
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-mediated preparation of a heterometallic [2 × 2] grid via a 1D metal–organic template with extraordinary acid/base-resistance†
Yi Hana,
Hao Zhenga,
Huijun Li*b,
Hongli Wanga,
Shi-Min Wang*c,
Yanling Genga and
Lei Wang *a
*a
aKey Laboratory of Eco-Chemical Engineering, Ministry of Education, Inorganic Synthesis and Applied Chemistry, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: inorchemwl@126.com
bCollege of Chemistry and Chemical Engineering, Henan Polytechnic University, Jiaozuo, Henan 454000, P. R. China. E-mail: lihuijunxgy@hpu.edu.cn
cDepartment of Material and Chemistry Engineering, Henan Institute of Engineering, Henan 450007, P. R. China. E-mail: wsmhaue@163.com
First published on 17th January 2017
Abstract
A new 1D coordination polymer, [FeIIL(H2O)]·2H2O (1) (H2L = 2,6-bis[3-(pyrazin-2-yl)-1,2,4-triazoly]pyridine) with extraordinary acid/base-resistance was synthesized. It was found that the 1D metal–organic chain can serve as a template to prepare a heterometallic [2 × 2] grid [FeIII2Na2L4(H2O)4]·8H2O (2) through a solvent-mediated strategy by immersing the fresh crystals of 1 in a saturated MeOH solution of NaN3. The transformation was accompanied by the complete dissolution of the original crystals, and the cleavage/regeneration of coordination bonds. The final structure of 2 was unambiguously characterized by means of a single-crystal X-ray diffraction (SCXRD) study. Notably, L2− in 1 exhibits an “acute triangle” configuration, while an “obtuse triangle” configuration is observed in 2. In addition, treatments of 1 with saturated MeOH solutions of NaF, NaCl, NaBr, NaI, and NaN3 in DMF, EtOH, and H2O were also respectively checked. The results did not show any appreciable indication, probably implying that the transformation kinetics is influenced not only by a sodium source but also by a solvent-exclusive effect. Significantly, the resulting grid of 2 could not be accessed by a direct one-pot self-assembly synthetic approach.
Introduction
In the past decades, coordination polymers (CPs) composed of metal-containing nodes and organic ligands through coordination bonds have experienced a rapid development.1 Due to the nature of the coordination bond, namely “labile and reversible”, fruitful reports about single-crystal to single-crystal (SC–SC) transformation-mediated modifications of MOFs have been documented, accompanied by the cleavage and regeneration of coordination bonds, resulting in changes in terms of coordination environment, framework dimensionality and structural interpenetration, etc.2 In this context, the parent crystals can be regarded as a template to fabricate new crystals, which are usually difficult to access by a direct one-pot self-assembly synthetic approach. Among the transformations, solvent-mediated process involving dissolution of the initial MOFs, followed by the reassembly of new phase in solution is relative rare.3 Furthermore, the final structures encounter difficulty in resolution by means of single-crystal X-ray diffraction (SCXRD) studies, due to the tiny sizes of newly formed crystals. Consequently, it remains a great challenge to determine the exact structures of the transformed products.Supramolecular coordination complexes (SCCs), particularly [n × n] grids, that are composed of homo- or heterogeneous transition metal ions with ordered arrangements, have been found to possess significant degree of predictability and have the promising candidates for structure-dependent behavior and data storage.4 The most popular approach to the grid-type arrays is the “self-assembly” strategy. This involves using an organic ligand encoded with suitable coordination information to match the transition metal ions with stereochemical and bonding preferences. For example, if appropriate transition metal ions and a symmetric tritopic bidentate–tridentate–bidentate segmental ligand, 2,6-bis[3-(pyrazin-2-yl)-1,2,4-triazoly]pyridine (H2L) or its analogues were selected, [3 × 3] grids were prone to access.4a–c,5 In other words, given the information recognition between metal ion and ligand, it is difficult to anticipate the preservation of potential coordination sites, thereby achieving a mononuclear complex or [2 × 2] grid from the symmetric tritopic ligand (H2L).
Recently, the exploration of interconversion between SCCs or metal–organic polyhedra (MOPs) and metal–organic frameworks are well-documented and emerging one of the most promising fields.6 In 2013, we demonstrated that grid could also be employed as a metal-containing node to obtain extended networks.5a However, no attempts to study the conversion of a CP into a grid have been described previously. Herein, we present an unusual solvent-mediated pathway for the preparation of a heterometallic [2 × 2] grid [FeIII2Na2L4(H2O)4]·8H2O (denoted as 2) via a 1D metal–organic chain [FeIIL(H2O)]·2H2O (denoted as 1) as the template for the first time. Significantly, the resulting grid could not be access by a direct one-pot self-assembly synthetic approach. Furthermore, the crystal structure of the grid was unambiguously characterized by SCXRD, and the conversion was monitored by both powder X-ray diffraction (PXRD) and optical microscopy.
Experimental section
Materials and physical measurements
All chemicals were obtained from commercial sources and used as received without purification. IR spectra were recorded from KBr pellets in the range 4000–400 cm−1 on a Shimadzu IR435 spectrometer. A FLASH EA 1112 analyzer was performed for the elemental analysis. Thermogravimetric analysis (TGA) was recorded on a Netzsch STA 449C thermal analyzer between 30 and 800 °C and a heating rate of 10 °C min−1 in atmosphere. Powder X-ray diffraction (PXRD) data were measured on the PANalytical X'Pert PRO diffractometer using Cu-Kα radiation (λ = 1.541874 Å). Energy-dispersive X-ray spectrometry (EDS) was performed on a Bruker ISMNM 761 scanning electron microscope.Synthesis of [FeIIL(H2O)]·2H2O (1)
A mixture of FeSO4·7H2O (14 mg, 0.05 mmol), H2L (18.4 mg, 0.05 mmol), KSCN (9.7 mg, 0.1 mmol) and water (10 mL) was heated at 160 °C for 96 h. After cooling the mixture to RT, yellow block crystals of 1 were harvested (10.5 mg, 57% yield based on H2L). Elemental analysis (%) for 1 C17H14FeN11O3: calcd C 42.88, H 2.96, N 32.35; found C 43.05, H 3.39, N 32.03.Synthesis of [FeIII2Na2L4(H2O)4]·8H2O (2)
Freshly as-prepared samples (10 mg) of 1 were immersed in saturated MeOH solution (3 mL) of NaN3. About 5 days later, black block crystals were isolated (8 mg, 80% yield). Elemental analysis (%) for 2 C68H60Fe2N44Na2O12: calcd C 44.31, H 3.28, N 33.44; found C 45.05, H 3.01, N 32.88. The bulk product can be obtained by a vigorous stirring a freshly as-prepared 1 immersed in saturated MeOH solution of NaN3.Crystal data collection and refinement
Data of 1 (CCDC-1512713) was collected on a Rigaku Saturn 724 CCD diffractometer (Mo Kα, λ = 0.71073 Å) at room temperature. Absorption corrections were applied by using multiscan program. The data were corrected for Lorentz and polarization effects. The structures were solved by direct methods and refined with a full-matrix least-squares technique based on F2 with the SHELXL-97 crystallographic software package.7 The hydrogen atoms were assigned with common isotropic displacement factors and included in the final refinement by using geometrical restrains. Data collection for 2 (CCDC-1512712) was carried on an Agilent Technologies SuperNova Single Crystal Diffractometer using Cu Kα radiation (λ = 1.54178 Å) at 100 K. The structure was solved using SHELXS-97 and was refined with SHELXL-97.7 The hydrogen atoms were included in the structure-factor calculations at idealized positions by using a riding model and were refined isotropically. Crystal data and the selected bond lengths and angles for 1 and 2 are summarized in Tables S1 and S2.†Results and discussion
Synthesis, general characterization, and crystal structure of 1
Yellow crystals of 1 were obtained by hydrothermal reaction of FeSO4·7H2O with H2L in the presence of KSCN at 160 °C for 96 h, and the phase purity of 1 is confirmed by PXRD analysis. Thermogravimetric analysis (TGA) reveals 1 could be stable up to 400 °C after a weight loss of guest and coordinated H2O molecules in the region 30–198 °C (observed, 11.29%; calculated, 11.43%) (Fig. 3), and its intact framework was further confirmed by PXRD (Fig. S3†). SCXRD reveals that 1 crystallizes in the monoclinic space group C2 and has a one-dimensional (1D) chain structure (Fig. 1a). There exist two independent FeII ions in the asymmetric unit with both occupancies of 0.5. Notably, both of the two FeII ions are located on the special positions towards twofold axis. The octahedral coordination sphere around Fe1 is completed by six N atoms from two of the L2− ligands, affording a FeN6 environment. The Fe1–N bond lengths are ranging from 2.145(4) to 2.240(4) Å. While Fe2 is octahedrally coordinated by two L2− ligands through the pyrazinyl and triazole N atoms and two axial aqua ligands. The average Fe2–N and Fe2–O bond lengths are 2.230(4) and 2.044(7) Å, respectively. The coordination mode adopted by L2− ligand is shown in Scheme 1d. Notably, rotations of the C–C axis were observed in triazole-pyrazinyl moieties, leading to the distortion of L2− ligand with an “acute triangle” configuration (Fig. 1b and c). In addition, as-synthesized 1 was determined to be stable in H2O and common organic solvents. Significantly, after being soaked in aqueous solutions with pH = 1 and 13 (prepared with HCl and NaOH solution respectively) for 24 h, the unaltered PXRD patterns of 1 indicate the robustness of the framework under harsh chemical conditions (Fig. 4a and S5†). To the best of our knowledge, the nature of extraordinary acid/base-resistance observed in 1 is uncommon in the field of metal–organic materials.8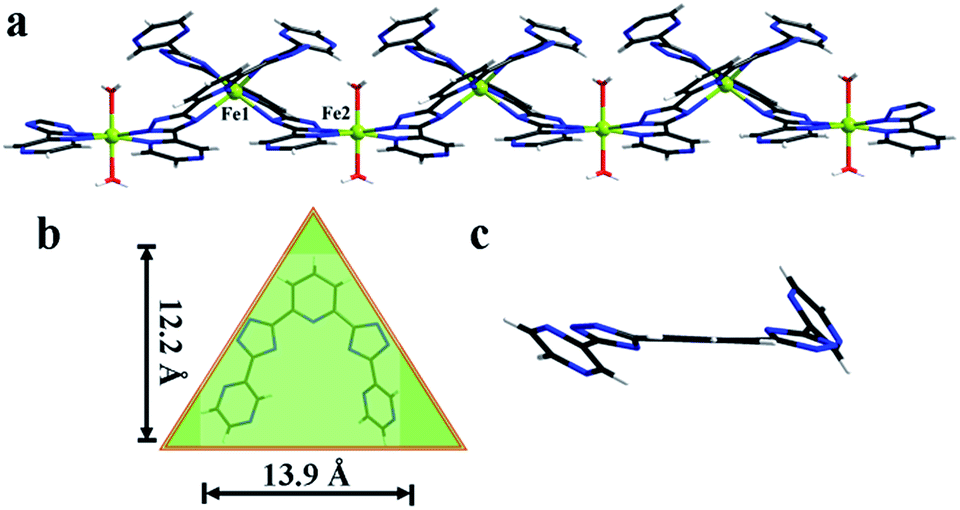 | ||
| Fig. 1 Single crystal X-ray structures of the 1D metal–organic chain for 1 (a), the “acute triangle” configuration of H2L in 1 (b), the side view of H2L in 1 (c). Color scheme: FeII green, N blue, C black, O red, H white. | ||
 | ||
| Fig. 2 Single crystal X-ray structures of the [2 × 2] grid for 2 (a), the “obtuse triangle” configuration of H2L in 2 (b), the side view of H2L in 2 (c). Color scheme: FeIII brown, Na pink, N blue, C black, O red. | ||
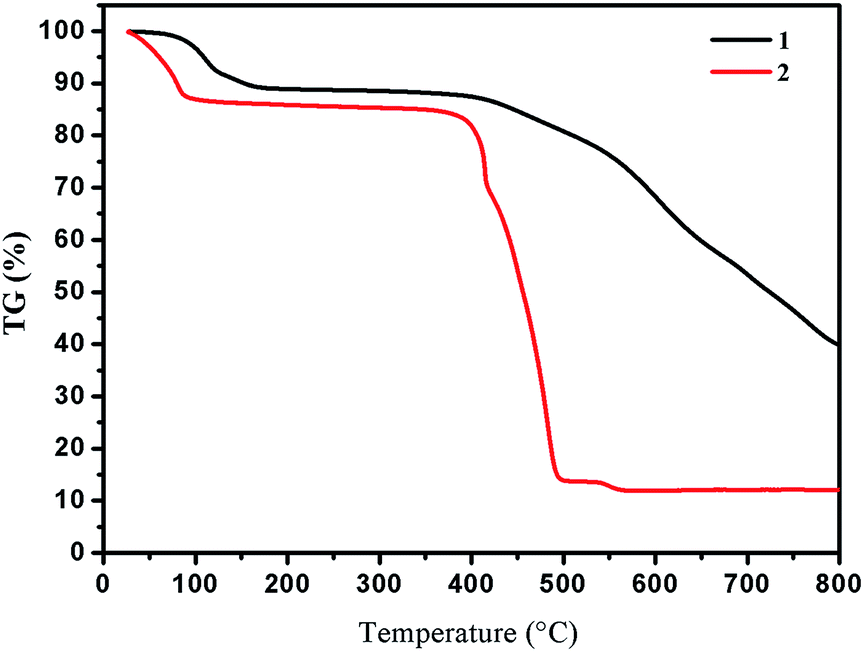 | ||
| Fig. 3 TGA curves of as-synthesized 1 and 2. | ||
 | ||
| Scheme 1 Coordination modes of H2L in 1 (d) and 2 (e and f). Types of a–c can be seen in the ref. 5a. | ||
 | ||
| Fig. 4 PXRD patterns of 1 after immersion in aqueous solutions with different pH values and NaX (X = F, Cl, Br and I) (a) and 2 (b). | ||
Solvent-mediated preparation of the heterometallic [2 × 2] grid of 2 from 1, and general characterization, crystal structure of 2
In some cases, the coordination bonds between metal ions and coordinated solvent molecules are slightly labile so that coordinated solvent molecules can be easily removed or replaced by inorganic/organic ligands.9 Given the characteristics of the FeII–H2O moieties in 1, we considered that the terminal H2O ligand may be substituted by halide anions in terms of F−, Cl−, Br−, I−, and N3− to further realize the potential structure tailoring. Inspired by this, fresh crystals of 1 were respectively immersed in saturated MeOH solutions of NaF, NaCl, NaBr, NaI, and NaN3 at room temperature. It was found that the exclusive color change from colorless to deep brown was observed in the presence of N3−, accompanied by the complete dissolution of the original crystals (Fig. S6†). Furthermore, homogenous black crystalline solids appeared in a couple of days. PXRD analysis confirmed that the samples of 1 in saturated MeOH solutions of NaF, NaCl, NaBr and NaI still keep the framework structure intact (Fig. 3a), while a distinct pattern from that of 1 was achieved from the newly generated solid, suggesting a new phase was present (Fig. 3b). This observed result may arise from the different pH values of the above saturated MeOH solutions of the corresponding sodium sources, that is, the dissociation accompanied by the cleavage of Fe–N and Fe–O coordination bonds and further dissolution of 1 should require a relative high pH value.SCXRD demonstrates that the new black crystalline phase has the chemical formula of [FeIII2Na2L4(H2O)4]·8H2O (2). In 2, a weight loss of 12.17% (calculated, 11.26%) in the range 30–105 °C could be assigned to the loss of guest and coordinated H2O molecules; above 400 °C, a series of decomposition steps commence (Fig. 3). 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and displays a rare heterometallic FeIII/Na-based [2 × 2] grid (Fig. 2a). The crystallographically asymmetric unit of 2 contains one FeIII ion, one Na ion and two deprotonated L2− ligands. Significantly, FeIII ion should be generated from the oxidation of FeII in alkaline condition during the self-assembly of 2. The FeIII ion displays a distorted octahedral geometry, binding with six N atoms from two L2− ligands, and Na ion is octahedrally coordinated by four N atoms from two independent L2− ligands and two O atoms from terminal aqua ligands. The Fe–N bond lengths are ranging from 1.924(4) to 1.942(4) Å, indicative of characteristic FeIII–N bond lengths observed in the reported FeIII-based coordination polymers.10 And further confirmation of oxidation state of Fe ions could also be provided by the charge balance. In 2, L2− ligands do not show obvious distortion and adopt two kinds of coordination modes, to connect one FeIII and one Na ions (Fig. 2c, Scheme 1e and f), resulting in an “obtuse triangle” configuration (Fig. 2b). Compared with the previously reported heterometallic grids,11 which are usually constructed by “one-pot synthesis” or stepwise synthetic procedures by introducing metal ions sequentially in the order of increasing coordination lability, 2 is the first grid by a solvent-mediated preparation and the first heterometallic grid-type example including main metal ion. In addition, fresh single crystals of 2 can be insoluble in MeOH, EtOH, MeCN, but readily dissolve in DMF, DMA and DMSO (Fig. S7†). Notably, attempts to a direct one-pot self-assembly synthesis of 2 failed.
and displays a rare heterometallic FeIII/Na-based [2 × 2] grid (Fig. 2a). The crystallographically asymmetric unit of 2 contains one FeIII ion, one Na ion and two deprotonated L2− ligands. Significantly, FeIII ion should be generated from the oxidation of FeII in alkaline condition during the self-assembly of 2. The FeIII ion displays a distorted octahedral geometry, binding with six N atoms from two L2− ligands, and Na ion is octahedrally coordinated by four N atoms from two independent L2− ligands and two O atoms from terminal aqua ligands. The Fe–N bond lengths are ranging from 1.924(4) to 1.942(4) Å, indicative of characteristic FeIII–N bond lengths observed in the reported FeIII-based coordination polymers.10 And further confirmation of oxidation state of Fe ions could also be provided by the charge balance. In 2, L2− ligands do not show obvious distortion and adopt two kinds of coordination modes, to connect one FeIII and one Na ions (Fig. 2c, Scheme 1e and f), resulting in an “obtuse triangle” configuration (Fig. 2b). Compared with the previously reported heterometallic grids,11 which are usually constructed by “one-pot synthesis” or stepwise synthetic procedures by introducing metal ions sequentially in the order of increasing coordination lability, 2 is the first grid by a solvent-mediated preparation and the first heterometallic grid-type example including main metal ion. In addition, fresh single crystals of 2 can be insoluble in MeOH, EtOH, MeCN, but readily dissolve in DMF, DMA and DMSO (Fig. S7†). Notably, attempts to a direct one-pot self-assembly synthesis of 2 failed.
Furthermore, treatments of 1 with NaN3 in DMF, EtOH and H2O were also checked, respectively. The results did not show any appreciable indication (Fig. S8†), probably due to the extremely slow dissociation or dissolution kinetics of the reaction,12 even though the corresponding saturated DMF, EtOH or H2O solvents of NaN3 possess relative high pH values.
Conclusions
In conclusion, we have demonstrated that a metal–organic chain can serve as a template to prepare a heterometallic [2 × 2] grid through a solvent-mediated strategy. The transformation kinetics is influenced not only by a sodium source- but also by a solvent-exclusive effect. This system exhibits a number of features which are remarkable in a 1D coordination polymer, including outstanding resistance to acid/base and conversion into a heterometallic [2 × 2] grid under a special condition. The realizations in this work will enable the extensive exploration of new aspects and potential applications towards low-dimensional coordination polymers, and further a new method for the access to grid-type arrays.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21601102, 21601050, 51372125, 21571112, 51572136 and 61405054), Natural Science Foundation of Shandong Province (No. 2016ZRB01A60) and Research Fund for the Doctoral Program of Henan Institute of Engineering (No. D2013005).References
- (a) S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, RSC Publishing, 2009 Search PubMed; (b) L. R. MacGillivray, Metal–Organic Frameworks: Design and Application, Wiley & Sons, 2010 Search PubMed.
- (a) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef CAS; (b) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC; (c) Y. Han, J.-R. Li, Y. Xie and G. Guo, Chem. Soc. Rev., 2014, 43, 5952–5981 RSC; (d) P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- (a) H. Yang, L. Li, J. Wu, H. Hou, B. Xiao and Y. Fan, Chem.–Eur. J., 2009, 15, 4049–4056 CrossRef CAS PubMed; (b) F. Pan, J. Wu, H. Hou and Y. Fan, Cryst. Growth Des., 2010, 10, 3835–3837 CrossRef CAS; (c) X. Cui, A. N. Khlobystov, X. Chen, D. H. Marsh, A. J. Blake, W. Lewis, N. R. Champness, C. J. Roberts and M. Schröder, Chem.–Eur. J., 2009, 15, 8861–8873 CrossRef CAS PubMed; (d) X. Li, Z. Yu, X. Li and X. Guo, Chem.–Eur. J., 2015, 21, 16593–16600 CrossRef CAS PubMed; (e) X. Li, Y. Gong, H. Zhao and R. Wang, CrystEngComm, 2014, 16, 8818–8824 RSC.
- (a) L. K. Thompsona, O. Waldmann and Z. Xua, Coord. Chem. Rev., 2005, 249, 2677–2690 CrossRef; (b) G. N. Newton, T. Onuki, T. Shiga, M. Noguchi, T. Matsumoto, J. S. Mathieson, M. Nihei, M. Nakano, L. Cronin and H. Oshio, Angew. Chem., Int. Ed., 2011, 50, 4844–4848 CrossRef CAS PubMed; (c) T. Shiga, T. Matsumoto, M. Noguchi, T. Onuki, N. Hoshino, G. N. Newton, M. Nakano and H. Oshio, Chem.–Asian J., 2009, 4, 1660–1663 CrossRef CAS PubMed; (d) W.-Q. Lin, J.-D. Leng and M.-L. Tong, Chem. Commun., 2012, 48, 4477–4479 RSC; (e) M. Barboiu, G. Vaughan, R. Graff and J.-M. Lehn, J. Am. Chem. Soc., 2003, 125, 10257–10265 CrossRef CAS PubMed.
- (a) Y. Han, N. F. Chilton, M. Li, C. Huang, H. Xu, H. Hou, B. Moubaraki, S. K. Langley, S. R. Batten, Y. Fan and K. S. Murray, Chem.–Eur. J., 2013, 19, 6321–6328 CrossRef CAS PubMed; (b) L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Inorg. Chem., 2009, 48, 3323–3341 CrossRef CAS PubMed.
- (a) G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403–9453 RSC; (b) M. J. Zaworotko, Nat. Chem., 2009, 1, 267–268 CrossRef CAS PubMed; (c) T.-F. Liu, Y.-P. Chen, A. A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 17358–17361 CrossRef CAS PubMed; (d) J.-R. Li, D. J. Timmons and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368–6369 CAS; (e) A. Schoedel, L. Wojtas, S. P. Kelley, R. D. Rogers, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2011, 50, 11421–11424 CrossRef CAS PubMed; (f) A. Schoedel, A. J. Cairns, Y. Belmabkhout, L. Wojtas, M. Mohamed, Z. Zhang, D. M. Proserpio, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2013, 52, 2902–2905 CrossRef CAS PubMed; (g) A. Schoedel, W. Boyette, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 14016–14019 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXTL 2008/4, Structure Determination Software Suite, Bruker AXS, Madison, Wisconsin, USA Search PubMed.
- (a) X. Zhang, X. Zhang, J. A. Johnson, Y.-S. Chen and J. Zhang, J. Am. Chem. Soc., 2016, 138, 8380–8383 CrossRef CAS PubMed; (b) T.-F. Liu, D. Feng, Y.-P. Chen, L. Zou, M. Bosch, S. Yuan, Z. Wei, S. Fordham, K. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 413–419 CrossRef CAS PubMed; (c) D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed; (d) H.-L. Jiang, D. Feng, K. Wang, Z.-Y. Gu, Z. Wei, Y.-P. Chen and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed; (e) D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed; (f) X. Li, H. Xu, F. Kong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 13769–13773 CrossRef CAS PubMed.
- (a) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed; (b) X.-N. Cheng, W.-X. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2007, 129, 15738–15739 CrossRef CAS PubMed; (c) M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2009, 131, 7524–7525 CrossRef CAS PubMed; (d) H. J. Park, Y. E. Cheon and M. P. Suh, Chem.–Eur. J., 2010, 16, 11662–11669 CrossRef CAS PubMed; (e) X.-L. Wang, C. Qin, S.-X. Wu, K.-Z. Shao, Y.-Q. Lan, S. Wang, D.-X. Zhu, Z.-M. Su and E.-B. Wang, Angew. Chem., Int. Ed., 2009, 48, 5291–5295 CrossRef CAS PubMed; (f) Y.-Q. Lan, S.-L. Li, H.-L. Jiang and Q. Xu, Chem.–Eur. J., 2012, 18, 8076–8083 CrossRef CAS PubMed.
- (a) P. J. van Koningsbruggen, Y. Maeda and H. Oshio, Top. Curr. Chem., 2004, 233, 259–324 CrossRef CAS; (b) D. J. Hardinga, P. Hardinga and W. Phonsrib, Coord. Chem. Rev., 2016, 313, 38–61 CrossRef; (c) N. Ortega-Villar, A. Y. Guerrero-Estrada, L. Pineiro-López, M. C. Muñoz, M. Flores-Álamo, R. Moreno-Esparza, J. A. Real and V. M. Ugalde-Saldívar, Inorg. Chem., 2015, 54, 3413–3421 CrossRef CAS PubMed.
- (a) X. Bao, W. Liu, L.-L. Mao, S.-D. Jiang, J.-L. Liu, Y.-C. Chen and M.-L. Tong, Inorg. Chem., 2013, 52, 6233–6235 CrossRef CAS PubMed; (b) A. R. Stefankiewicz, J. Harrowfield, A. Madalan, K. Rissanen, A. N. Sobolev and J.-M. Lehn, Dalton Trans., 2011, 40, 12320–12332 RSC; (c) Z. Xu, L. K. Thompson, C. J. Matthews, D. O. Miller, A. E. Goeta and J. A. K. Howard, Inorg. Chem., 2001, 40, 2446–2449 CrossRef CAS PubMed; (d) S. R. Parsons, L. K. Thompson, S. K. Dey, C. Wilson and J. A. K. Howard, Inorg. Chem., 2006, 45, 8832–8834 CrossRef CAS PubMed; (e) J. Wu, L. Zhao, L. Zhang, X.-L. Li, M. Guo and J. Tang, Inorg. Chem., 2016, 55, 5514–5519 CrossRef CAS PubMed.
- X. Song, S. Jeong, D. Kim and M. S. Lah, CrystEngComm, 2012, 14, 5753–5756 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and bond angles, additional figures. CCDC 1512712 and 1512713. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26412c |
| This journal is © The Royal Society of Chemistry 2017 |