DOI:
10.1039/C6RA27916C
(Paper)
RSC Adv., 2017,
7, 12576-12585
Rapid detection of blaNDM-1 in multidrug-resistant organisms using a novel electrochemical biosensor
Received
8th December 2016
, Accepted 16th February 2017
First published on 22nd February 2017
Abstract
Traditional detection methods of multidrug-resistant organisms (MDROs) include phenotype-based screening for carbapenem-hydrolyzing enzymes, such as the paper disk diffusion assay, the broth trace dilution assay, a modified Hodge assay, and sequencing analysis. In the present study, a novel electrochemical biosensor was successfully constructed, and highly specific locked nucleic acid (LNA) probes were used for the marker-free, direct, and rapid detection of the drug-resistance gene blaNDM-1 in MDROs in complex clinical specimens. The highly specific LNA probes were designed for the direct and fast detection of blaNDM-1 DNA in clinical bacterial specimens without requiring culturing or amplification. The linearity range was 10 pg L−1 to 100 μg L−1, and the limit of detection was as low as 1 pg L−1. The mean coefficient of variation values of intra-day and inter-day reproducibility were 3.93% and 5.96%, respectively. The time for biosensor incubation and scanning detection was as short as 30 min. In addition, the immunity of the electrochemical biosensor against interference was excellent. In the present study, highly specific LNA probes, fast detection, and the simultaneous acquisition and analysis of data were combined to create a novel marker-free direct detector of bacterial drug-resistance genes, which is crucial for the fast diagnosis of multidrug-resistant organisms and holds promise for the fast accurate detection of other drug-resistant bacteria.
1. Introduction
In August 2010, “superbacteria” were reported by social media causing worldwide panic.1,2 Superbacteria are bacteria carrying a new multi-drug-resistance gene.3 This gene encodes a new metallo-β-lactamase that can hydrolyze virtually all β-lactam antibiotics except aztreonam, which explains the multidrug resistance observed in superbacteria. The discoverers of this gene believed New Delhi, India to be the source of the superbacteria and thus termed this novel metallo-β-lactamase New Delhi metallo-β-lactamase-1 (blaNDM-1).4–6 The full name of the drug-resistant bacteria carrying blaNDM-1 is type-I New Delhi metallo-β-lactamase producing enterobacteria, which include Escherichia coli (E. coli), Klebsiella pneumoniae, Enterobacter cloacae and proteus.7–9 Although these blaNDM-1-containing bacteria exhibit virulence similar to other enterobacteria, they are resistant to a wide spectrum of β-lactam antibiotics, such as penicillins, cephalosporins, and carbapenems.10 To prevent the prevalence of blaNDM-1-producing multidrug-resistant organisms (MDROs), it is critical to develop a fast specific detection method for the early screening and treatment of these organisms.11
Currently, blaNDM-1 detection methods include phenotype-based screening for carbapenem-hydrolyzing enzymes, such as the paper disk diffusion assay, the broth trace dilution assay, and a modified Hodge assay.12 After the bacteria are screened, carbapenem-hydrolyzing enzyme-positive strains are subject to a PCR assay using target primers for blaNDM-1, followed by sequencing analysis to identify blaNDM-1-producing bacteria. The activity of the metalloenzyme depends on the presence of Zn2+.13 Therefore, the phenotypic detection of metalloenzyme-producing bacteria is based on chelation with metal-chelating agents and Zn2+, which inactivates the metalloenzyme and widens the inhibition zone. Based on phenotypic screening, a double-disk synergy assay is used to reconfirm the production of the metalloenzyme. In brief, imipenem (IPM) and EDTA-impregnated paper disks are placed 10–15 mm apart on a plate covered with bacteria.14,15 If the imipenem inhibition zone widens in the direction of the EDTA disks, the bacteria are judged to produce the metalloenzyme. PCR is the gold standard of metalloenzyme detection. It was reported that quantitative fluorescence PCR can detect the blaNDM-1 gene extracted from a single colony within 1 h.14,16 Nevertheless, these methods have drawbacks such as being time-consuming, being complex, and having low sensitivity and specificity, as well as high costs; therefore, they are not ideal for the fast clinical detection of blaNDM-1-producing MDROs.
Unlike the detection methods described above, electrochemical biosensors have advantages such as short analysis time, device miniaturization, high specificity and sensitivity, and low limits of detection.17,18 An electrochemical biosensor is typically composed of biological molecule recognition elements and a signal transformer. The potential difference between the electrode surface and electrolyte solution changes when the electrochemical reactions of the chemical elements in the base electrolyte solution change. The sensor's signal transformer transforms such change into analyte concentration-related signal output, which is then processed and displayed by an electric signal acquisition and analysis system.19
The development of a method for direct bacteria detection without amplification is a long-term goal. However, low sensitivity is the main problem for the direct detection of a low abundance of NDM+ multi drug resistant bacteria. In contrast, specificity is another important concern for NDM+ multidrug-resistant bacteria. Meanwhile, most biosensors are immobilized with DNA probes, causing two problems: unspecific hybridization and less sensibility. To solve these problems, the target sequences underwent PCR amplification before detection with biosensors; however, PCR is laborious and time consuming. Therefore, the design of specific detection probes should be the focus of the electrochemical detection of NDM+ multidrug-resistant bacteria. In the present study, we designed a pair of 1ocked nucleic acid (LNA) probes aimed at the entire genome of blaNDM-1. LNA nucleotides contain a methylene bridge that connects the 2′-oxygen of ribose with the 4′-carbon.20 This bridge could reduce the conformational flexibility of the ribose and increase the local organization of the phosphate backbone.21,22 The 5′-terminus of the probe was modified with a sulfhydryl group, and the probe was fixed onto the Au electrode surface via a Au–S bond. The probe was partially complementary to the target gene. In the presence of the target gene in the hybridization solution, the LNA probe and its complementary sequence formed a nucleic acid complex, which produced redox reactions on the biosensors surface and gave rise to electrochemical signal changes. This method did not require DNA purification or PCR amplification and allowed for the accurate detection of drug-resistance genes in clinical bacterial specimens. We studied the repeatability and specificity of the electrochemical biosensors. We also detected the presence of blaNDM-1 DNA and using the electrochemical biosensor. The response current change values (ΔI) clearly increased for the blaNDM-1 DNA detected sample when compared to the negative control. In contrast, the response current change values (ΔI) remained largely unchanged for the sample without detected blaNDM-1 DNA detected when compared to the negative control. The highlight of this approach is direct, rapid, specific and effective detection. Scheme 1 shows the electrochemical detection device and its detection process and rationale.
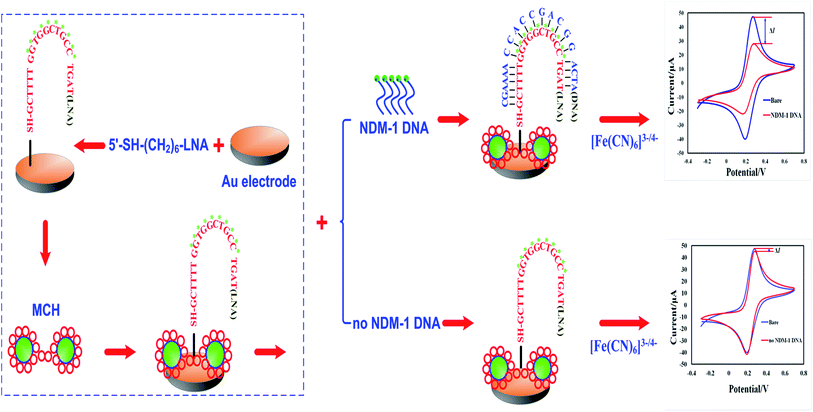 |
| | Scheme 1 Schematic diagram of the electrochemical biosensor fabrication process and the LNA probes hybridized to a target DNA molecule. | |
2. Materials and methods
2.1 Reagents
LNA probe 5′-SH-(CH2)6-GCTTTTG*G*T*G*G*C*T*G*C*C*TGAT (SH denotes the thiol group, the asterisk-marked (*) bases represent the LNA) were purchased from Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China). The probes were purified by reverse-phase HPLC and characterized by MALDI-TOF spectrometry. Absolute alcohol, nitric acid, acetone, potassium ferricyanide, PBS powder, and potassium ferrocyanide were commercially available analytically pure products. [Fe(CN)6]3−/4− was purchased from the Chongqing Dongfang Reagents Factory (Chongqing, China). The clinical bacterial specimens used in this study were provided by the Laboratory of Microbiology of our hospital's Department of Laboratory Medicine, and the control, a blaNDM-1 plasmid, was donated by Prof. Bo Zhang from the Hospital Infection Control Department of Southwest Hospital.
2.2 Apparatus
A model ZD-2 automatic electric potential titrimeter (Shanghai Jingke Leici Co., Ltd, China), a model KQ-5200B ultrasound washer (Kunshan Ultrasound Instruments Co., Ltd, Jiangsu, China), and a pH meter (Mettler-Toledo, Switzerland) were used in this study. A CHI760D electrochemical workstation was purchased from Shanghai Chenhua Instruments Co., Ltd. (Shanghai, China). An electrochemical biosensor detection system was fabricated using a homemade LNA-modified Au electrode as the working electrode, a saturated calomel electrode as reference electrode, and a platinum electrode as the counter electrode. The electrochemical properties of the biosensors were characterized by cyclic voltammetry (CV) at various stages of modification. An electrochemical biosensor was fabricated using the LNA probe-modified Au-electrode as a working electrode. The supporting electrolyte buffer (SEB) of the LNA–DNA hybridization reaction solution (PBS, pH 7.4) contained 2.0 mmol L−1 [Fe(CN)6]3−/4− and 5.0 mmol L−1 KCl. Atomic force microscope (AFM) images of the biosensors, the LNA probes, and the LNA–DNA hybridization complex were collected by employing an AFM IPC-208B (Chongqing University, China). The AFM IPC-208B measurement was performed under the following conditions: scan range of 10.5 nm × 10.5 nm, tungsten probes (force constant 0.06 Nm), tapping mode imaging, scanning point by point at room temperature, and data processed by G3DR software.
2.3 Preparation of the LNA electrochemical biosensors
The Au electrode (3 mm in diameter) was burnished first with 0.3 μm and then with 0.05 μm Al2O3 powder. Between burnishes, the electrodes were washed first with purified water and then in an ultrasonic bath with nitric acid, acetone and ultrapure water each for 5 min. After each wash, the electrodes were air-dried. Then, twenty microliters of various concentrations of the sulfhydryl-modified LNA probe solution was dripped onto the activated electrochemical biosensor surface and air-dried at room temperature. Subsequently, 10 μL of 1 mmol L−1 6-mercapto-1-hexanol (MCH) solution was added to block the non-specific absorptive sites on the biosensor surface. After 1 h, the Au electrode surface was washed with superpure water to complete the processing of the LNA probe-modified electrochemical biosensor. Prior to use, the Au electrodes were stored at 4 °C in the dark.
2.4 Hybridization reaction
The blaNDM-1 target sequence solution (10−5, 10−4, 10−3, 10−2, 10−1, 1.0, 101, and 102 μg L−1) was added into different electrochemical biosensors, respectively, and hybridization lasted for 45 min at 37 °C. In the presence of the target gene in the hybridization solution, the LNA probe specifically bound to the target sequence to form the LNA–DNA complex, which deters electron transfer on the electrode surface and increases the response current change of the electrochemical biosensor. Such a signal change (ΔI) correlates to the level of nucleic acid hybridization on the biosensor surface. The higher the ΔI value is, the larger the number of analytes bound to the LNA probes is. The original reduction peak current of the LNA probe-modified biosensor was deemed I0, and the reduction peak current following nucleic acid hybridization was recorded as I. The signal change was determined by the following equation: ΔI = |I − I0|.
2.5. Sensitivity, specificity, and reproducibility of biosensors
The current differences before and after the hybridization and the reaction time were determined for all of the various final concentrations of the target sequence solution (10−5, 10−4, 10−3, 10−2, 10−1, 1.0, 101, and 102 μg L−1). Linear regression analysis was performed for the response current differences and the blaNDM-1 concentrations. The assay was repeated 6 times, and the mean value of the measurements was calculated.
A blaNDM-1 probe-modified biosensor was incubated 30 min with the following solutions: solution a, containing 100 μg L−1 of the blaNDM-1 control plasmid; solution b, containing 100 μg L−1 of the blaNDM-1 control plasmid and an interfering substance (1 mg L−1 E. coli, 1 mg L−1 Staphylococcus aureus); solution c, containing an interfering substance (1 mg L−1 E. coli, 1 mg L−1 Staphylococcus aureus, determined to contain no blaNDM-1 gene); and solution d, a negative control (PBS solution). Following incubation, cyclic voltammetric scanning was performed at room temperature, and the response current values were recorded. The measured peak current values were compared to evaluate the specificity of the electrochemical biosensors.
To validate the assay reproducibility of the biosensors, intra-day (6 assays for each concentration) and inter-day (3 assays for each concentration for 6 consecutive days) precision assays were performed using 10, 100, and 1000 μg L−1 of the target sequence solution. The response current values were recorded, and the ΔI ± SD and the coefficient of variation were calculated to evaluate the reproducibility of the electrochemical biosensors.
3. Results and discussion
3.1 Electrochemical characterization of the stepwise modified biosensor
Cyclic voltammetry was performed to characterize the electrochemical characteristics of the biosensor at various stages of modification. Fig. 1 is the cyclic voltammogram showing the real-time detection of the electrochemical biosensor. The abscissa represents the working potential of scanning, and the ordinate represents the current response. Due to the absence of redox-active substances in the system, within the scanning range from −0.3 V to 0.7 V, no obvious redox peaks appeared when the LNA probe-modified electrode was immersed into a PBS solution (Fig. 1, curve a); curve b is the cyclic voltammogram of the bare Au electrode incubated with the SEB, and curve c is the cyclic voltammogram of the LNA probe-modified electrode incubated with the SEB. As a non-conductive substance, the LNA probe deters the electron transfer of the conductive ions in the solution on the surface of the electrochemical biosensor to a certain degree, resulting in a decrease in the redox peak current; curve d is the cyclic voltammogram following blockage with MCH, indicating that MCH deterred electron transfer while blocking the non-specific absorptive sites and led to a further decrease in the peak current value, curve e is the cyclic voltammogram of the modified electrochemical biosensor incubated with a solution containing the interfering substance only (no blaNDM-1 gene-free E. coli or Staphylococcus aureus). Unlike curve d, curve e shows that the current-response of the electrochemical biosensor was virtually unchanged before and after incubation. Curve f is the cyclic voltammogram of the LNA probe-modified electrochemical biosensor incubated with a solution containing 100 μg L−1 of blaNDM-1 DNA. On the biosensor surface, the nucleic acid hybridization complex is a biomacromolecule without electroactivity, which further deters electron transfer. Therefore, the redox peak current was lower in curve f than in curves d and e. These results demonstrate that we successfully constructed an LNA-modified electrochemical biosensor for the direct detection of blaNDM-1.
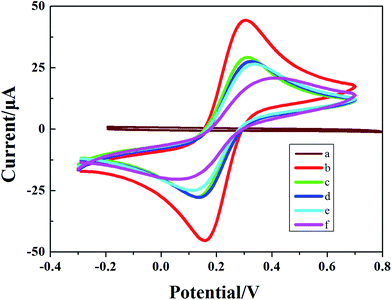 |
| | Fig. 1 Cyclic voltammogram scans of different electrochemical biosensors made by the modified fabrication processes. (a) A bare Au electrode in PBS (pH 7.4, 0.05 mol L−1); (b) a bare Au electrode; (c) LNA; (d) MCH/LNA; (e) interfering substances/MCH/LNA; and (f) a blaNDM-1/MCH/LNA-modified electrode in a solution of PBS (pH 7.4, 0.05 mol L−1) with 2.0 mmol L−1 [Fe(CN)6]3−/4− at a scan rate of 50 mV s−1. | |
3.2 Electrochemical impedance spectroscopy of the stepwise modified electrode
Electron-transfer resistance (Ret) at the electrode is an important parameter, thus electrochemical characteristics of the electrochemical biosensor were investigated by electrochemical impedance spectroscopy (EIS) measurements. It is well known that the semicircle diameter of EIS is equal to Ret in the Nyquist diagram. Fig. 2 illustrates the EIS of different electrodes. The bare Au electrode has a small semicircle in the high-frequency section (curve a). Then, following loading of the LNA probe on the Au electrode, Ret increased compared to the bare Au electrode (curve b). The reason for this may be that the LNA probe formed some barrier obstructing the electron transfer. When blocked with MCH, the semicircle increased markedly (curve c) because the captured MCH with its non-electroactive properties obstructs electron transfer. Ret further increased following successive blaNDM-1 adsorption onto the electrode surface (curve d), which attributed to the inhibition effect of the blaNDM-1/MCH/LNA biomacromolecules on electron transfer.
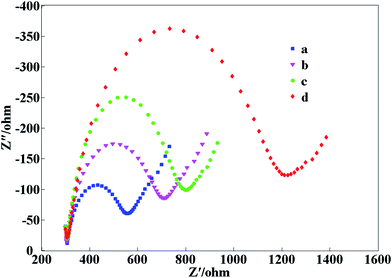 |
| | Fig. 2 Electrochemical impedance spectroscopy (EIS) scans of different electrochemical biosensors made by the modified fabrication processes. (a) A bare Au electrode; (b) LNA; (c) MCH/LNA; (d) a blaNDM-1/MCH/LNA-modified electrode in a solution of PBS (pH 7.4, 0.05 mol L−1) with 2.0 mmol L−1 [Fe(CN)6]3−/4− at a scan rate of 50 mV s−1. | |
3.3 AFM characterization of the stepwise-modified electrodes
The IPC-208B high-resolution AFM, which integrates the techniques of scanning tunnel microscopy (STM) and AFM, was independently developed by Chongqing University.23 This microscope is used to detect the vertical motion of a microcantilever and can offer a maximal precision of 0.1 nm horizontally and 0.01 nm vertically.24,25 Colored graphics of the analytical results of the Au electrode fabrication and the application of the electrodes using the AFM IPC-208B were obtained. The AFM IPC-208B technique was employed to characterize the antisense oligonucleotide binding to a dextran polymer from the spatial arrangement of atoms.23,24 This study provides not only a novel route for the characterization of the spatial structure of the nucleotide hybridization complex using AFM IPC-208B, but also a new method to confirm the intermolecular hydrogen bonding between the LNA probes and the blaNDM-1 DNA. After every modified step, the thickness and surface morphology of the modified films were obviously clearly altered. A colored graphic of the analytical results of the locations of various elements are marked by different pixels. Fig. 3A shows an image of the LNA probes modified on the surface of the electrochemical biosensor, which were irregularly wrinkled and exhibited a root mean square (RMS) roughness of 14.296 nm (Fig. 3C). Fig. 3B shows an image of the LNA probes and the blaNDM-1 DNA hybridization complex, which were double helix structures and exhibited a RMS roughness of 17.423 nm (Fig. 3D). In Fig. 3A and B, white square pixels represent C-atoms, black circle pixels represent O-atoms, and black star shapes represent P-atoms. The upper-right corner of Fig. 3A and B shows a schematic diagram of the LNA probes and the blaNDM-1 DNA hybridization complex, respectively. These of these results appear to further support the successful fabrication and potential application of the proposed biosensor.
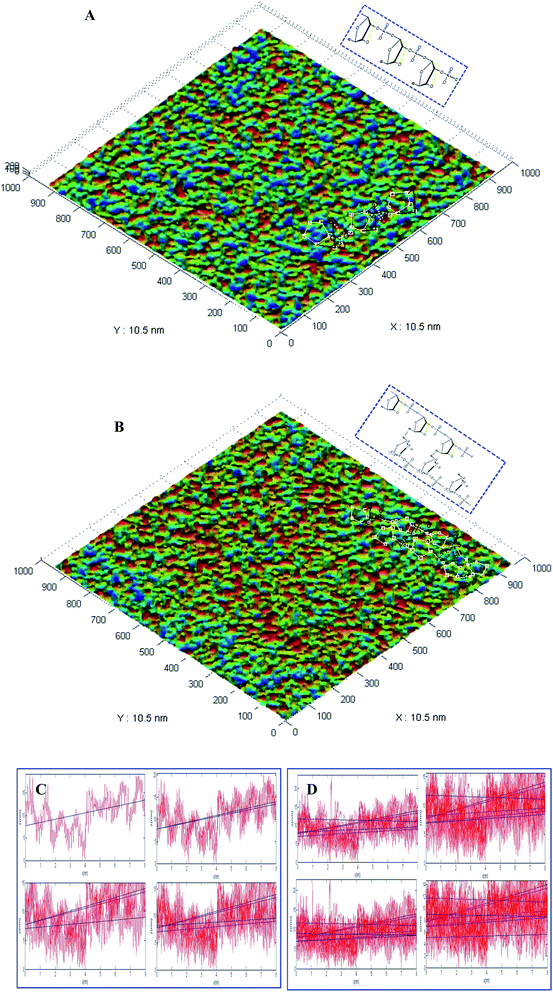 |
| | Fig. 3 AFM images and the root mean square (RMS) roughness of the different electrochemical biosensors made by the modified fabrication processes. AFM IPC-208B images of (A) the LNA probe C-atoms ( ), O-atoms ( ), O-atoms ( ), and P-atoms ( ), and P-atoms ( ), and (B) the LNA probes and the blaNDM-1 DNA hybridization complex. The upper-right corner of Fig. 3A and B shows a schematic diagram of the LNA probes and the blaNDM-1 DNA hybridization complex, respectively. The root mean square (RMS) roughness of (C) the LNA probes and (D) the LNA probes and the blaNDM-1 DNA hybridization complex modified on the surface of electrochemical biosensors. ), and (B) the LNA probes and the blaNDM-1 DNA hybridization complex. The upper-right corner of Fig. 3A and B shows a schematic diagram of the LNA probes and the blaNDM-1 DNA hybridization complex, respectively. The root mean square (RMS) roughness of (C) the LNA probes and (D) the LNA probes and the blaNDM-1 DNA hybridization complex modified on the surface of electrochemical biosensors. | |
3.4 Optimization of the LNA probe concentration
To determine the optimal concentration of the LNA probe, we performed an LNA probe optimization assay. In brief, 0.5, 1.0, 1.5, 2.0, and 2.5 μM of LNA probe solution were prepared and immobilized on the surface of the working electrode of the biosensor. The response current was then determined by cyclic voltammetry. As shown in Fig. 4, the cyclic voltammograms before and after LNA probe modification in the SEB differed significantly, suggesting that the specific probes were immobilized successfully onto the surface of the electrochemical biosensor and influenced the electrochemical signals on the modified biosensor. On the electrochemical biosensor, the current response change value first increased and then decreased with the increasing LNA probe concentration. When the LNA probe concentration increased from 0.5 μM to 1.5 μM, the response current change value tended to increase. When the LNA probe concentration increased from 1.5 μM to 2.5 μM, the response current change value tended to decrease. When the LNA probe concentration was 1.5 μM, the response current change value reached a peak. Therefore, the optimal concentration of the LNA probe was 1.5 μM.
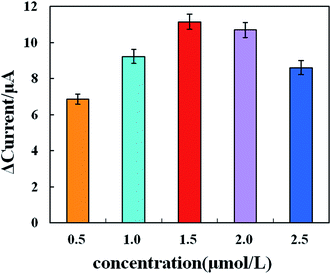 |
| | Fig. 4 CV scans of different concentrations of probe-modified electrochemical biosensors. The error bars represent the standard deviations of three parallel samples at each target concentration. | |
The response current change value (ΔI) increased significantly with LNA probe concentrations ranging from 0.5 μM to 1.5 μM and then decreased from 1.5 μM to 2.5 μM. The reason for this decrease may be that the high concentration of the LNA probe modified too much of the electrode surface and generated a high steric-hindrance effect, which may negatively affect LNA–DNA hybridization. The spatial arrangement of the LNA probe molecules may explain the probe-concentration-dependent behavior. When the LNA probe concentration rises above the threshold, the LNA probe molecules are densely packed on the Au electrode surface and provide limited space for the target sequences to enter and hybridize. In addition to this spatial restriction, the spatial arrangement of the high concentration LNA probes and electrostatic repulsion between DNA fragments may also to decrease the hybridization efficiency.26,27
3.5 Specificity of the biosensors
Specificity is an important measure of the performance of a biosensor's detection.28 We detected blaNDM-1 DNA and an interfering substance (a blaNDM-1 gene-free E. coli/Staphylococcus aureus solution) using the electrochemical biosensor. As shown in Fig. 5, the response current change values (ΔI) before and after incubation in solution “a” and solution “b” were 10.16 and 9.79 μA, respectively. In contrast, the response current values remained largely unchanged before and after incubation in solution “c”, which contained the interfering substance only, and in solution “d”, a negative control. The results indicate that even if the concentration of the interfering substance was 10 times the blaNDM-1 DNA concentration, the detection results were not significantly affected, suggesting excellent immunity to interference of the blaNDM-1 electrochemical biosensor that we constructed. Therefore, the biosensor exhibited good selectivity for blaNDM-1 and could be used for the accurate quantification of blaNDM-1.
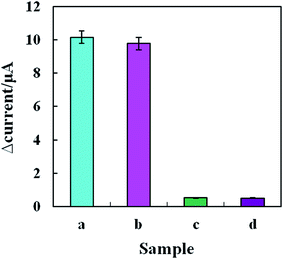 |
| | Fig. 5 Specificity of the LNA-modified electrochemical biosensors. (a) 100 μg L−1 blaNDM-1 DNA; (b) 100 μg L−1 blaNDM-1DNA and 1 mg L−1 of interfering substances; (c) 1 mg L−1 of interfering substances; (d) negative control (error bars: SD, n = 6). | |
3.6 Reproducibility of the biosensors
To assess the assay reproducibility, the same person performed repeated assays on a stable specimen at short time intervals under the same operating conditions, and a mean value, standard deviation, and coefficient of variation were then calculated for these assay results. The mean value reflects the average of the measurements, and the standard deviation suggests the degree of dispersion of the measurements in relation to the mean value. The intra-day reproducibility assay required multiple assays on the same specimen within a single day, and the inter-day reproducibility assay required one assay per day for 6 consecutive days on the same specimen. Table 1 shows the results of the reproducibility assay of the LNA-modified electrochemical biosensor. At 10 μg L−1 of blaNDM-1 DNA, the coefficient of variation values of intra-day and inter-day reproducibility assays were 3.61% and 5.28%, respectively; at 100 μg L−1 of blaNDM-1 DNA, the coefficient of variation values of intra-day and inter-day precision assays were 4.06% and 6.08%, respectively; and at 1000 μg L−1 of blaNDM-1 DNA, the coefficient of variation values of intra-day and inter-day precision assays were 4.11% and 6.52%, respectively. For these three blaNDM-1 DNA concentrations, the mean coefficient of variation values of intra-day and inter-day precision assays were 3.93% and 5.96%, respectively, both of which are less than 10%. These results demonstrate good reproducibility of the electrochemical biosensor.
Table 1 Reproducibility of the electrochemical biosensor
| blaNDM-1 DNA concentration (μg L−1) |
Intra-assaya (n = 6) |
Inter-assayb (n = 6) |
| Mean (μA) |
S.D. (μA) |
Coefficient of variation (%) |
Mean (μA) |
S.D. (μA) |
Coefficient of variation (%) |
| Intra-assay: for one sample, tests were repeated 6 times in 1 day. Inter-assay was conducted on 6 sequential days, with 3 runs per day and with 3 replicate tubes per day at every target DNA concentration. |
| 10 |
9.15 |
0.33 |
3.61 |
9.09 |
0.48 |
5.28 |
| 100 |
10.58 |
0.43 |
4.06 |
10.85 |
0.66 |
6.08 |
| 1000 |
8.75 |
0.36 |
4.11 |
8.89 |
0.58 |
6.52 |
3.7 Calibration curves
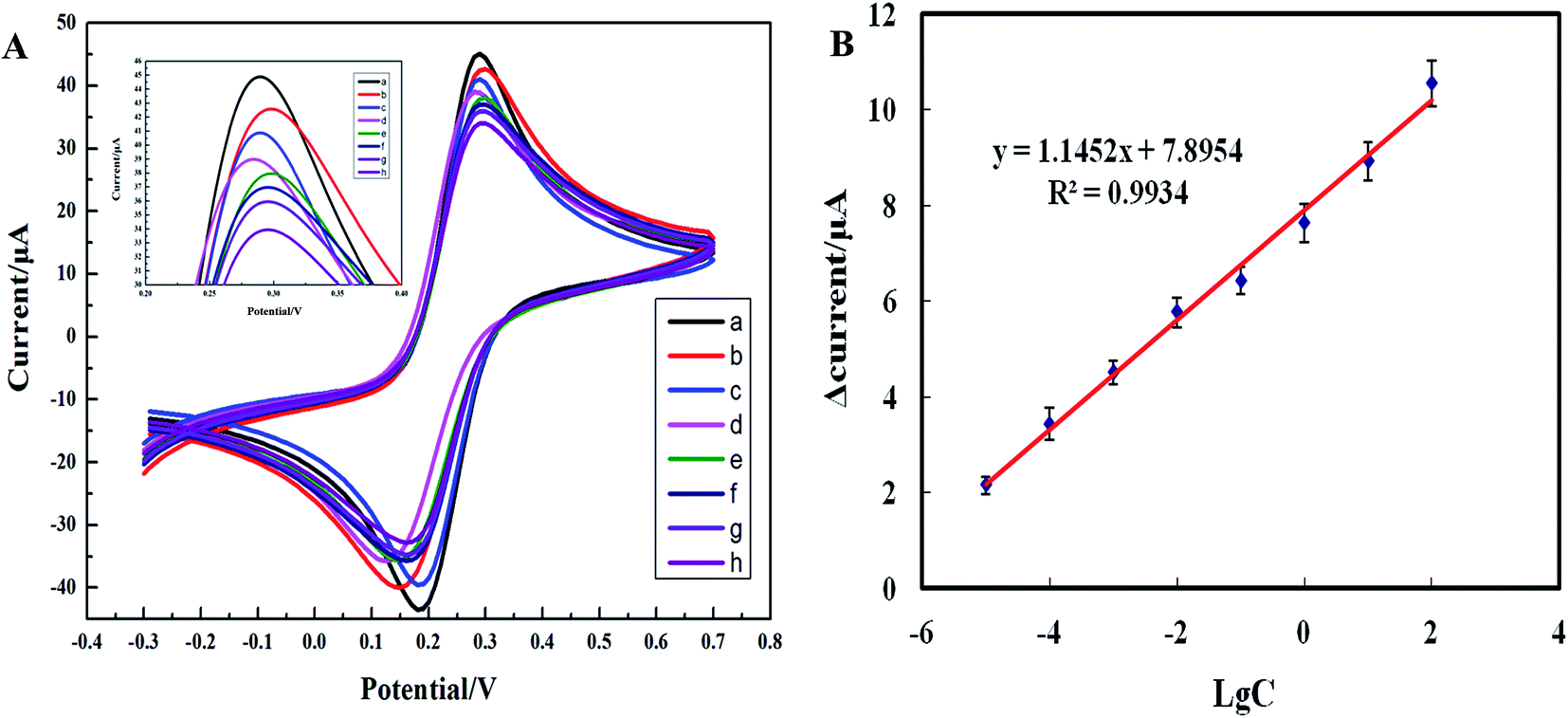
Under the optimal assay conditions, the electrochemical biosensor was used to assay various concentrations of the blaNDM-1 plasmid DNA (1 pg L−1, 10 pg L−1, 100 pg L−1, 1 ng L−1, 10 ng L−1, 100 ng L−1, 1 μg L−1, 10 μg L−1, 100 μg L−1, and 1000 μg L−1) using cyclic voltammetry (Fig. 6A). At the initial stage of hybridization, the redox peak current value clearly decreased when compared with the blank control, as the large nucleic acid hybridization complexes partially blocked the electron-transfer channels on the biosensor surface. When the blaNDM-1 plasmid DNA concentration increased from 10 pg L−1 to 100 μg L−1, hybridization caused the phase change value to increase first to saturation and then finally decrease, with 100 μg L−1 as the blaNDM-1 saturation point. As shown in Fig. 6B, between 10 pg L−1 and 100 μg L−1, the logarithmic concentration values of the blaNDM-1 DNA and the peak current values showed good linearity. The linear regression equation was y = 1.1452x + 7.8954, and the coefficient of correlation was 0.9934. Therefore, with the increasing blaNDM-1 DNA concentrations between 10 pg L−1 and 100 μg L−1, the LNA probes immobilized on the biosensor surface bound to an increasing amount of blaNDM-1 DNA, resulting in a gradual increase in the current response change value. When the blaNDM-1 concentration increased to 100 μg L−1, the binding of the LNA probes and blaNDM-1 became saturated. Therefore, with a subsequent blaNDM-1 DNA concentration increase, no larger response current change value was observed. Similarly, with a blaNDM-1 DNA concentration lower than 10 pg L−1, the accurate detection of the blaNDM-1 DNA was impossible because the LNA probes that were immobilized onto the biosensors surface were not able to bind to adequate amounts of the blaNDM-1 DNA. The results demonstrate that the electrochemical biosensors had a wide linear range and a low limit of detection.
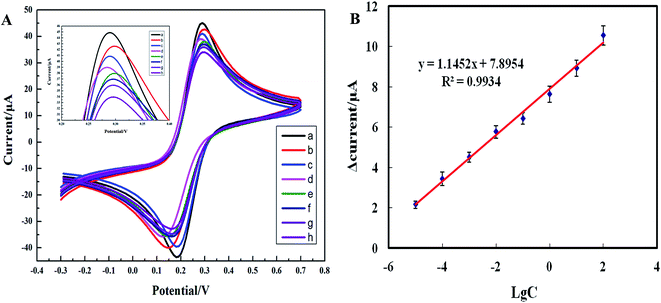 |
| | Fig. 6 (A) Cyclic voltammograms of the electrochemical biosensors in the presence of the different concentrations blaNDM-1 plasmid DNA (from a to g: 10 pg L−1, 100 pg L−1, 1 ng L−1, 10 ng L−1, 100 ng L−1, 1 μg L−1, 10 μg L−1, and 100 μg L−1) in a solution of PBS (pH 7.4, 0.05 mol L−1) with 2.0 mmol L−1 [Fe(CN)6]3−/4− and 5.0 mmol L−1 KCl. (B) The calibration curves of the electrochemical biosensors in the presence of the different concentrations blaNDM-1 plasmid DNA (10 pg L−1 to 100 μg L−1) (C, the blaNDM-1 concentration (μg L−1); LgC, blaNDM-1 denary logarithm). The plots show the current response (μA) on the y-axis as a function of increasing blaNDM-1 DNA concentration on the x-axis. Each data point represents an average of at least three measurements ± standard deviation. | |
3.8 Clinical applications
To evaluate the clinical superiority of the electrochemical biosensor in a specimen assay, clinical bacterial specimens containing blaNDM-1 DNA (n = 30) and blaNDM-1 DNA-free clinical specimens from healthy volunteers (n = 10), which had been validated by the modified Hodge assay, the IPM-EDTA double-disk synergy test, a gel electrophoresis assay, and PCR, were assayed using our electrochemical biosensors. The results of the electrochemical biosensor detection matched 100% with those of the PCR detection. To determine the linear range and the lowest limit of detection of the electrochemical biosensor for detection of complex clinical specimens, one clinical bacterial specimen was diluted in a two-fold dilution series with ddH2O to prepare a series of specimens containing various concentrations of blaNDM-1 DNA. The specimens were added into the detection chambers and incubated for 20 min and finally analyzed by cyclic voltammetry (Fig. 7A). The blaNDM-1 DNA concentrations in the specimens were then determined using the standard curve. For instance, 65 μg L−1 of blaNDM-1 DNA corresponded to a 9.5 μA current response change value on the standard curve. As shown in Fig. 7B, with a blaNDM-1 DNA concentration between 0.00065 and 65 μg L−1, the logarithm of the blaNDM-1 DNA concentration in the clinical specimens and the peak response current of the electrochemical biosensor showed good linearity. The linear regression equation was y = 0.96x + 7.3424, and the coefficient of correlation was 0.9867. These results demonstrate that the electrochemical biosensor could be used to detect trace blaNDM-1 DNA in clinical specimens.
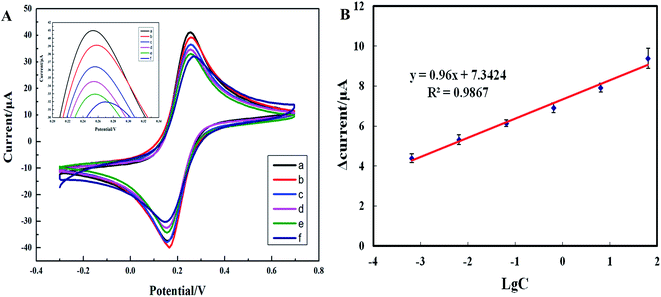 |
| | Fig. 7 Cyclic voltammograms (A) and calibration curves (B) of the electrochemical biosensors in the presence of the different concentrations of clinical bacterial specimens with blaNDM-1 DNA (from a to f: 0.00065, 0.0065, 0.065, 0.65, 6.5, 65 μg L−1) in a solution of PBS (pH 7.4, 0.05 mol L−1) with 2.0 mmol L−1 [Fe(CN)6]3−/4− and 5.0 mmol L−1 KCl (C, the blaNDM-1 concentration (μg L−1); LgC, blaNDM-1 denary logarithm). The plots show the current-response (μA) on the y-axis as a function of increasing blaNDM-1 DNA concentration on the x-axis. Each data point represents an average at least three measurements ± standard deviation. | |
3.9 Method comparison
To evaluate the superiority of the electrochemical biosensor in the direct detection of drug-resistance genes in clinical MDROs, we performed a methodological comparison of this detection method with other detection methods reported in the literature. As shown in Table 2, the linearity range was 10 pg L−1 to 100 μg L−1, and the limit of detection was as low as 1 pg L−1 for the electrochemical biosensor for the detection of the blaNDM-1 control plasmid. Therefore, our method was clearly superior to the other methods. Moreover, the detection results with our electrochemical biosensor were accurate and specific; the detection process was simple and fast, with incubation and detection requiring only 30 min. Therefore, our method is ideal for the bedside real-time detection of blaNDM-1 DNA in clinical bacterial specimens.
Table 2 Comparison of the detection methods of multidrug-resistant organisms and drug-resistance genes with various signal amplification methods
| No. |
Methods |
Object (gene) |
Linear range |
Detection limit |
Ref. |
| LAMP: loop-mediated isothermal amplification. LC-MS/MS: liquid chromatography-tandem mass spectrometry. |
| 1 |
Real-time PCR |
Stool (NDM-1) |
— |
1 × 101 CFU/100 mg |
29 |
| 2 |
LAMPa |
A. baumannii (NDM-1) |
— |
10.70 pg μL−1 |
30 |
| 3 |
LC-MS/MSb |
Enterobacteriaceae/non-enterobacteriaceae (IMP/VIM, KPC/NDM/OXA) |
— |
0.04 μg mL−1 |
31 |
| 4 |
LAMP |
Bacterial strains (L1 metallo-β-lactamase) |
— |
3.79 pg μL−1 |
32 |
| 5 |
Electrochemical |
Bacterial strains (NDM-1) |
10 pg L−1 to 100 μg L−1 |
1 pg L−1 |
This work |
4. Conclusions
In the present study, we successfully constructed an electrochemical biosensor and established a fast electrochemical detection method for blaNDM-1 drug-resistance genes in MDROs based on LNA probes. The constructed electrochemical biosensor greatly increased both the capability of electron transfer on the biosensor surface and the detection sensitivity. The LNA probes enhanced the interference immunity of the biosensor, greatly increasing the detection specificity.33 In conclusion, the constructed electrochemical biosensor exhibits a very wide linearity range, a very low limit of detection, and very high sensitivity for the direct detection of blaNDM-1 DNA in clinical bacterial specimens. Furthermore, this biosensor can used to detect trace blaNDM-1 DNA without requiring amplification.
Acknowledgements
The authors thank Professor Yun Zhang (English Department, Third Military Medical University, PR China) for critically reading the manuscript and kindly giving precious advice. This study was supported financially by the National Natural Science Foundation of China (81370049), the Application Development Projects of Chong Qing CSTC (cstc 2014yykf B10004), the Transformation of Scientific and Technological Achievements of the Third Military Medical University (2014 XZH 05, 2015 XZH 10), and the Science and Technology Development Fund of China Academy of Engineering Physics (wss-2014-03).
References
- K. K. Kumarasamy, M. A. Toleman, T. R. Walsh, J. Bagaria, F. Butt, R. Balakrishnan, U. Chaudhary, M. Doumith, C. G. Giske, S. Irfan, P. Krishnan, A. V. Kumar, S. Maharjan, S. Mushtaq, T. Noorie, D. L. Paterson, A. Pearson, C. Perry, R. Pike, B. Rao, U. Ray, J. B. Sarma, M. Sharma, E. Sheridan, M. A. Thirunarayan, J. Turton, S. Upadhyay, M. Warner, W. Welfare, D. M. Livermore and N. Woodford, Lancet Infect. Dis., 2010, 10, 597–602 CrossRef CAS PubMed.
- A. U. Khan and P. Nordmann, Scand. J. Infect. Dis., 2012, 44, 531–535 CrossRef PubMed.
- T. I. Huo, J. Chin. Med. Assoc., 2010, 73, 557–558 CrossRef PubMed.
- P. Deshpande, C. Rodrigues, A. Shetty, F. Kapadia, A. Hedge and R. Soman, J. Assoc. Physicians India, 2010, 58, 147–149 Search PubMed.
- P. Nordmann, L. Poirel, T. R. Walsh and D. M. Livermore, Trends Microbiol., 2011, 19, 588–595 CrossRef CAS PubMed.
- L. Dortet, L. Poirel and P. Nordmann, BioMed Res. Int., 2014, 2014, 249856 Search PubMed.
- D. Yong, M. A. Toleman, C. G. Giske, H. S. Cho, K. Sundman, K. Lee and T. R. Walsh, Antimicrob. Agents Chemother., 2009, 53, 5046–5054 CrossRef CAS PubMed.
- G. Cornaglia, H. Giamarellou and G. M. Rossolini, Lancet Infect. Dis., 2011, 11, 381–393 CrossRef CAS PubMed.
- S. Chen, W. Feng, J. Chen, W. Liao, N. He, Q. Wang, F. Sun and P. Xia, Ann. Clin. Microbiol. Antimicrob., 2014, 13, 42 CrossRef PubMed.
- P. W. Thomas, M. Zheng, S. Wu, H. Guo, D. Liu, D. Xu and W. Fast, Biochemistry, 2011, 50, 10102–10113 CrossRef CAS PubMed.
- D. J. Diekema and M. A. Pfaller, Clin. Infect. Dis., 2013, 56, 1614–1620 CrossRef PubMed.
- A. Y. Guh, B. M. Limbago and A. J. Kallen, Expert Rev. Anti-Infect. Ther., 2014, 12, 565–580 CrossRef CAS PubMed.
- S. Rai, V. Manchanda, N. P. Singh and I. R. Kaur, Indian J. Med. Microbiol., 2011, 29, 275–279 CrossRef CAS PubMed.
- P. Nordmann, L. Poirel, A. Carrer, M. A. Toleman and T. R. Walsh, J. Clin. Microbiol., 2011, 49, 718–721 CrossRef PubMed.
- J. Jena, N. K. Debata, R. K. Sahoo and E. Subudhi, Burns, 2015, 41, 1758–1763 CrossRef PubMed.
- A. Kruttgen, S. Razavi, M. Imohl and K. Ritter, Med. Microbiol. Immunol., 2011, 200, 137–141 CrossRef PubMed.
- M. Mir, S. Martinez-Rodriguez, O. Castillo-Fernandez, A. Homs-Corbera and J. Samitier, Electrophoresis, 2011, 32, 811–821 CrossRef CAS PubMed.
- D. Zhang and Q. Liu, Biosens. Bioelectron., 2016, 75, 273–284 CrossRef CAS PubMed.
- L. Pires, K. Sachsenheimer, T. Kleintschek, A. Waldbaur, T. Schwartz and B. E. Rapp, Biosens. Bioelectron., 2013, 47, 157–163 CrossRef CAS PubMed.
- D. A. Braasch and D. R. Corey, Chem. Biol., 2001, 8, 1–7 CrossRef CAS PubMed.
- B. Vester and J. Wengel, Biochemistry, 2004, 43, 13233–13241 CrossRef CAS PubMed.
- R. N. Veedu and J. Wengel, Mol. BioSyst., 2009, 5, 787–792 RSC.
- M. Wen, B. Li, W. Bai, S. Li and X. Yang, Mol. Vision, 2008, 14, 114–117 CAS.
- B. Li, M. Wen, W. Li, M. He, X. Yang and S. Li, Int. J. Pharm., 2011, 408, 91–96 CrossRef CAS PubMed.
- D. Chen, H. Gan, X. Huang, Q. Shen, X. Du, W. Tang and X. Yang, Ther. Apheresis Dial., 2012, 16, 173–180 CrossRef CAS PubMed.
- Q. Xu, K. Chang, W. Lu, W. Chen, Y. Ding, S. Jia, K. Zhang, F. Li, J. Shi, L. Cao, S. Deng and M. Chen, Biosens. Bioelectron., 2012, 33, 274–278 CrossRef CAS PubMed.
- S. Mishra, S. Ghosh and R. Mukhopadhyay, Langmuir, 2014, 30, 10389–10397 CrossRef CAS PubMed.
- X. Zhu, J. Li, H. He, M. Huang, X. Zhang and S. Wang, Biosens. Bioelectron., 2015, 74, 113–133 CrossRef CAS PubMed.
- T. Naas, A. Ergani, A. Carrer and P. Nordmann, Antimicrob. Agents Chemother., 2011, 55, 4038–4043 CrossRef CAS PubMed.
- W. Liu, D. Zou, Y. Li, X. Wang, X. He, X. Wei, C. Shao, X. Li, W. Shang, K. Yu, D. Liu, Y. Li, J. Guo, Z. Yin and J. Yuan, J. Clin. Microbiol., 2012, 50, 1580–1585 CrossRef CAS PubMed.
- M. V. Kulkarni, A. N. Zurita, J. S. Pyka, T. S. Murray, M. E. Hodsdon and D. R. Peaper, J. Clin. Microbiol., 2014, 52, 2500–2505 CrossRef CAS PubMed.
- Z. Yang, W. Liu, Q. Cui, W. Niu, H. Li, X. Zhao, X. Wei, X. Wang, S. Huang, D. Dong, S. Lu, C. Bai, Y. Li, L. Huang and J. Yuan, Front. Microbiol., 2014, 5, 692 Search PubMed.
- C. Briones and M. Moreno, Anal. Bioanal. Chem., 2012, 402, 3071–3089 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a




 ), O-atoms (
), O-atoms ( ), and P-atoms (
), and P-atoms ( ), and (B) the LNA probes and the blaNDM-1 DNA hybridization complex. The upper-right corner of Fig. 3A and B shows a schematic diagram of the LNA probes and the blaNDM-1 DNA hybridization complex, respectively. The root mean square (RMS) roughness of (C) the LNA probes and (D) the LNA probes and the blaNDM-1 DNA hybridization complex modified on the surface of electrochemical biosensors.
), and (B) the LNA probes and the blaNDM-1 DNA hybridization complex. The upper-right corner of Fig. 3A and B shows a schematic diagram of the LNA probes and the blaNDM-1 DNA hybridization complex, respectively. The root mean square (RMS) roughness of (C) the LNA probes and (D) the LNA probes and the blaNDM-1 DNA hybridization complex modified on the surface of electrochemical biosensors.