
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of the removal and adsorption mechanisms of cadmium and lead from aqueous solution by activated carbons prepared from Typha angustifolia and Salix matsudana
Chunfang Tang *a,
Yan Shua,
Riqing Zhangb,
Xin Lic,
Jinfeng Songa,
Bing Lia,
Yuting Zhanga and
Danling Oua
*a,
Yan Shua,
Riqing Zhangb,
Xin Lic,
Jinfeng Songa,
Bing Lia,
Yuting Zhanga and
Danling Oua
aCollege of Environmental Science and Engineering, Central South University of Forestry and Technology, Changsha 410004, China. E-mail: cshntcf@163.com; Fax: +86 731 85623372; Tel: +86 139 75116986
bCollege of Forestry, Central South University of Forestry and Technology, Changsha 410004, China
cCollege of Environmental Science and Engineering, Hunan University, Changsha 410082, China
First published on 13th March 2017
Abstract
Activated carbon adsorption is a promising technique for treatment of low-concentration heavy metal pollutants with high efficiency, simple operation, and flexibility; however, the cost of commercial activated carbon is too high. Therefore, much attention has been paid to activated carbon derived from low-cost agricultural and forest wastes. In this work, adsorption of Cd and Pb in aqueous solutions were conducted through batch experiments using Typha angustifolia carbon (TC) and Salix matsudana carbon (SC) prepared by phosphoric acid activation, and the adsorbents were characterized using SEM, FTIR, XPS, elemental analyzer, BET surface area, and zeta potential measurements. Both TC and SC mainly encompassed mesopores and bonds of O–H, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and PO/P–O–C. The BET surface area and isoelectric point (pHIEP) of TC and SC was 130.42 and 234.42 m2 g−1 and 3.45 and 4.38, respectively. The adsorption capacity of Cd and Pb for TC and SC both relied positively on the pH and humic acid but negatively on NaCl in the media. The adsorption process was spontaneous and endothermic, involved physicochemical reactions, and fitted well with the pseudo second-order and Langmuir models. Both the intraparticle diffusion and the chemical reaction were the rate limiting steps during adsorption. The maximum sorption capacity of Cd and Pb was 48.08 and 61.73 mg g−1 (TC) and 40.98 and 58.82 mg g−1 (SC), respectively, at pH 5 and 25 °C. The adsorption–desorption results showed that both activated carbons had good regeneration and reusability properties. The results suggested that both TC and SC had potential applications in the removal of Cd and Pb from aqueous solutions.
O, and PO/P–O–C. The BET surface area and isoelectric point (pHIEP) of TC and SC was 130.42 and 234.42 m2 g−1 and 3.45 and 4.38, respectively. The adsorption capacity of Cd and Pb for TC and SC both relied positively on the pH and humic acid but negatively on NaCl in the media. The adsorption process was spontaneous and endothermic, involved physicochemical reactions, and fitted well with the pseudo second-order and Langmuir models. Both the intraparticle diffusion and the chemical reaction were the rate limiting steps during adsorption. The maximum sorption capacity of Cd and Pb was 48.08 and 61.73 mg g−1 (TC) and 40.98 and 58.82 mg g−1 (SC), respectively, at pH 5 and 25 °C. The adsorption–desorption results showed that both activated carbons had good regeneration and reusability properties. The results suggested that both TC and SC had potential applications in the removal of Cd and Pb from aqueous solutions.
1. Introduction
Heavy metal pollution can result in great environmental issues due to their persistence and toxicity, among which Cd and Pb are coexistent, human carcinogenic, and should be paid more attention.1,2 Physical and chemical methods are commonly used to remove heavy metals from wastewater.3,4 However, the cost of these methods such as ion exchange, membrane separation, electroanalysis, and air floatation are expensive and sometimes unsuitable for the treatment of low-level metal pollutants.5–7 Adsorption methods provide the advantages of low cost, simple operation, and low secondary environmental pollution, and can also be used to remove heavy metal pollutants at less than 100 mg L−1, when other methods cannot work effectively.8Activated carbon is the most widely used adsorbent currently, but the price of commercial activated carbon produced from coal and wood raw materials is very high, which limits its application, especially in countries lacking these resources. Thus, currently the activated carbons prepared from agricultural and forestry residues have received increasing attention.9–13 The adsorption capacity of activated carbon depends on its raw materials and preparation methods. Chemical activation has a lower energy consumption and higher productivity than physical activation. Usually H3PO4 is used as an activator due to lower environmental toxicity than ZnCl2 and a lower activated temperature than KOH.9,13
Cattails are perennial, herbage plants with a high adaptability and large biomass. They are widely used in constructed wetland for removing nitrogen, phosphorous, and organics from wastewater.14 However, casually discarding the withered aerial part of cattails will not only waste resources but also cause the eutrophication of water and swamping of lakes. Researchers have proved that cattail-based activated carbon has a good adsorption ability on some organics,15 as well as the advantages of rich resource, low cost, waste recycling, resource saving, and environment friendliness.9–13 Among which, Typha angustifolia provides not only a high biomass and but also pore structure due to its vascular property. Willow plants like Salix matsudana possess many advantages for manufacturing activated carbon including various species, easy reproduction, fast growth, high biomass, and tolerance to adverse environments.16 Therefore, both T. angustifolia and S. matsudana are potential low cost materials for preparing activated carbon and were comprehensively discussed to exploit their new application for environmental protection in this paper.
Overall, to obtain cost-effective activated carbon requires more research including optimizing activating and adsorption conditions. Based on the pre-experiments of the adsorption of Cd and Pb with activated carbons prepared from T. augustifolia and S. matsudana impregnated by H3PO4, HNO3, and KOH, H3PO4 was chosen for the preparation of T. augustifolia and S. matsudana carbons (TC and SC). The TC and SC are presumed to have encompassed phosphoric acid functional group and efficiently adsorbed Cd and Pb from aqueous solution. In a quest for a promising cattail and willow carbons, this study aimed to prepare a low cost TC and WC, evaluate their sorption properties for Cd and Pb, and physicochemically characterize TC and WC before and/or after adsorption experiments.
2. Materials and methods
2.1 Preparation of carbons
The withered aerial part of T. angustifolia and branches of S. matsudana were sampled from Yanghu Wetland in Changsha and non-flooding area of Dongting Lake in Yueyang, China, respectively. Cleaned cattail samples were dried at 80 °C to a constant weight and grounded with a micro-crusher (FZ102, Taisite, Tianjing). Sawdust of cleaned willow branches was also dried to a constant weight. After screened with a 60 mesh sieve, both samples were kept in sealed plastic bags individually for preparing carbons.10.0000 g of samples were first pre-incubated with 30 mL of 40% (v/v) H3PO4 for 12 h in a series of 200 mL porcelain crucibles wrapped with a foil, then carbonized at 200 °C for 0.5 h and activated at 450 °C for 1 h in a muffle. After natural cooling, all carbons were washed several times with deionized water until the pH turned to 6–7; then dried at 105 °C for 24 h, polished, and sieved through a 100 mesh (particle size < 150 mm).
2.2 Analysis of physical–chemical characteristics of carbons
The physical and chemical properties of carbons were characterized by scanning electron microscopy (SEM) (QUANTA 450, China), Fourier transform infrared spectrophotometry (FTIR) (Shimadzu IRAffinity-1, Japan), automatic surface area and pore size analysis (Quadrasorb SI-3MP, America), zeta potential analysis (Nano ZS ZEN3600, Britain), and X-ray photoelectron spectroscopy (XPS) (K-Alpha, America), respectively. The elemental compositions of both activated carbons were analyzed with elemental analyzer (Elementar, Germany).2.3 Adsorption experiments
A series of 250 mL flasks containing 50 mL solution of Cd and Pb with a given level (40–200 mg L−1) and initial pH (3–8) were added a given dose of TC and SC (0.02–2 g), and shaken at a given temperature (15–40 °C) and time (2–120 min) in a vibrator. NaCl (0.01–0.50 mol L−1) and humic acid (HA) (5–25 mg L−1) were used as competitive ion and dissolved organic compound, respectively, for studying their effects on the adsorption of Cd and Pb. Commercial activated carbon (CAC) was used as a reference to compare the maximum adsorption capacity. Except the tests of parameter effects on the adsorption, the adsorption time, initial pH value, temperature, initial metal ion concentration, dose of carbons in solutions, and vibrating rate were 60 min, 5, 25 °C, 50 mg L−1, 0.1000 g (2 g L−1), and 180 rpm, respectively. The initial values of pH in solutions were adjusted with 0.1 mol L−1 solution of HCl and NaOH. The levels of Cd and Pb in filtrate after adsorption were analyzed by atom adsorption spectrometer (AAnalyst 300, Perkin-Elmer, Germany).2.4 Adsorption and desorption cycles
The desorption conditions for Cd and Pb were similar to those of adsorption studies. 0.1 g of TC or SC was treated with 50 mL solution containing 50 mg L−1 of Cd or Pb (pH 5). The solution was centrifuged after 60 min adsorption. The spent TC or SC was resuspended in 50 mL of 1.0 mol L−1 HCl for 12 h. After centrifugal separation the TC or SC was washed several times with deionized water in order to completely remove Cl−, and then reused to adsorb 50 mg L−1 Cd and Pb in 50 mL solution. The above procedure was repeated four times using the same method, and all the levels of Cd and Pb in the supernate after centrifugation were measured.2.5 Statistics and analysis of data
The results were the means of two duplicate tests.The adsorption capacity (mg g−1) and removal efficiency (%) were figured by formulas (1) and (2), respectively:
 | (1) |
 | (2) |
Gibbs eqn (3)–(5) of changes of standard free energy (ΔG0, kJ mol−1), enthalpy (ΔH0, kJ mol−1), and entropy (ΔS0, J mol−1 K−1) were used to analyze adsorption thermodynamics:
ΔG0 = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKc lnKc
| (3) |
 | (4) |
 | (5) |
Adsorption isotherms were assayed by Langmuir (6) and Freundlich (7) models, and adsorption feasibility was judged by formula (8):
 | (6) |
 | (7) |
| RL = 1/(1 + KaC0) | (8) |
Equations of pseudo-first (9) and pseudo-second order (10) were used to evaluated adsorption kinetics:
|
ln(qe − qt) = lnqe + k1t
| (9) |
 | (10) |
Diffusion mechanism was analyzed with models of liquid film diffusion (11) and intra-particle diffusion (12), respectively:
 | (11) |
| qt = kidt1/2 + C | (12) |
3. Results and discussion
3.1 Characterization of carbons
The physicochemical characteristics of activated carbon are most important factors for adsorption capacity. Table 1 gives the compositions of C, H, O, N, and P of both activated carbons obtained from elemental analysis and XPS (data inside the parenthesis) except H, which showed a similar trend. The composition difference was probably due to the different analysis mechanism. The XPS measures only a few atomic layers on the material surface, whereas the elemental analysis presents the average composition of the entire sample. The fraction of O in TC was higher than that of SC, but SC contained more C element. It was probably attributed to the fact that T. angustifolia is a vascular herbaceous plant whereas S. matsudana belongs to woody plants.| Carbon | C (%) | H (%) | O (%) | N (%) | P (%) |
|---|---|---|---|---|---|
| TC | 43.73 (73.71) | 3.30 | 30.77 (20.05) | 0.25 (1.53) | 1.89 (2.14) |
| SC | 72.89 (89.51) | 3.21 | 14.76 (8.60) | 0.05 (0.24) | 1.12 (1.65) |
Both TC and SC mainly had mesopores, and their BET specific surface area, total pore volume, and average pore diameter were 130.42 and 234.42 m2 g−1, 13.29 and 24.36 mL g−1, and 4.14 and 3.45 nm, respectively. The micro-morphology of two carbons is presented in Fig. 1. TC had an irregular cylindrical structure with a rough surface and uneven pores, while SC presented a rough surface with layered structure and fine granular. The physical characteristics of SC were better than those of TC, which helped more with the physical adsorption of Cd and Pb.
 | ||
| Fig. 1 SEM micrograph of Typha angustifolia activated carbon (TC) and Salix matsudana activated carbon (SC). | ||
The zeta potential of colloid particle is the potential difference between adsorptive and diffusive layers adhered on the solid surface in a liquid–solid system. The isoelectric point (pHIEP) of solid is the negative logarithm of potential ions, and is the pH value of solution when the zeta potential becomes zero and if H+ acts as a potential ion. The zeta potential of TC and SC declined from 9.57 to 23 mV and 15.89 to 20.11 mV when the solution pH increased from 3 to 9, and their pHIEP values were 3.45 and 4.38, respectively (Fig. 2). At the same solution pH, the zeta potential and pHIEP of TC were smaller than those of SC. This was more helpful for the adsorption of Cd and Pb by ion exchange or electrostatic attraction and complexation when the initial solution pH was lower or higher than the pHIEP, respectively.
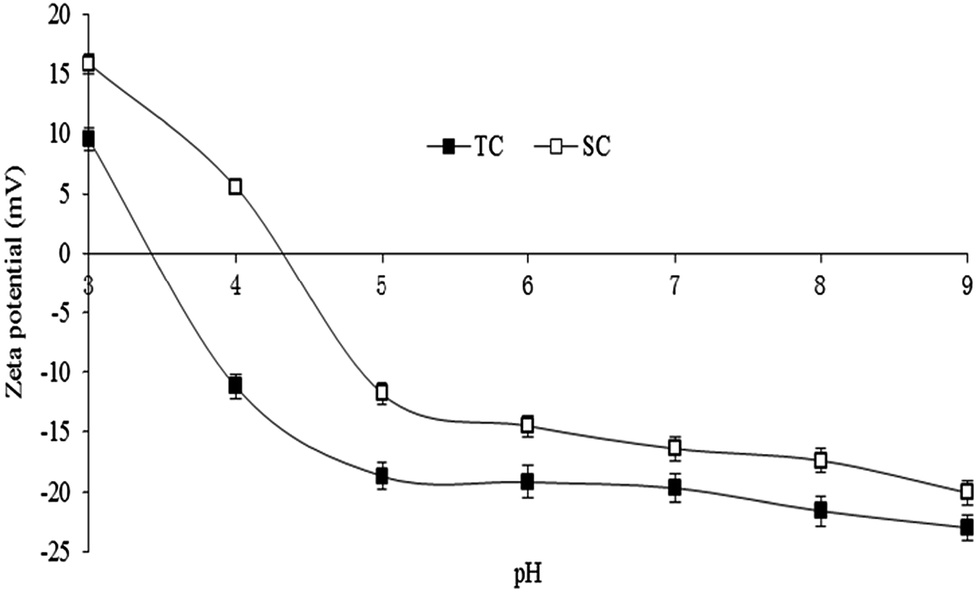 | ||
| Fig. 2 Zeta potential of Typha angustifolia activated carbon (TC) and Salix matsudana activated carbon (SC). Error bar is the standard deviation of the mean. | ||
As shown in Fig. 3, both TC and SC contained functional groups with oxygen. Among adsorption peaks of TC, the obvious broad peaks at bands of 3424 and 3183 cm−1 were attributed to the stretch vibration of hydroxyl (O–H); the peak at 1633 cm−1 was due to the stretch vibration of carbonyl (CO). The peak at 1398 cm−1 was assigned to the stretch vibration of C–H and also the intra-surface bending vibration of C–O–H. The stretch vibration of PO caused an adsorption at 1072 cm−1. While in the FTIR spectrum of SC, O–H resulted in an obvious broad adsorption peak at 3460 cm−1, and C–O produced an absorption peak at 1355 cm−1; the adsorption peaks appearing at bands of 1650 and 1520 cm−1 were assigned to the stretch vibration of CO, and PO and P–O–C produced an adsorption peak at 1080 cm−1.
 | ||
| Fig. 3 FTIR spectrum of Typha angustifolia activated carbon (TC) and Salix matsudana activated carbon (SC). | ||
The band shift and disappearance of some functional groups after the adsorption of Cd and Pb indicated that the corresponding groups participated in the adsorption to some extent.17,18 The peak area of oxygen containing groups of both carbons obviously decreased after the adsorption of Pb, which probably resulted from the ion exchange between Pb2+ and H+ contained in –OH, –COOH, –CHO, and the formation of complexes due to the electrostatic attraction between Pb2+ and deprotonation of –OH.19 The absorption peak between 2000 and 2500 cm−1 was probably contributed to the release of CO2 during the analysis of infrared spectra.18 The areas of some adsorption peaks of TC were bigger than those of SC, suggesting that TC contained more functional groups which were more helpful to the chemical adsorption of Cd and Pb.
The XPS is an effective approach to analyze the surface functional groups of adsorbents, and may provide more useful information on the adsorption mechanism than the FTIR, thermodynamics, and isotherms data. In the whole spectra of XPS (Fig. 4 TC-a and SC-a), the presence of P2p peaks suggested the successful modification of TC and SC by H3PO4. Cd2p and Pb4f peaks appeared after their adsorption, which indicated that Cd and Pb were successfully adsorbed by TC and SC. The high revolution of the C1s spectra yielded peaks at approximately 284.72, 285.78 and 286.51, 288.79, and 291.41 and 292.86 eV, representing groups of C–C/CC/C–H, C–O/C–OH, CO, and O–CO, respectively (Fig. 4 TC-b); and peaks at 284.75 and 284.77, 286.23, 286.86, 288.76, and 291.34 eV corresponded to C–C/CC/C–H, C–O/C–OH, C–O–C, CO, and O–CO, respectively (Fig. 4 SC-b).20 The P2p peak contained two main peaks at 133.33 and 134.08 eV (Fig. 4 TC-c) and 132.95 and 134.12 eV (Fig. 4 SC-c), which was attributed to PO and P–O–C, respectively. The P atom in TC and SC mainly presented on the carbon surface by bonding to O atoms, which fitted well to the result of the FTIR analysis.21 Groups containing O played a key role on Cd and Pb adsorption. Before adsorption, the high resolution O1s spectra contained three main peaks centered at 531.41, 533.01, and 536.05 eV (Fig. 4 TC-d) and 530.92, 532.75, and 536.53 eV (Fig. 4 SC-d), which was attributed to groups of C–OH/O–H, C–O–C, and –COOH, respectively.21 After sorption, the new peaks appeared at 530.31 eV (CO), 533.93, 534.47 eV (–COOH) (Fig. 4 TC-f), 532.43 eV (C–OH), and 533.14 eV (C–O–C) (Fig. 4 SC-f), which indicated their reactions with Pb. The disappearance of peak at 536.53 eV (Fig. 4 SC-e) and the shift of some peaks assigned to groups containing O (Fig. 4 TC-e and SC-e) also suggested the occurrence of adsorption reaction between TC and SC with Cd and Pb.
 | ||
| Fig. 4 X-ray photoelectron spectroscopy of Typha angustifolia activated carbon (TC) and Salix matsudana activated carbon (SC). | ||
3.2 Effect of contact time on adsorption and kinetics
The study of adsorption kinetics can demonstrate the diffusion rate of adsorbate in solid–liquid adsorption systems, and help determine optimum parameters for the design of adsorption devices. The adsorption of Cd and Pb onto TC reached 95% of qe in 2 min, and 90% of qe onto SC in 8 min. After 10 min of contact, the adsorption of both ions by both carbons approached the value of qe and tended to remain in a stable stage (Fig. 5a). There were more Cd and Pb adsorbed on TC than on SC, but both had a similar adsorption equilibrium time, indicating a faster adsorption rate for TC than SC. | ||
| Fig. 5 Influence of initial solution pH, time, activated carbon dose, temperature, NaCl, and HA on the adsorption of Cd and Pb on Typha angustifolia activated carbon (TC) and Salix matsudana activated carbon (SC). Error bar is the standard deviation of the mean. | ||
The adsorption process can be divided into three stages: film mass transfer, intraparticle diffusion, and chemical reaction on adsorbent.22 The concentration difference of adsorbate between adsorbent and solution is the main diffusion force of adsorbate. The initial rapid adsorption of Cd and Pb on TC and SC was due to rich adsorptive sites and large adsorption energy of carbons, large external diffusion rate of Cd and Pb, and also a small diameter of Cd and Pb ions.23 The reason for the followed slow adsorption was attributed to adsorption transfer from extra diffusion to internal one,24 and the final saturated adsorption was due to the electrostatic repulsion between metal ions and adsorbent and thus restrict the function of the remaining activated sites.23
The normal pseudo-first and second order models were used in this work to describe adsorption kinetics. The values of qe, k1, k2, and R2 calculated from the slopes and intercepts of fitted lines of adsorption data are shown in Table 2. The values of R12 from the pseudo-first model ranked from 0.47 to 0.93, and the values of q1e,cal were far less than the corresponding values of qe,exp. It indicated that the pseudo-first order equation could not describe the whole adsorption process, which was similar to the results in Ho et al.25 Although the pseudo-second order adsorption model could not definitely reflect the adsorption mechanism, it could fit well with heavy metal ion adsorption on most activated carbons, and had been widely used in the analysis of adsorption kinetics.23 The values of R22 based on pseudo-second model were 0.99, and the q2e,cal was fitted well with the corresponding qe,exp. It suggested that the pseudo-second order model could describe the whole adsorption process of Cd and Pb on TC and SC; there were similar results in studies of Bouhamed et al.7 and Hamissa et al.26
| Carbon | Ion | qe,exp (mg g−1) | Pseudo first-order | Pseudo second-order | ||||
|---|---|---|---|---|---|---|---|---|
| q1e,cal (mg g−1) | k1 (min−1) | R12 | q2e,cal (mg g−1) | k2 (g mg−1 min−1) | R22 | |||
| TC | Cd | 19.183 | 6.140 | 0.032 | 0.47 | 19.342 | 1.990 | 0.99 |
| Pb | 21.489 | 2.067 | 0.043 | 0.93 | 21.739 | 0.450 | 0.99 | |
| SC | Cd | 9.534 | 1.007 | 0.051 | 0.84 | 10.299 | 0.150 | 0.99 |
| Pb | 19.356 | 1.674 | 0.046 | 0.78 | 20.000 | 0.217 | 0.99 | |
3.3 Effect of initial solution pH on adsorption
The initial solution pH is a key factor to determining adsorption capacity, and is greatly related to the surface charges and electricity of both adsorbent and adsorbate, including protonation or deprotonation of activated carbon and hydration, hydrolysis, and precipitation of metal ions.22,24,27–29 With the increase of initial pH from 3 to 8, the adsorption capacity of Cd and Pb on TC increased from 15.91 and 16.93 mg g−1 to 20.02 and 24.01 mg g−1, and their removal efficiencies changed from 63.64 and 67.72% to 88.01 and 96.04%, respectively. However, the Cd and Pb adsorption capacity on SC increased from 7.98 and 15.14 mg g−1 to 17.38 and 23.93 mg g−1, and their removal efficiency changed from 31.9 and 60.55% to 69.5 and 95.93%, respectively (Fig. 5b).When the initial solution pH is lower than the pHIEP of carbon, carbon will be positively charged, and the adsorption mainly includes physical adsorption and ion exchange.28 The adsorption of Cd and Pb was largely attributed to the ion exchange between Cd(OH)+, Pb2+, Pb(OH)+, Pb2OH3+, and Pb4(OH)44+ in solutions and H+ on TC and SC, as well as between H+ in solutions and the ions of Cd and Pb absorbed on both carbons.24,29 The repulsion of Cd and Pb ions between activated carbon and solution would also decrease their adsorption capacity.
The surface of TC and SC would be negatively charged when the initial pH in the solution was bigger than the pHIEP of TC or SC. Accordingly, the adsorption of Cd and Pb was mainly due to the electrostatic attraction between Cd2+, Cd(OH)+, Pb2+, Pb(OH)+, Pb2OH3+, and Pb4(OH)44+ and the negatively charged surface of TC and SC. With the increase of initial solution pH, the level of H+ in the solution decreased, and the competition adsorption between H+ and the ions of Cd and Pb due to ion exchange also decreased. Furthermore, the net negative charge on the surface of TC and SC went up with the increase of pH in the solution, revealing that adsorption sites combined with cations increased, and thus there was an increase in the adsorption of Cd and Pb. The precipitation of Cd and Pb on the surface of carbons including the combination of phosphate group with Cd and Pb on the carbon surface also caused an increase in their adsorption capacity.
The adsorption capacity of Pb was better than that of Cd, especially when the initial solution pH was more than 6, which was probably attributed to the stronger electrostatic attraction and ion exchange between Pb and TC and SC. This was also contributed to the combination between the large amount of Pb precipitation and TC and SC by the hydrogen bond.24,30 However, only a few Cd deposited until pH value reached to 8, and Cd sinked completely at pH value of 11 in the solution.31 Compared to SC, the adsorption capacity of Cd and Pb on TC was obviously higher, which was probably due to the lower pHIEP of TC. The adsorption ability of activated carbon was so greatly dependent on the solution pH as to achieve better adsorption effectiveness and prevent precipitation of Cd and Pb, the initial pH value in the solution was designated as 5 in the remaining experiments based on pre-adsorption experiments.
3.4 Effect of dose of activated carbons on adsorption
The removal efficiency of Cd and Pb from the solution significantly increased with the addition of both carbons from 0.02 to 2 g. The removal rates for Pb by both carbons and Cd by TC were more than 90% at carbon dosage of 0.5 g. On the other hand, the adsorption capacity of Cd and Pb on TC decreased from 54.38 and 61.19 mg g−1 to 1.20 and 1.23 mg g−1, and on SC declined from 29.25 and 54.50 mg g−1 to 1.13 and 1.22 mg g−1 (Fig. 5c).The increase of removal efficiency of Cd and Pb with carbon dose resulted from the increase of total surface area and activated function groups on carbons. However, the removal did not reach 100%, indicating that the competed adsorption of Cd and Pb between carbon and solution occurred, and the adsorption and desorption coexisted in the system. On the contrary, the obvious decrease of adsorption capacity of Cd and Pb with the dose of carbons could be explained by (1) at low levels of activated carbons in the solution, Cd and Pb ions had a relatively high content, and competed adsorption sites of carbons, and thus reached a high adsorption capacity; whereas, at high doses of carbons, the adsorption of Cd and Pb did not reach saturation on carbons due to a rather low content of Cd and Pb, or there were excess activated sites on carbons; (2) although the adsorptive sites for a given quality of carbon were constant, carbon particles would coagulate with each other with the increase of carbon content in a given volume of solution, causing the decrease of effective specific surface areas, and thus the increase of diffusion distance of adsorbates on the surface or inside carbons.30 Considering the removal efficiency and adsorption capacity together, 0.1000 g (2 g L−1) of carbon was chosen for the rest experiments.
3.5 Effect of initial temperature in solution on adsorption thermodynamics
The increase of solution temperature increased the adsorption capacity of Cd and Pb on TC and SC (Fig. 5d), indicating that the adsorption process was endothermic,23 which was in accordance with ΔH0 > 0 (Table 3). The reasons were as follows: (1) the increased temperature increased the sites on the surface of carbons, decreased the thick of double electric layer of carbons, and thus the diffusion resistance of adsorbates to adsorbents; (2) the increased temperature helped to the dehydration for Cd and Pb ions, and the diameter of free ions is less than that of hydrated ones (the diameter of free ions of Cd is 0.97 Å, and Pb is 1.20 Å; hydrated ions of Cd 4.26 Å, Pb 4.01 Å).32 Therefore, free ions could be easily adsorbed by ion exchange and diffuse to the micropores of carbons, causing an increased adsorption capacity in a given volume pore.23| Ion | T (K) | lnKc |
ΔG0 (kJ mol−1) | ΔH0 (kJ mol−1) | ΔS0 (J mol−1 K−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| TC | SC | TC | SC | TC | SC | TC | SC | ||
| Cd | 288 | 1.219 | −0.905 | −2.918 | 2.167 | 5.767 | 34.440 | 29.974 | 112.222 |
| 293 | 1.225 | −0.708 | −2.984 | 1.725 | |||||
| 298 | 1.259 | −0.331 | −3.119 | 0.820 | |||||
| 303 | 1.314 | −0.136 | −3.310 | 0.343 | |||||
| 308 | 1.356 | 0.136 | −3.471 | −0.349 | |||||
| 313 | 1.400 | 0.152 | −3.642 | −0.396 | |||||
| Pb | 288 | 1.901 | 0.535 | −4.552 | −1.281 | 5.535 | 46.556 | 34.963 | 166.180 |
| 293 | 1.904 | 0.782 | −4.638 | −1.906 | |||||
| 298 | 1.992 | 1.224 | −4.936 | −3.032 | |||||
| 303 | 2.021 | 1.66 | −5.092 | −4.196 | |||||
| 308 | 2.033 | 1.876 | −5.207 | −4.805 | |||||
| 313 | 2.076 | 1.957 | −5.402 | −5.092 | |||||
The adsorption of Cd and Pb on SC was more sensitive to temperature than on TC, indicating that chemical adsorption on SC needed more thermo and was less easier to occur than on TC. At an identical temperature, the adsorption capacity of Pb was higher than that of Cd, indicating that more thermal energy or a higher temperature was needed for the same adsorption capacity of Cd as Pb. An initial solution temperature of 25 °C was chosen as an adsorption temperature for the rest of the experiments based on the non-obvious effects of temperature on adsorption.
Analysis of adsorption thermodynamics helps determine the values of ΔG0, ΔH0, and ΔS0, and thus the characteristics of adsorption. For the adsorption of Cd and Pb on TC and Pb on SC, the ΔG0 values were below zero and decreased with the increase of the temperature, revealing that the initiative and feasibility of adsorption increased with the temperature.33 That the values of ΔG0 of Cd adsorption on SC were bigger than zero did not mean that the adsorption was non-spontaneous, but rather this meant that an energy barrier existed during adsorption, and ΔG0 would drop below zero with the increase of temperature. However, the values of ΔG0 also depend on the models of thermodynamics chosen, which can turn a negative ΔG0 to a positive one.24 The ΔG0 values of TC less than those of SC revealed that the adsorption of Cd and Pb on TC was more spontaneous, feasible, and effective than that on TC. That the values of ΔG0 of Pb adsorption were lower than those of Cd indicated that the adsorption of Pb had a higher spontaneity and larger feasibility, which was one of the reasons for the greater adsorption capacity of Pb than Cd.
ΔH0 can indicate an energy barrier during adsorption, and ΔH0 > 0 suggests that adsorption is endothermic and the increase in temperature helps adsorption.24 That the ΔH0 of Cd and Pb adsorption on TC and SC was greater than zero suggested that (1) to obtain a larger adsorption capacity the hydrated metal ions should be dehydrated all or part of combined water to decrease diameter, which was an endothermic process,32,34 and (2) both the diffusion of Cd and Pb from the liquid phase to the solid one and the broken of ion bond for Cd and Pb ion exchange adsorption needed energy or thermo.34,35 TC had a smaller ΔH0 than that of SC, so TC needed less energy to reach a same sorption capacity.
The values of ΔS0 more than zero in this study demonstrated the increase in the random of adsorption, which was probably due to (1) the structure change of carbons, Cd, and Pb during adsorption, the replacement of water molecules on carbons by ions of Cd and Pb, and the dehydration of Cd and Pb hydrated ions for diffusing into the micropores of carbons;33,36 (2) the release of H+ from carbon could also produce a positive value of ΔS0. Whereas ΔS0 less than zero reveals that the order of adsorption increased during adsorption.17 The ΔS0 of TC was less than that of SC, so the adsorption of Cd and Pb on TC was more ordered.
3.6 Effect of sodium chloride and humic acid
In practically, the adsorption of heavy metals by activated carbon can be affected by several environmental factors. In this study, NaCl and HA were used as competitive ion and dissolved organic compound, respectively. The adsorption of Cd and Pb on both TC and SC decreased with the increase in NaCl from 0.01 to 0.5 mol L−1 (Fig. 5e), but was positively affected by the existence of HA ranging from 5 to 5 mg L−1 (Fig. 5f).The decrease in adsorption of Cd and Pb by NaCl might be resulted from the changes in the properties of electrical double layer of adsorbent. According to the surface chemistry theory,37 when solid adsorbent is in contact with adsorbate species in solution, they are bound to be surrounded by an electrical diffused double layer, the thickness of which is significantly expanded by the presence of electrolyte. Such expansion inhibits the adsorbent particles and metal species from approaching each other more closely and leads to the lower adsorption through the weakened electrostatic attraction. The competition adsorption between Na+ and ions of Pb and Cd for adsorption sites of TC or SC might also impair the adsorption of Pb and Cd on TC and SC. At low pH values, the negatively charged HA was easily adsorbed onto TC and SC, and the interaction of Cd and Pb ions with surface adsorbed HA on TC and SC was stronger than that of Cd and Pb with bare TC and SC. So, the presence of HA, a dissolved organic compound, in the environment could be a sink of Cd and Pb pollutants.
3.7 Evaluation of adsorption isotherms
Adsorption occurring in a solid–liquid system is a dynamical equilibrium process. Adsorption isotherm can demonstrate the distribution of adsorbate between solid and liquid phases at adsorption equilibrium, and offer a theoretic maximum adsorption capacity, which are of great significance for the optional design of adsorption device and the analysis of adsorption mechanism. The mostly used adsorption isotherm models of Langmuir and Freundlich were used in this study. The values of R2 obtained from both Langmuir and Freundlich isotherms for TC and SC ranked from 0.98–0.99 and 0.96–0.99, respectively, revealing that both isothermal models could fit the data at adsorption equilibrium, especially the Langmuir model.The parameters obtained from the slopes and intercepts of isotherms are presented in Table 4. The maximum adsorption capacity Qm of TC for Cd and Pb adsorption was 48.08 and 61.73 mg g−1 and of SC was 40.98 and 58.82 mg g−1, respectively, at initial pH of 5 and 25 °C in the solution. These values were more than the figures reported in the study of Momčilović et al.12 However, the Qm of both carbons was lower than the Qm values in the studies of Wang et al.10 and Xu et al.22 The Qm of Cd on bean husk-based carbon activated by HNO3 (ref. 38) and the Qm of Pb on lotus stalk carbon activated by H3PO4 (ref. 39) reached 180 and 139 mg g−1, respectively, which were far higher than the values of Qm in the present study. Furthermore, the Qm of TC and SC for Cd and Pb was less than 56.82 and 70.42 mg g−1 of the commercial activated carbon (Table 4). These different adsorption efficiencies were probably caused by different physicochemical properties of adsorbents due to different raw materials and methods for the preparation of activated carbons and adsorption parameters. Similar to the result in Youssef et al.,3 TC and SC presented low specific surface areas but high values of Qm, indicating that surface functional groups and pHIEP of carbons played a key role in Cd and Pb adsorption. The phosphate-containing functions in TC and SC could also help improve the adsorption capacity.
| Carbon | Ion | T (°C) | Langmuir | Freundlich | |||||
|---|---|---|---|---|---|---|---|---|---|
| Qm (mg g−1) | Ka (L mg−1) | RL | R2 | KF (mg g−1) | 1/n | R2 | |||
| TC | Cd | 25 | 48.08 | 0.059 | 0.252 | 0.99 | 9.022 | 0.339 | 0.99 |
| Pb | 25 | 61.73 | 0.036 | 0.357 | 0.99 | 6.252 | 0.461 | 0.97 | |
| SC | Cd | 25 | 40.98 | 0.027 | 0.423 | 0.98 | 3.008 | 0.488 | 0.96 |
| Pb | 25 | 58.82 | 0.009 | 0.689 | 0.99 | 5.352 | 0.476 | 0.97 | |
| CAC | Cd | 25 | 56.82 | 0.035 | 0.362 | 0.99 | 5.747 | 0.459 | 0.97 |
| Pb | 25 | 70.42 | 0.036 | 0.363 | 0.99 | 6.072 | 0.505 | 0.97 | |
1/n represents the curve of isotherm or the energy distribution of adsorption site. 1/n < 1 indicates a more heterogeneous adsorption site and energy distribution and a feasible adsorption, and 2 < n < 10 means an easier adsorption.14 The values of 1/n in both carbons ranged between 0.1 and 0.5 in this work, indicating that the adsorption of both metal ions on both carbons could occur easily. RL values based on Langmuir isotherm can also be used to assess the difficulty of adsorption. Adsorption is unfavorable, linear, favorable, or irreversible when RL > 0, RL = 1, 0 < RL < 1, or RL = 0, respectively.40 The values of RL ranging from 0 to 1 meant that the adsorption was favorable in the present study.
Comparatively, TC possessed a better adsorption ability for both ions than SC, and the adsorption capacity of Pb was larger than that of Cd on both carbons. TC had a smaller pHIEP compared to SC, and thus more negative charges at the same low initial solution pH, which helped more with electrostatic attraction and complexing adsorptions. Similar to the results in Shi et al.,24 the Qm of Pb was obviously greater than that of Cd, indicating a better affinity of Pb on both carbons. There were three reasons: (1) the free energy and diameter of hydrated ions are main factors in determining adsorption capacity. Hydrated ions with smaller diameters can easily diffuse inside carbon particles. Ions with low and high hydrated energy tend to be combined with carbon and water, respectively; so inside carbon particulates, ions with low hydrated energy are easy to dehydrate, and thus adsorbed inside carbons. Therefore, the Qm of Pb was more than that of Cd due to a lower diameter (the diameter of hydrated ions of Cd 4.26 Å, Pb 4.01 Å) and free energy of hydrated ion of Pb.32 (2) Metal ions with a large diameter tend to be electrostatically adsorbed, so hydroxy metal ions are more easily adsorbed compared to free metal ions. The hydrolysis constant of pK1 of Pb is 7.9 less than 10.1 of Cd, indicating Pb could be easily hydrolyzed, and thus was readily adsorbed by electrostatic attraction.41 (3) Metal with a larger electric negativity can easily receive electrons and vice versa. The electric negativity of Pb is 2.33 more than 1.69 of Cd, which was also one of the reasons for higher Qm of Pb; furthermore, the greater the electric negativity of metals, the more acidity there is, so Pb tended to combine with hydroxyl radical, carboxyl, and phosphate groups on activated carbons surface according to the theory of hard and soft acid and base.42
3.8 Adsorption mechanism and types
In this study, all the values of R2 obtained from intraparticle diffusion equation were more than 0.96%, indicating that the intraparticle diffusion was a rate-limiting step in adsorption. However, if intraparticle diffusion is the only rate-controlling process, the fitted curve will start from the origin; if film diffusion and intraparticle diffusion occur independently, there should be two or more fitted lines, the first one representing boundary diffusion, and the second one representing intraparticle diffusion.43 In the present research, there was only one fitted line which did not start at the origin (Fig. 6); moreover, the values of R2 obtained from the film diffusion model were less than 0.85 (data not provided in this paper), suggesting that both film and intraparticle diffusions occurred simultaneously, but intraparticle diffusion was the main rate control step; similar results were presented in studies of Youssef et al.3 and Xu and Liu.22 Research also showed that the formation of a chemical bond was also a rate control process.44 The values of R2 resulted from the pseudo-second order model were 0.99 in this experiment, revealing that chemical reaction played an important role during adsorption of Cd and Pb on TC and SC, and was also a rate limiting step.22 | ||
| Fig. 6 Intraparticle diffusion model of Cd and Pb adsorption on Typha angustifolia activated carbon (TC) and Salix matsudana activated carbon (SC). Error bar is the standard deviation of the mean. | ||
ΔG0, ΔH0, and ΔS0 also provide a way to judge adsorption mechanism. ΔG0 < 0 reveals that adsorption process is controlled by the change of entropy not enthalpy; ΔG0 > 0 indicates a common phenomenon of ion exchange adsorption, the formation of metal ion complexing compound on adsorptive sites of adsorbent.44 ΔH0 > 0 shows there is a chemical or bonding reaction, and ΔS0 > −10−12 kJ K−1 mol−1 means there is a dissociation or deprotonation during adsorption.24 In the present study, most ΔG0 < 0 suggested that the change of entropy controlled the adsorption process; for SC, a small part of ΔG0 > 0 referred to that ion exchange occurred during the adsorption of Cd on SC; ΔH0 > 0 and ΔS0 > 0 indicated that the adsorption contained chemical reaction and dissociation, respectively. Furthermore, the R2 values from isothermal models ranged from 0.96 to 0.99, suggesting that there were both single molecular layer chemical reactions and multiple molecular layer physical reactions during adsorption.45
After the adsorption of Cd and Pb, the appearance of some new peaks at 530.31 eV (CO), 533.93, 534.47 eV (–COOH) (Fig. 4 TC-f), 532.43 eV (C–OH), and 533.14 eV (C–O–C) (Fig. 4 SC-f), the disappearance of peak at 536.53 eV (Fig. 4 SC-e), and the shift of some peaks assigned to groups containing O further suggested the occurrence of chemical reactions between TC and SC with Cd and Pb.
3.9 Adsorption and desorption cycle
Desorption studies of metals are helpful for the recycling of adsorbents and the recovery of metal resources. The results of the multiple adsorption/desorption cyclic tests are presented in Table 5. The adsorption capacity of TC for Cd and Pb was reduced by 27.94 and 20.2% from 19.19 to 14.2 mg g−1 and 20.78 to 16.62 mg g−1, respectively. While for SC, it declined by 33.50 and 22.08%, respectively. Thus, it was feasible for the recovery of Pb and Cd and the regeneration of TC and SC after adsorption equilibrium, which would effectively reduce the overall cost for the adsorbents. The reason for the small fraction of heavy metal ions adsorbed by TC and SC not recoverable by desorption was probably that the metal ions were bound to the TC and SC through strong interaction, and thus desorption capacity reduced during successive cycles. For the more precision reason of desorption, further study is required to know whether HCl provides an ability to change the structure of the adsorbent.| Carbon | TC | SC | ||
|---|---|---|---|---|
| Ion | Cd | Pb | Cd | Pb |
| Adsorption/desorption (mg g−1) | 19.19/18.14 | 20.78/20.05 | 11.82/11.07 | 19.88/18.92 |
| 17.89/16.35 | 19.84/18.61 | 10.77/9.70 | 18.78/17.44 | |
| 16.02/14.11 | 18.44/16.72 | 9.42/8.18 | 17.31/15.49 | |
| 14.02/11.70 | 16.62/14.35 | 7.86/6.49 | 15.49/13.34 | |
4. Conclusions
The adsorption efficiency relied positively on the pH and humic acid but negatively on NaCl in the media. Cd and Pb adsorption from solution was spontaneous and endothermic and contained physico-chemical reactions. The intra-particle diffusion and chemical reaction were the adsorption rate control steps. The pseudo second order and Langmuir models fitted the adsorption process very well. The Qm of Cd and Pb was 48.08 and 61.73 mg g−1 on TC and 40.98 and 58.82 mg g−1 on SC, respectively. Both activated carbons provided good regenerating and reusing properties. The results indicated that both TC and SC were potential adsorbents for Cd and Pb from aqueous solution. Yet, further researches are required to optimize the preparation and maximize the adsorption capacity.Acknowledgements
This work was funded by the Project of Education Department of Hunan Province of China (Grant No. 16A221) and the National Natural Science Foundation of China (Grant No. 51579097). We are thankful to the Editors and Referees for their precious comments on our manuscript.References
- R. A. Goyer, Environ. Health. Perspect., 1993, 100, 177–187 CrossRef CAS PubMed.
- G. Van Maele-Fabry, N. Lombaert and D. Lison, Environ. Int., 2016, 86, 1–13 CrossRef CAS PubMed.
- A. M. Youssef, T. El-Nabarawy and S. E. Samra, Colloids Surf., A, 2004, 235, 153–163 CrossRef CAS.
- Z. Li, B. Huang, J. Huang, G. Chen, C. Zhang, X. Nie and G. Zeng, RSC Adv., 2015, 5, 90588–90595 RSC.
- G. Issabayeva, M. K. Aroua and N. M. N. Sulaiman, Bioresour. Technol., 2006, 97, 2350–2355 CrossRef CAS PubMed.
- I. Ghodbane, L. Nouri, O. Hamdaoui and M. Chiha, J. Hazard. Mater., 2008, 152, 148–158 CrossRef CAS PubMed.
- F. Bouhamed, Z. Elouear and J. Bouzid, J. Taiwan Inst. Chem. Eng., 2012, 43, 741–749 CrossRef CAS.
- M. A. P. Cechinel and A. A. U. D. Souza, J. Cleaner Prod., 2014, 65, 342–349 CrossRef CAS.
- C. K. Singh, J. N. Sahu, K. K. Mahalik, C. R. Mohanty and B. R. Mohan, J. Hazard. Mater., 2008, 153, 221–228 CrossRef CAS PubMed.
- L. Wang, J. Zhang, R. Zhao, Y. Li and C. Li, Bioresour. Technol., 2010, 101, 5808–5814 CrossRef CAS PubMed.
- K. Q. Li, Z. Zheng and Y. Li, J. Hazard. Mater., 2010, 181, 440–447 CrossRef CAS PubMed.
- M. Momčilović, M. Purenović, A. Bojić, A. Zarubica and M. Ranđelović, Desalination, 2011, 276, 53–59 CrossRef.
- A. M. Bahri, L. Calvo, M. A. Gilarranz and J. J. Rodriguez, Biochem. Eng. J., 2012, 203, 348–356 Search PubMed.
- M. Maddisona, T. Mauring, K. Remm, M. Lesta and U. Mander, Ecol. Eng., 2009, 35, 258–264 CrossRef.
- L. Ren, J. Zhang, Y. Li and C. Zhang, Biochem. Eng. J., 2011, 168, 553–561 CAS.
- G. Berndes, F. Fredrikson and P. Börjesson, Agric., Ecosyst. Environ., 2004, 103, 207–223 CrossRef CAS.
- M. A. Z. Abidin, A. A. Jalil, S. Triwahyono, S. H. Adam and N. H. N. Kamarudin, Biochem. Eng. J., 2011, 54, 124–131 CrossRef CAS.
- V. K. Gupta and A. Nayak, Biochem. Eng. J., 2012, 180, 81–90 CAS.
- S. Orsetti, M. D. L. M. Quiroga and E. M. Andrade, Chemosphere, 2006, 65, 2313–2321 CrossRef CAS PubMed.
- Y. Li, Q. Yue and B. Gao, J. Hazard. Mater., 2010, 178, 455–461 CrossRef CAS PubMed.
- M. Li, Y. Liu, G. Zeng, S. Liu, X. Hu, D. Shu, L. Jiang, X. Tan, X. Cai and Z. Yan, J. Colloid Interface Sci., 2017, 485, 269–279 CrossRef CAS PubMed.
- T. Xu and X. Liu, Chin. J. Chem. Eng., 2008, 16, 401–406 CrossRef CAS.
- D. Doulia, C. Leodopoulos, K. Gimouhopoulos and F. Rigas, J. Colloid Interface Sci., 2009, 340, 131–141 CrossRef CAS PubMed.
- T. Shi, S. Jia, Y. Chen, Y. M. Wen and C. Du, J. Hazard. Mater., 2009, 169, 838–846 CrossRef CAS PubMed.
- Y. S. Ho, G. McKay, D. A. J. Wase and C. F. Forster, Adsorpt. Sci. Technol., 2000, 18, 639–650 CrossRef CAS.
- A. M. B. Hamissa, A. Lodi, M. Seffen, E. Finocchio and R. Botter, Biochem. Eng. J., 2010, 159, 67–74 Search PubMed.
- B. I. Olu-Owolabi and E. I. Unuabonah, Appl. Clay Sci., 2011, 51, 170–173 CrossRef CAS.
- L. D. Pablo, M. L. Chávez and M. Abatal, Biochem. Eng. J., 2011, 171, 1276–1286 Search PubMed.
- W. S. W. Ngah, L. C. Teong, R. H. Toh and M. A. K. M. Hanafiah, Biochem. Eng. J., 2013, 223, 231–238 Search PubMed.
- A. Örnek, M. Özacar and İ. A. Şengil, Biochem. Eng. J., 2007, 37, 192–200 CrossRef.
- D. Mohan, C. U. Pittman, M. Bricka, F. Smith and B. Yancey, J. Colloid Interface Sci., 2007, 310, 57–73 CrossRef CAS PubMed.
- E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef CAS.
- E. Eren, J. Hazard. Mater., 2009, 165, 63–70 CrossRef CAS PubMed.
- X. L. Wu, D. Zhao and S. T. Yang, Desalination, 2011, 269, 84–91 CrossRef CAS.
- T. S. Anirudhan and P. S. Suchithra, J. Ind. Eng. Chem., 2010, 16, 130–139 CrossRef CAS.
- X. Li, Y. Li and Z. Ye, Biochem. Eng. J., 2011, 178, 60–68 CAS.
- L. Osipow, Soil Chemical: Theory and Industrial Applications, Krieger, NewYork, 1972 Search PubMed.
- L. Chávez-Guerrero, R. Rangel-Méndez, E. Muñoz-Sandoval, D. A. Cullen, D. J. Smith, H. Terrones and M. Terrones, Water Res., 2008, 42, 3473–3479 CrossRef PubMed.
- H. Liu, P. Dai, J. Zhang, C. Zhang, N. Bao, C. Cheng and L. Ren, Chem. Eng. J., 2013, 228, 425–434 CrossRef CAS.
- T. Depci, A. R. Kul and Y. Önal, Biochem. Eng. J., 2012, 200, 224–236 Search PubMed.
- D. S. Lin, Y. M. Xu, G. H. Sun and X. H. Dai, J. Agro-Environ. Sci., 2007, 26, 510–515 CAS.
- Z. T. Zhou, J. Agro-Environ. Sci., 1981, 2, 68–73 Search PubMed.
- Y. S. Ho, Water Res., 2003, 372, 323–2330 Search PubMed.
- T. S. Anirudhan, P. S. Suchithra and S. Rijith, Colloids Surf., A, 2008, 326, 147–156 CrossRef CAS.
- T. Karthikeyan, S. Rajgopal and L. R. Miranda, J. Hazard. Mater., 2005, 124, 192–199 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |