant facets, respectively. The rate of O2 evolution on Pt/WO3 and PbOx/WO3 is 479.1 μmol h−1 g−1 and 599.0 μmol h−1 g−1, respectively. Loading PbOx on the dominant (002) facets of WO3 can enhance the photocatalytic O2 evolution rate further. However, the O2 evolution rate on Pt/PbOx/WO3 is 929.7 μmol h−1 g−1. The photocatalytic water oxidation activity of Pt/PbOx/WO3 is nearly double than that of Pt/WO3, indicating that PbOx nanoparticles located on the dominant facets of square-like WO3 have a positive effect on water oxidation reaction.
 | ||
| Fig. 9 Photocatalytic water oxidation performances of Pt/WO3 and Pt/PbOx/WO3. | ||
As demonstrated in Fig. 10, the UV-vis diffuse reflectance spectra of the square-like WO3 loaded with different cocatalysts show similar absorption property to pure WO3 with a strong absorption range from 200 to 350 nm, but slightly weakened the absorption intensity due to that the cocatalysts shadow the photocatalyst absorbing the light.50 It can be deduced from the adsorption edge that all the samples has a band gap of 2.78 eV, indicating that the loaded of PbOx and Pt on the surface of square-like WO3 did not change its band gap. The photoluminescence spectra (Fig. 11) of the photocatalysts give more information of the density of recombination of photogenerated electron–hole pairs. Pt/WO3 and PbOx/WO3 exhibit weaker emission peak intensity than pure WO3 and a significantly decreased PL intensity is observed for Pt/PbOx/WO3 in the wavelength range of 300–800 nm, suggesting that the lower electron–hole recombination rate in Pt/PbOx/WO3.51 The result of PL is in good agreement with that of photocatalytic performances (Fig. 9). Such a decreased electron–hole recombination might be ascribed to selective deposition of a suitable dual-cocatalysts onto the needed facets of square-like WO3 leading accelerated transfer of the photogenerated electrons and holes from the surfaces of WO3.
 | ||
| Fig. 10 PL spectra of WO3, Pt/WO3, PbOx/WO3 and Pt/PbOx/WO3. | ||
 | ||
| Fig. 11 UV-vis diffuse reflectance spectra of WO3, Pt/WO3, PbOx/WO3 and Pt/PbOx/WO3. | ||
It has been reported that the deposition of suitable dual-cocatalysts on the needed facets of semiconductor-based crystals (e.g., BiVO4, TiO2) resulted in a remarkable synergetic effect in the photocatalytic performance, which could be attributed to the spatial separation of the photogenerated charges between facets.13,16,22 Our recent research works reveal that light-induced preferential flow of photogenerated holes and electrons to the dominant (002) facets and edged facets of square-like WO3, on which oxidation and reduction occurs, respectively.23 Here we proposed the reaction mechanism of visible-light-driven O2 evolution over the square-like WO3 loaded with Pt and PbOx dual cocatalysts as depicted in Fig. 12. PbOx nanoparticles dispersed on the hole-rich (002) facets were function as oxidation cocatalysts for trapping holes where as Pt nanoparticles dispersed on the electron-rich (020) and (200) facets were function as reduction cocatalysts for trapping electrons. In general, light-induced spatial separation of the photogenerated electrons and holes toward the edged facets and the dominant crystal facets of square-like WO3 nanoplates respectively, which results in hole-rich (002) major top facets and electron-rich (020) and (200) facets.23 The oxidation reaction of water to O2 evolution takes place on the hole-rich (002) major top facets, while the reduction reaction takes place on the electron-rich (020) and (200) facets. At the same time, PbOx cocatalysts loaded on the dominant facets act as oxidation sites for O2 evolution to catalyze the four-electron water oxidation to produce O2, Pt loaded on the edged facets are to offer reduction sites for Ag+ and catalyze the reduction reactions, namely the reduction of Ag+ to Ag. The simultaneous existence of Pt and PbOx cocatalysts is supposed to be synergistically working for the efficient separation and transfer of the photoexcited electrons and holes, thus contributing to the photocatalytic performance of Pt/PbOx/WO3.
 | ||
| Fig. 12 Proposed mechanism of photocatalytic water splitting O2 evolution over Pt/PbOx/WO3 photocatalyst. | ||
4. Conclusion
In conclusion, square-like WO3 nanoplates with specific exposed crystal facets were synthesized by hydrothermal method. Pt and PbOx dual cocatalysts were selectively deposited on the edged (200) and (020) facets and the dominant (002) facets of square-like WO3 by photo-deposited, respectively. The WO3 deposited with dual cocatalysts show remarkable enhanced photocatalytic activities than that WO3 loaded with single cocatalyst. The enhanced photocatalytic performances are due to light-induced spatial separation of the photogenerated electrons and holes toward different facets of square-like WO3 nanoplates and selectively deposition of suitable dual-cocatalysts onto the needed facets of square-like WO3.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21273157) for the financial support.References
- B. Ma, J. Kim, T. Wang, J. Li, K. Lin, W. Liu and S. Woo, RSC Adv., 2015, 5, 79815–79819 RSC.
- J. Zhang, Z. Liu and Z. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 9684–9691 CAS.
- X. Xiang, L. Zhang, L. Chou and X. Li, CrystEngComm, 2014, 16, 5180 RSC.
- M. Liu, H. Li and Y. Zeng, J. Nanomater., 2015, 2015, 1–7 Search PubMed.
- H. Widiyandari, A. Purwanto, R. Balgis, T. Ogi and K. Okuyama, Chem. Eng. J., 2012, 180, 323–329 CrossRef CAS.
- K. Kalantar-zadeh, J. Z. Ou, T. Daeneke, A. Mitchell, T. Sasaki and M. S. Fuhrer, Applied Materials Today, 2016, 5, 73–89 CrossRef.
- J. Z. Ou, R. A. Rani, S. Balendhran, A. S. Zoolfakar, M. R. Field, S. Zhuiykov, A. P. O'Mullane and K. Kalantar-zadeh, Electrochem. Commun., 2013, 27, 128–132 CrossRef CAS.
- Y. Feng, C. Liu, H. Che, J. Chen, K. Huang, C. Huang and W. Shi, CrystEngComm, 2016, 18, 1790–1799 RSC.
- W. Shi, H. Lv, S. Yuan, H. Huang, Y. Liu and Z. Kang, Sep. Purif. Technol., 2017, 174, 282–289 CrossRef CAS.
- P. Wang, Y. Xia, P. Wu, X. Wang, H. Yu and J. Yu, J. Phys. Chem. C, 2014, 118, 8891–8898 CAS.
- G. W. Busser, B. Mei, P. Weide, P. C. K. Vesborg, K. Stührenberg, M. Bauer, X. Huang, M.-G. Willinger, I. Chorkendorff, R. Schlögl and M. Muhler, ACS Catal., 2015, 5, 5530–5539 CrossRef CAS.
- D. Noureldine, D. H. Anjum and K. Takanabe, Phys. Chem. Chem. Phys., 2014, 16, 10762–10769 RSC.
- L. Mu, Y. Zhao, A. Li, S. Wang, Z. Wang, J. Yang, Y. Wang, T. Liu, R. Chen, J. Zhu, F. Fan, R. Li and C. Li, Energy Environ. Sci., 2016, 9, 2463–2469 CAS.
- Y. Ma, R. Chong, F. Zhang, Q. Xu, S. Shen, H. Han and C. Li, Phys. Chem. Chem. Phys., 2014, 16, 17734–17742 RSC.
- Q. Zhang, Z. Li, S. Wang, R. Li, X. Zhang, Z. Liang, H. Han, S. Liao and C. Li, ACS Catal., 2016, 6, 2182–2191 CrossRef CAS.
- Q. Zhang, R. Li, Z. Li, A. Li, S. Wang, Z. Liang, S. Liao and C. Li, J. Catal., 2016, 337, 36–44 CrossRef CAS.
- T. Tanabe, W. Miyazawa, T. Gunji, M. Hashimoto, S. Kaneko, T. Nozawa, M. Miyauchi and F. Matsumoto, J. Catal., 2016, 340, 276–286 CrossRef CAS.
- E. Bae and T. Ohno, Appl. Catal., B, 2009, 91, 634–639 CrossRef CAS.
- L. Zhang, W. Wang, S. Sun, D. Jiang and E. Gao, Appl. Catal., B, 2015, 162, 470–474 CrossRef CAS.
- C. Zhen, J. C. Yu, G. Liu and H. M. Cheng, Chem. Commun., 2014, 50, 10416–10419 RSC.
- A. Meng, J. Zhang, D. Xu, B. Cheng and J. Yu, Appl. Catal., B, 2016, 198, 286–294 CrossRef CAS.
- R. Li, H. Han, F. Zhang, D. Wang and C. Li, Energy Environ. Sci., 2014, 7, 1369 CAS.
- H. Gong, R. Ma, F. Mao, K. Liu, H. Cao and H. Yan, Chem. Commun., 2016, 52, 11979–11982 RSC.
- K. Teramura, K. Maeda, T. Saito, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2005, 109, 21915–21921 CrossRef CAS PubMed.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed.
- S. Anandan and M. Miyauchi, Chem. Commun., 2012, 48, 4323–4325 RSC.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390 CAS.
- C. S. Chua, D. Ansovini, C. J. Lee, Y. T. Teng, L. T. Ong, D. Chi, T. S. Hor, R. Raja and Y. F. Lim, Phys. Chem. Chem. Phys., 2016, 18, 5172–5178 RSC.
- F. Ma, Y. Jiao, G. Gao, Y. Gu, A. Bilic, S. Sanvito and A. Du, ACS Appl. Mater. Interfaces, 2016, 8, 25667–25673 CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46(8), 1900–1909 CrossRef CAS PubMed.
- M. Long, W. Cai, J. Cai, B. Zhou, X. Chai and Y. Wu, J. Phys. Chem. B, 2006, 110(41), 20211–20216 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren and J. R. McKone, et al., Chem. Rev., 2010, 110(11), 6446–6473 CrossRef CAS PubMed.
- C. Iwakura and H. Tamura, Int. J. Hydrogen Energy, 1982, 7, 857 CrossRef.
- L. Liu, X. Gu, C. Sun, H. Li, Y. Deng, F. Gao and L. Dong, Nanoscale, 2012, 4, 6351 RSC.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- A. K. M. Farid Ul Islam, R. Islam and K. A. Khan, J. Mater. Sci.: Mater. Electron., 2005, 16(4), 203–207 CrossRef CAS.
- E. Ayalew, K. Janssens and K. D. Wael, Anal. Chem., 2016, 88(3), 1564–1569 CrossRef CAS PubMed.
- Y. Zhou, J. Long, Q. Gu, H. Lin, H. Lin and X. Wang, Inorg. Chem., 2012, 51, 12594–12596 CrossRef CAS PubMed.
- M. T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, M. M. Müller, H. J. Kleebe, K. Rachut, J. Ziegler, A. Klein and W. Jaegermann, J. Phys. Chem. C, 2013, 117, 22098–22110 CAS.
- B. H. Tan, E. Ye and W. Y. Fan, Adv. Mater., 2006, 18, 619–623 CrossRef.
- A. K. Goel, G. Skorinko and F. H. Pollak, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 7342 CrossRef CAS.
- J. M. Thomas and M. J. Tricker, J. Chem. Soc., Faraday Trans. 2, 1975, 71, 329–336 RSC.
- Y. Liu, H. Liu, J. Ma and J. Li, Electrochim. Acta, 2011, 56, 1352–1360 CrossRef CAS.
- M. Xu, Z. Wang, F. Wang, P. Hong, C. Wang, X. Ouyang, C. Zhu, Y. Wei, Y. Hun and W. Fang, Electrochim. Acta, 2016, 201, 240–250 CrossRef CAS.
- X. Ma, R. Wang, W. Guo, H. Yang, Z. Liang and C. Fan, Int. J. Electrochem. Sci., 2012, 7, 6012–6024 CAS.
- L. Lubenov, M. Bojinov and T. Tzvetkoff, J. Solid State Electrochem., 2007, 11, 1613–1620 CrossRef CAS.
- K. S. Kim, T. J. O'Leary and N. Winograd, Anal. Chem., 1973, 45(13), 2214–2218 CrossRef CAS.
- K. I. B. Eguiluz, G. R. P. Malpass, M. M. S. Pupo, G. R. Salazar-Banda and L. A. Avaca, Energy Fuels, 2010, 24, 4012–4024 CrossRef CAS.
- K. Wenderich, A. Klaassen, I. Siretanu, F. Mugele and G. Mul, Angew. Chem., Int. Ed., 2014, 53, 12476–12479 CAS.
- C. Young, T. M. Lim, K. Chiang, J. Scott and R. Amal, Appl. Catal., B, 2008, 78, 1–10 CrossRef CAS.
- Y. Zhang, F. Mao, H. Yan, K. Liu, H. Cao, J. Wu and D. Xiao, J. Mater. Chem. A, 2015, 3, 109–115 CAS.
| This journal is © The Royal Society of Chemistry 2017 |