
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix isolation FTIR study of hydrogen-bonded complexes of methanol with heterocyclic organic compounds†
Xiaotong Jiang,
Shijie Liu,
Narcisse T. Tsona ,
Shanshan Tang,
Lei Ding,
Hailiang Zhao and
Lin Du*
,
Shanshan Tang,
Lei Ding,
Hailiang Zhao and
Lin Du*
Environment Research Institute, Shandong University, Shanda South Road 27, 250100 Shandong, China. E-mail: lindu@sdu.edu.cn
First published on 12th January 2017
Abstract
Acting as hydrogen bond acceptors, heterocyclic compounds could interact with a hydrogen bond donor, where either the heteroatom or the π system is the bonding site. The hydrogen bonded complexes of heterocyclic compounds with methanol (MeOH) were studied using matrix isolation FTIR spectroscopy and theoretical calculations based on density functional theory. Four heterocyclic compounds, furan (Fu), 2,5-dihydrofuran (DHF), pyrrole (Py) and thiophene (Th) were selected as representative examples for acceptors. For each of the MeOH–Fu, MeOH–DHF and MeOH–Th complexes, the O–H⋯π and O–H⋯Y (Y![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, S) hydrogen bonded structures were obtained, while an N–H⋯O instead of O–H⋯N hydrogen bonded conformer was found in the MeOH–Py complex. The measured OH-stretching transitions of the complexes in the IR spectra were assigned to the O–H⋯O bonded MeOH–Fu (b) and MeOH–DHF (b) conformers and the N–H⋯O bonded Py–MeOH (b) conformer, respectively. However, it was hard to assign the spectra to the exact MeOH–Th conformer, because all the hydrogen bond characteristic features obtained for different MeOH–Th conformers are too close. DHF forms a stronger O–H⋯O hydrogen bond than furan, and the O–H⋯O hydrogen bonded MeOH–Fu complex is more stable than the O–H⋯S bonded MeOH–Th complex. Atoms in molecules analysis was also performed to understand the nature of interaction in the MeOH complexes. This analysis allowed us to characterize the new bond critical point generated in the complexes. The present results help to evaluate the atmospheric behavior of some heterocyclic compounds, and indicate their importance in the pre-nucleation mechanism at the molecular level.
O, S) hydrogen bonded structures were obtained, while an N–H⋯O instead of O–H⋯N hydrogen bonded conformer was found in the MeOH–Py complex. The measured OH-stretching transitions of the complexes in the IR spectra were assigned to the O–H⋯O bonded MeOH–Fu (b) and MeOH–DHF (b) conformers and the N–H⋯O bonded Py–MeOH (b) conformer, respectively. However, it was hard to assign the spectra to the exact MeOH–Th conformer, because all the hydrogen bond characteristic features obtained for different MeOH–Th conformers are too close. DHF forms a stronger O–H⋯O hydrogen bond than furan, and the O–H⋯O hydrogen bonded MeOH–Fu complex is more stable than the O–H⋯S bonded MeOH–Th complex. Atoms in molecules analysis was also performed to understand the nature of interaction in the MeOH complexes. This analysis allowed us to characterize the new bond critical point generated in the complexes. The present results help to evaluate the atmospheric behavior of some heterocyclic compounds, and indicate their importance in the pre-nucleation mechanism at the molecular level.
1. Introduction
The hydrogen bond plays an important role in atmospheric chemistry, especially in the field of aerosol formation and growth.1–4 Many experimental and theoretical investigations have been performed to understand intermolecular hydrogen bonds, with a focus on structural, spectroscopic and energetic issues.5–8 Hydrogen bonded complexes usually have small equilibrium constants, which make them difficult to study under atmospheric conditions. Therefore, these complexes are often studied experimentally under some form of cooling to favor complex formation.9–15Owing to the low temperature, matrix isolation infrared spectroscopy study is considered as a useful analysis technique for observing and characterizing hydrogen bonded interactions in complexes. Matrix isolation Fourier transform infrared spectroscopy (FTIR) coupled with ab initio calculations has been employed for the isolation and characterization of a wide range of molecular complexes and reactive Criegee intermediates. The study of intermolecular interactions between sulfuric acid and several molecules of atmospheric interest have been carried out using matrix isolation spectroscopic methods.10,16–18 For instance, the interactions in the H2SO4/CH3OH/H2O system have been studied by matrix isolation FTIR spectroscopy in conjunction with density functional theory calculations. Several new bands observed from the FTIR spectra indicate the existence of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 H2SO4/CH3OH/H2O cycle complex.18 The effect of hydrogen bond on the anharmonicity and intensity of OH oscillators in MeOH trapped in neon matrix has been investigated using near-infrared (NIR) and middle infrared (MIR) spectroscopy. The anharmonicity parameters were determined from the positions and intensities of the dimer bands of MeOH.19 For the purpose of discriminating between O–H⋯N and O–H⋯OC complexes, a deep research of matrix isolation FTIR spectra and the hydrogen bonded complexes with MeOH in argon has been taken.20 It was concluded that hydrogen bonds between weak acids such as MeOH tends to be formed at the carbonyl group. Gas phase reaction of phosphorus trichloride (PCl3) and MeOH was carried out at different ratios of PCl3/MeOH and the products were identified using matrix isolation infrared spectroscopy.21 Infrared vibrational spectra of matrix isolated PCl3, MeOH and their complexes were analyzed and assigned by comparing the observed and calculated frequencies and intensities. The experimental evidence for the noncovalent P⋯O interaction in PCl3–MeOH adducts was provided.
:1:1 H2SO4/CH3OH/H2O cycle complex.18 The effect of hydrogen bond on the anharmonicity and intensity of OH oscillators in MeOH trapped in neon matrix has been investigated using near-infrared (NIR) and middle infrared (MIR) spectroscopy. The anharmonicity parameters were determined from the positions and intensities of the dimer bands of MeOH.19 For the purpose of discriminating between O–H⋯N and O–H⋯OC complexes, a deep research of matrix isolation FTIR spectra and the hydrogen bonded complexes with MeOH in argon has been taken.20 It was concluded that hydrogen bonds between weak acids such as MeOH tends to be formed at the carbonyl group. Gas phase reaction of phosphorus trichloride (PCl3) and MeOH was carried out at different ratios of PCl3/MeOH and the products were identified using matrix isolation infrared spectroscopy.21 Infrared vibrational spectra of matrix isolated PCl3, MeOH and their complexes were analyzed and assigned by comparing the observed and calculated frequencies and intensities. The experimental evidence for the noncovalent P⋯O interaction in PCl3–MeOH adducts was provided.
MeOH is one of the most abundant group of oxygenated volatile organic compounds,22,23 and is one of the simplest stable organic compounds capable of forming intermolecular hydrogen bonded complexes. A number of studies have been carried out to investigate the red shifts of OH-stretching vibrational frequency in MeOH clusters since they provide sensitive probes for the hydrogen bond interaction.7,24–27 The OH-stretching transition for the interaction between two constituent MeOH molecules has a 111 cm−1 red shift in the jet-cooled gas phase, which has been augmented with information on its diagonal anharmonicity.27 The study of hydrogen bonded MeOH complexes has been performed by vapor phase infrared spectroscopy. In order to obtain experimental evidence for NIR transitions in complexes, the spectroscopic studies of hydrogen bond in the vapor phase involving MeOH and trimethylamine (TMA) have been performed. Upon complexation to TMA, the OH-stretching frequency of MeOH was red shifted by 186 cm−1 relative to the MeOH monomer. The first OH-stretching overtone transition of the complex is tentatively assigned at 6450 cm−1.26 Gas phase vibrational spectra of the bimolecular complex formed between MeOH and dimethylamine (DMA)/TMA have also been measured to determine the enthalpy of complexation for the MeOH–DMA and MeOH–TMA complexes in the temperature range 298–358 K. The spectroscopic infrared results show the enthalpy of formation to be −35.8 ± 3.9 and −38.2 ± 3.3 kJ mol−1 for MeOH–DMA and MeOH–TMA, respectively.24 Furthermore, the detection of the O–H⋯π interactions between MeOH and alkenes has also caused much attention in recent years. An FTIR spectroscopic study of small isolated clusters of MeOH and ethene has been performed with experimental red shifts of 45 cm−1 from the MeOH monomer fundamental position at 3686 cm−1, which confirms the formation of weak intermolecular hydrogen bond to a π cloud.7 A systematic study of the methylation of anisole to change the π cloud attractivity relative to the oxygen side for the docking of MeOH has been undertaken by supersonic jet-FTIR. The spectroscopic method of determining the docking preference of MeOH to anisole is found to reveal that the subtle balance between these two structures can be varied by one order of magnitude through single to triple methylation of the aromatic ring and introduction of a single tert-butyl substituent.25
Heterocyclic organic compounds containing oxygen, nitrogen, or sulfur, are an important group of atmospheric pollutants and they are the basic units of brown carbon, which is well known to be an important component in the atmosphere where it affects climate forcing and carbon cycling.28–30 Four heterocyclic compounds were selected as the representative examples for acceptors in this study: furan (Fu), 2,5-dihydrofuran (DHF), pyrrole (Py) and thiophene (Th). Among them, furan and its derivatives, such as, 2,5-dihydrofuran, are both primary and secondary pollutants in the atmosphere, and their emission contributes to the formation of ultrafine particles and ground-level ozone.31,32 Besides, furans can react with the atmospheric radicals and affect the oxidative budget of the lower troposphere.33 The atmospheric source of furan includes emissions from agricultural processes, atmospheric oxidation reactions of 1,3-butadiene and 1,3-pentadiene, tropical forest burning, food processing and direct emissions from plant material.28,34,35 Pyrrole was detected in biomass and biofuel burning, and it was identified as a potential light-absorption brown carbon. Atmospheric thiophene mainly comes from fossil fuel combustion.36 In order to evaluate the atmospheric behavior of these heterocyclic compounds, it is important to study their interaction with other atmospheric molecules, such as MeOH.
The formation of new particles in the ambient atmosphere is a complex phenomenum, but the detailed molecular level processes and the exact participating molecules still remain largely unknown. MeOH is believed to be a common hydrogen bond donor to drive gas to particle conversion,37,38 and heterocyclic organic compounds are quite possible to be candidate species to stabilize MeOH clusters via hydrogen bond. In this work, the hydrogen bond between MeOH and four heterocyclic compounds including furan, DHF, pyrrole and thiophene were investigated using matrix isolation FTIR spectroscopy. Quantum chemical calculations of the MeOH–Fu, MeOH–DHF, MeOH–Th, and MeOH–Py complexes were performed to help determine the most stable conformers and then interpret the spectra.
2. Experimental and computational methods
2.1 Experimental details
All the experiments were performed with a closed-cycle helium compressor cooled cryostat (PT-SHI-4-5, Janis Research Company, USA) to achieve low temperature. The cryostat was housed in a vacuum chamber. A pressure gauge (WRG-NW25, Edward, UK) was used to monitor the pressure inside the cryostat chamber. The base pressure of 10−5 mbar was generally recorded at the beginning of an experiment. The gas phase mixtures were co-deposited onto a diamond substrate at 14 K. Temperatures at the cold surface were measured with a silicon diode sensor (DT-670, Lakeshore, USA), and the temperature was regulated by a temperature controller (model 22C, Cryocon, USA). The substrate was put in the sample beam of a FTIR spectrometer to allow spectral measurement. Argon (99.999%, Deyang special gas company) was used as the matrix gas. MeOH (99.9%, Aladdin), furan (99%, Aladdin), DHF (97%, Aladdin), pyrrole (99.7%, Aladdin), and thiophene (99%, Aladdin) were used without further purification. The argon/sample ratios ranged from 1/700 to 5/700.The infrared spectra of the deposited samples were recorded at 0.5 cm−1 resolution with a Vertex 80v FTIR spectrometer (Bruker) fitted with a KBr beam splitter and liquid nitrogen cooled MCT (Mercury-Cadmium-Telluride) detector. The precursors were then separately deposited onto the diamond substrate at 14 K. The deposition was annealed to 25 K, 30 K, 35 K and maintained at each temperature for 30 min, and then re-cooled to 14 K. Spectra were recorded after each warming cycle to monitor any changes caused by these processes.
2.2 Computational details
Geometries optimizations and vibrational frequency calculations on the monomers (MeOH, DHF, furan, pyrrole and thiophene) and the complexes (MeOH–Fu, MeOH–DHF, MeOH–Py, and MeOH–Th) were performed with Gaussian 09 computational program package using the “opt = very tight,” and “int = ultrafine” options.39 Density functional theory (DFT) methods provide an adequate compromise between the desired chemical accuracy and the computational cost. The DFT-based functionals, such as the Becke-3 Lee–Yang–Parr (B3LYP), have been shown to provide reliable trends of bonding energies even though the small energy differences can be overshadowed by the DFT quadrature errors.40 Furthermore, DFT methods based on the B3LYP functional have also been indicated to produce quite accurate results for heterocyclic compounds.41 Besides the geometries optimizations, the harmonic vibrational frequency calculations were also performed with DFT methods with the absence of imaginary frequencies for all the structures of the relevant monomer and complexes, which ensured that the optimized structures were minima on the potential energy surface. The quantum chemical calculations enabled us to characterize the nature of the stationary points and also to assign the observed wavenumbers in our matrix isolation experiments.17 For comparison, M06-2X, ωB97X-D, and B3LYP-D3 functionals were also used, and the data calculated with these functionals are shown in ESI.† We used the aug-cc-pVTZ basis set for compounds containing the H and O atoms, and the aug-cc-pV(T+d)Z basis sets for compounds containing the S atom. The extra tight d functions in the aug-cc-pV(T+d)Z basis set has previously been shown to improve results for the third period atoms such as S.42,43 Thermal contributions to the energies were calculated under the harmonic oscillator–rigid rotor approximation.44 Corrected by the zero point vibrational energy (ZPVE) corrections, the energies changes were obtained through subtracting the energies of the two isolated reactants from the energy of the formed complexes:| ΔE = Ecomplex − ∑Ereactants | (1) |
The theory of atoms in molecules (AIM) is a topological method based on the electron density, ρ(r), and is applied to investigate interactions between the two monomers forming the hydrogen bonded complexes. The AIM analysis was performed using the wave functions computed at the B3LYP/aug-cc-pVTZ level of theory. The positive sign of the Laplacian charge density at BCP ∇2ρ(BCP) corresponds to closed shell interactions that are responsible for bonding, where electronic charge is concentrated around each nucleus. The bond critical points (BCPs) were evaluated from the B3LYP/aug-cc-pVTZ level using AIM 2000 program package (version 2).45 Generally, weak van der Waals type interactions are characterized by small values of the electron density and the Laplacian charge density.46 In hydrogen bonded complexes, the electron density and the Laplacian charge density are in the range of 0.002–0.034 and 0.016–0.130 a.u.47 The charge transfer of the complexes were calculated by integrating the electron density of the MeOH within the complex.
3. Results and discussion
3.1 Optimized geometries
Optimized structures of the two most stable MeOH complex conformers (a, and b) with the B3LYP functional are shown in Fig. 1. MeOH forms hydrogen bonds with heterocyclic organic compounds in two ways, O–H⋯π and O–H⋯Y (YO, S). In the O–H⋯π bonded structures, MeOH docks onto one side instead of the center of the heterocyclic organic compound ring. Previous studies have shown that a competition between O–H⋯O and O–H⋯π binding sites exists when MeOH docks onto 2,3-benzofuran.48 Similarly, the heterocyclic organic compounds investigated in this study can offer two hydrogen bond acceptor sites to an incoming hydrogen bond donor: its heteroatom and its delocalized π electron system. This bonding pattern has been found for the MeOH–benzene and water–benzene complexes.37 The docking motifs of MeOH and anisole have been studied by applying a cold supersonic jet expansion of the two monomers in helium.49 The results of this study showed that MeOH prefers the oxygen of anisole over the π system. The MeOH forms a hydrogen bond with the oxygen of anisole, whereas docking onto the π system is at least 1 kJ mol−1 less attractive and therefore hardly observable in the jet spectra. A competition of the O–H⋯O versus O–H⋯π hydrogen bonds in 2,3-benzofuran clusters with water and MeOH molecules suggests that these two comparable conformers are almost equally stable.50 Two isomers having different types of hydrogen bonds are distinguished by applying fluorescence-detected infrared spectroscopy and dispersed fluorescence spectroscopy.
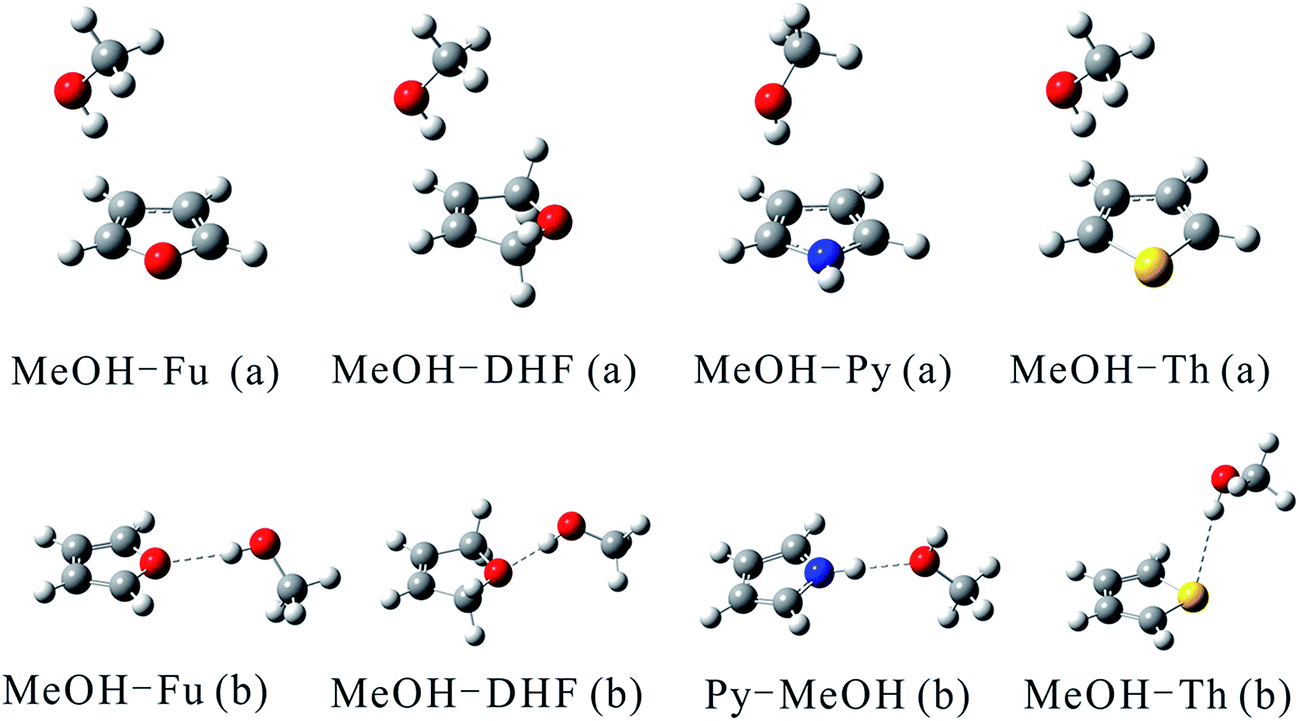 | ||
| Fig. 1 Structures of the MeOH complexes optimized at the B3LYP/aug-cc-pVTZ and B3LYP/aug-cc-pV(T+d)Z levels of theory for complexes containing O and N atoms and those containing S atom, respectively. | ||
The geometric parameters including the OH bond lengths and the changes upon complexation, the intermolecular hydrogen bond distance and the hydrogen bond angle calculated using the B3LYP functional are provided in Table 1. The O–H⋯O hydrogen bond lengths lie between 1.8877 and 2.0749 Å with bond angle varying from 167.1° to 173.4°. The intermolecular hydrogen bond distance in MeOH–Fu (b) complex is about 0.2 Å longer than that in the MeOH–DHF (b) complex, which indicates that MeOH–DHF (b) complex forms a stronger O–H⋯O hydrogen bond. The intermolecular hydrogen bond distance of the O–H⋯S bonded MeOH–Th (b) complex (2.7166 Å) is 0.6 Å longer than that of the MeOH–Fu (b) complex. The hydrogen bond distance in the MeOH–dimethyl ether (DME) and MeOH–dimethyl sulfide (DMS) complexes were reported to be 1.9085 Å and 2.4207 Å respectively,51 at the B3LYP/aug-cc-pVTZ level. Both are smaller than the O–H⋯O bonded MeOH–Fu (b) and O–H⋯S bonded MeOH–Th (b) complex. This suggests that linear compounds, like DME and DMS, can form stronger hydrogen bond than the aromatic heterocyclic compounds, such as furan and thiophene.
| Conformera | Bond type | R(OH)b | Δr(OH)c | R(HB)d | θ(HB)e |
|---|---|---|---|---|---|
| a The MeOH–Th complexes are calculated with the aug-cc-pV(T+d)Z basis set, and all other complexes with the aug-cc-pVTZ basis set, respectively.b OH bond length in the complex.c Change in the OH/NH bond length upon complexation.d The intermolecular hydrogen bond distance.e The intermolecular hydrogen bond angle.f In the Py–MeOH (b) complex, MeOH acts as hydrogen bond acceptor. R(NH) for pyrrole. R(NHb⋯O) and θ(NHb⋯O) for the Py–MeOH complexes. | |||||
| MeOH–Fu (a) | O–H⋯π | 0.9636 | 0.0028 | ||
| MeOH–Fu (b) | O–H⋯O | 0.9638 | 0.0030 | 2.0749 | 167.1 |
| MeOH–DHF (a) | O–H⋯π | 0.9636 | 0.0029 | ||
| MeOH–DHF (b) | O–H⋯O | 0.9702 | 0.0095 | 1.8877 | 173.4 |
| MeOH–Py (a) | O–H⋯π | 0.9652 | 0.0045 | ||
| Py–MeOH (b)f | N–H⋯O | 1.0109 | 0.0077 | 1.9844 | 178.7 |
| MeOH–Th (a) | O–H⋯π | 0.9634 | 0.0026 | ||
| MeOH–Th (b) | O–H⋯S | 0.9633 | 0.0025 | 2.7166 | 168.2 |
In the O–H⋯π bonded conformers, the OH bond length ranges from 0.9634 Å to 0.9752 Å. The change of OH bond lengths is more important in the O–H⋯O bonded conformers. The OH bond is lengthened by 0.0029 Å in the MeOH–DHF (a), and 0.0095 Å in the MeOH–DHF (b) conformers, respectively. In the MeOH–Th complexes, the changes of the OH bond length in the O–H⋯S and O–H⋯π bonded conformers are very similar. The OH bond lengths and the changes in the OH bond length upon complexation are very similar for the O–H⋯O/S bonded MeOH–Fu (b) and MeOH–Th (b) conformers, which suggests that the ability of furan to weaken the OH bond via hydrogen bond is equivalent to that of thiophene. This result is consistent with comparison between the MeOH–DME and MeOH–DMS complexes.51 Moreover, for O–H⋯O hydrogen bonded complexes, the OH bond length elongation upon complexation of MeOH–DME (0.0086 Å) is more important than that of MeOH–Fu (b) (0.0030 Å). A similar trend is also found in the O–H⋯S bonded MeOH–DMS (0.0091 Å) and MeOH–Th (b) (0.0025 Å) complexes. Among all the O–H⋯Y bonded MeOH complexes, MeOH–DHF (b) shows the most important bond length change Δr(OH) and the smallest hydrogen bond length r(HB). DHF forms the strongest hydrogen bond in the heterocyclic organic compounds. Generally, the hydrogen bond angles are expected to be close to 180° in an ideal hydrogen bonded complex.24,52 As seen in Table 1, the O–H⋯O/S hydrogen bond angles in MeOH–Fu and MeOH–Th deviate by 12° from the ideal linear orientation. For the O–H⋯O hydrogen bonded MeOH–DHF complexes, the angles only deviate by 6°. Generally, in similar hydrogen bonded MeOH complexes, smaller deviation of hydrogen bond angle represents more stable structure. As a result, MeOH–DHF (b) is the most stable complex of all the O–H⋯Y (YO, S) bonded complexes.
3.2 Topological analysis
AIM analysis was applied to describe the bonding in the MeOH complexes based on the topology of the electron density.53 Fig. 2 shows the AIM plots for the MeOH complexes with bond critical points (BCPs) and ring critical points (RCPs). The topological parameters, including electron density, Laplacian charge density, and change in atomic charge at the H atom Δq(H) are listed in Table 4. Two quantitative criteria have been suggested to characterize the strength of a hydrogen bond. The electron density is in the range of 0.002–0.0340 a.u. and the Laplacian charge density is in range of 0.016–0.130 a.u.47 The AIM analysis of these complexes shows only one BCP between the MeOH and each of the heterocyclic compounds. It is worth mentioning that, as shown in Table 2, the values of the electron density (0.0093–0.0295 a.u.) and the Laplacian charge density (0.0251–0.0865 a.u.) of the MeOH complexes were well within the range specified for the existence of the hydrogen bond. The positive and small values of the Laplacian charge density of the BCP's indicate the closed shell nature of the interactions similar to those found in hydrogen bonds and ionic interactions. | ||
| Fig. 2 AIM plots of the MeOH complexes with bond critical points (BCPs), ring critical points (RCPs) and electron density paths. | ||
| Conformer | Bond type | Δq(H) | ρ(BCP) | ∇2ρ(BCP) |
|---|---|---|---|---|
| a All values are in a.u. | ||||
| MeOH–Fu (a) | O–H⋯π | 0.0045 | 0.0098 | 0.0271 |
| MeOH–Fu (b) | O–H⋯O | 0.0298 | 0.0179 | 0.0651 |
| MeOH–DHF (a) | O–H⋯π | 0.0044 | 0.0104 | 0.0281 |
| MeOH–DHF (b) | O–H⋯O | 0.0510 | 0.0295 | 0.0865 |
| MeOH–Py (a) | O–H⋯π | 0.0111 | 0.0116 | 0.0314 |
| Py–MeOH (b) | N–H⋯O | 0.0504 | 0.0225 | 0.0791 |
| MeOH–Th (a) | O–H⋯π | 0.0071 | 0.0093 | 0.0266 |
| MeOH–Th (b) | O–H⋯S | 0.0041 | 0.0094 | 0.0251 |
The charge density ρ(BCP) at the BCP for the O–H⋯π interaction is about half of that for the O–H⋯O interaction. The Laplacian charge density at BCP is also smaller for the O–H⋯π interaction than for the O–H⋯O interaction. In addition, the loss of electronic charge on the hydrogen atom for O–H⋯O hydrogen bond is more important than in the O–H⋯π hydrogen bond, confirming that the O–H⋯O hydrogen bonded complex is more stable than the O–H⋯π bonded one. The charge density and Laplacian charge density of the O–H⋯π bonded MeOH–Th (a) is almost similar to the O–H⋯S bonded MeOH–Th (b), indicating that these two MeOH–Th isomers are almost equally stable.
3.3 Interaction energies
The calculated binding energy (BE), enthalpy of formation (ΔHθ) and Gibbs free energy of formation (ΔGθ) of the MeOH complexes are summarized in Table 3. As seen from Table 3, the ZPVE can be as large as 5.1 kJ mol−1. The BEs of O–H⋯π hydrogen bonded complexes range between −3.7 and −8.1 kJ mol−1. Compared to the O–H⋯O bonded conformers, the binding strengths in the O–H⋯π bonded conformers for MeOH–Fu and MeOH–DHF are weaker by 2.6 kJ mol−1 and 12.1 kJ mol−1, respectively. The BEs clearly indicate that the O–H⋯O bonded conformers are more strongly bound than the corresponding O–H⋯π hydrogen bonded conformers. Concerning for the MeOH–Py complexes, the BE of the N–H⋯O bonded Py–MeOH (b) is much less negative than that of O–H⋯π bonded MeOH–Py (a) complex. For the three other MeOH-complexes that are formed by furan, DHF and thiophene, the O–H⋯π bonded conformers (a) have less negative ΔHθ and larger ΔGθ than the corresponding conformers (b). The calculated BE and ΔHθ values of O–H⋯π and O–H⋯S bonded conformers of the MeOH–Th complex are very similar (differing by less than 1 kJ mol−1). However, the ΔGθ value of the MeOH–Th (b) conformer is smaller than the corresponding value of the MeOH–Th (a) conformer. Based on all the thermodynamic parameters presented above, the stability order of the two conformers of each MeOH-complex is as follows, MeOH–Fu (b) > MeOH–Fu (a), MeOH–DHF (b) > MeOH–DHF (a), Py–MeOH (b) > MeOH–Py (a) and MeOH–Th (b) ≈ MeOH–Th (a). Consequently, the MeOH–Fu (b), MeOH–DHF (b), Py–MeOH (b) and both O–H⋯π and O–H⋯S bonded MeOH–Th complexes are more likely to be formed in atmospheric environment. The nucleation trend shows a fundamental step in atmospheric new particle formation.| Conformera | Bond type | BEb | ZPVE | ΔHθ | ΔGθ |
|---|---|---|---|---|---|
| a The MeOH–Th complexes are calculated with the aug-cc-pV(T+d)Z basis set, and all other complexes with the aug-cc-pVTZ basis set, respectively.b BE corrected with ZPVE. All energies are given in kJ mol−1.c In the Py–MeOH (b) complex, MeOH acts as hydrogen bond acceptor. | |||||
| MeOH–Fu (a) | O–H⋯π | −4.5 | 2.5 | −1.7 | 22.5 |
| MeOH–Fu (b) | O–H⋯O | −7.1 | 3.3 | −4.7 | 20.6 |
| MeOH–DHF (a) | O–H⋯π | −3.7 | 2.7 | −0.9 | 22.1 |
| MeOH–DHF (b) | O–H⋯O | −15.8 | 5.1 | −14.3 | 16.3 |
| MeOH–Py (a) | O–H⋯π | −8.1 | 3.1 | −5.6 | 21.1 |
| Py–MeOH (b)c | N–H⋯O | −16.4 | 3.6 | −13.9 | 13.4 |
| MeOH–Th (a) | O–H⋯π | −4.6 | 2.4 | −1.6 | 22.1 |
| MeOH–Th (b) | O–H⋯S | −3.3 | 1.9 | 0.05 | 17.1 |
The BEs relevant for hydrogen bonding are also compared for the lowest energy conformers of every MeOH complexes. The maximum BE is predicted for the Py–MeOH (b) conformer with a value of −16.4 kJ mol−1. This is due to the presence of a strong N–H⋯O hydrogen bond. The second most stable complex is MeOH–DHF (b), which is 0.6 kJ mol−1 less stable than the Py–MeOH (b) conformer. The following less stable structure is the MeOH–Fu (b) conformer having a BEs of 7.1 kJ mol−1 with a weaker O–H⋯O hydrogen bond. The O–H⋯O bonded MeOH–Fu (b) has larger calculated BE, ΔHθ and ΔGθ values than the O–H⋯S bonded MeOH–Th (b) conformer. These computed hydrogen bond indicators nicely correlate with the previous work, which showed that O–H⋯S hydrogen bond is weaker than the O–H⋯O hydrogen bond.54 Besides, this difference in hydrogen bond strength is reflected in the OH-stretching vibrational frequencies calculated for the MeOH–Fu and MeOH–Th complexes.
3.4 IR spectra
The hydrogen bonds in the MeOH complexes were detected with matrix isolation FTIR. The argon matrix infrared spectra of MeOH complexes in the OH-stretching fundamental transition![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) OH region were measured after the matrix annealed to 14 K. In the spectra of the MeOH complexes, bands with new features that were not present in the two monomers were marked with asterisk, which indicates the formation of the complexes. The spectra of MeOH, furan, and the MeOH–Fu complex are presented in Fig. 3. Different mixing ratios of Fu/MeOH were applied to confirm the formation of the hydrogen-bonded complex. The two lower spectra in Fig. 3 are the monomers of furan (trace a) and MeOH (trace b). Trace c and d are spectra of the MeOH–Fu complexes with different Fu/MeOH mixing ratio, 1:1 for trace c and 5:1 for trace d. The fundamental OH-stretching frequency in MeOH is found to be ∼3666 cm−1. In the spectra of MeOH–Fu complexes, this band has also been detected. Features due to the red shifts (ΔOH) of the OH-stretching fundamental transition of MeOH were observed at ∼3616 cm−1 and ∼3635 cm−1 in the spectrum of the MeOH–Fu complex, it is assigned here to the OH-stretching transition frequency of the 1:1 MeOH–Fu (b) complex due to its stronger O–H⋯O hydrogen bond. The intensity of the OH-stretching frequency of the MeOH monomer decreases with increasing Fu/MeOH mixing ratio because part of MeOH takes part in the formation of MeOH–Fu complex forming the O–H⋯O hydrogen bond. Furthermore, since increasing the proportion of the furan leads to more MeOH-complex formation, the intensity of the OH-stretching frequency of the MeOH–Fu complex increases with the increment of adding proportion on furan. It should be mentioned that the 3530 cm−1 band which decreases slightly with increasing Fu/MeOH mixing ratio is recorded in all infrared spectra of the MeOH monomer and the corresponding MeOH complexes investigated in this study. We assign these features to the dimer of MeOH. Similar phenomenon has been found in previous studies.27,55 With the formation of MeOH complexes, MeOH tends to interact stronger with heterocyclic organic compounds forming hydrogen bonded complexes rather than forming MeOH dimer.
OH region were measured after the matrix annealed to 14 K. In the spectra of the MeOH complexes, bands with new features that were not present in the two monomers were marked with asterisk, which indicates the formation of the complexes. The spectra of MeOH, furan, and the MeOH–Fu complex are presented in Fig. 3. Different mixing ratios of Fu/MeOH were applied to confirm the formation of the hydrogen-bonded complex. The two lower spectra in Fig. 3 are the monomers of furan (trace a) and MeOH (trace b). Trace c and d are spectra of the MeOH–Fu complexes with different Fu/MeOH mixing ratio, 1:1 for trace c and 5:1 for trace d. The fundamental OH-stretching frequency in MeOH is found to be ∼3666 cm−1. In the spectra of MeOH–Fu complexes, this band has also been detected. Features due to the red shifts (ΔOH) of the OH-stretching fundamental transition of MeOH were observed at ∼3616 cm−1 and ∼3635 cm−1 in the spectrum of the MeOH–Fu complex, it is assigned here to the OH-stretching transition frequency of the 1:1 MeOH–Fu (b) complex due to its stronger O–H⋯O hydrogen bond. The intensity of the OH-stretching frequency of the MeOH monomer decreases with increasing Fu/MeOH mixing ratio because part of MeOH takes part in the formation of MeOH–Fu complex forming the O–H⋯O hydrogen bond. Furthermore, since increasing the proportion of the furan leads to more MeOH-complex formation, the intensity of the OH-stretching frequency of the MeOH–Fu complex increases with the increment of adding proportion on furan. It should be mentioned that the 3530 cm−1 band which decreases slightly with increasing Fu/MeOH mixing ratio is recorded in all infrared spectra of the MeOH monomer and the corresponding MeOH complexes investigated in this study. We assign these features to the dimer of MeOH. Similar phenomenon has been found in previous studies.27,55 With the formation of MeOH complexes, MeOH tends to interact stronger with heterocyclic organic compounds forming hydrogen bonded complexes rather than forming MeOH dimer.
 | ||
| Fig. 3 Infrared spectra of MeOH, furan, and the co-deposition of them at 14 K. (a) Fu/Ar = 1/700; (b) MeOH/Ar = 1/700; (c) MeOH/Fu/Ar = 1/1/700; (d) MeOH/Fu/Ar = 1/5/700. Bands marked with asterisk are new features from MeOH–Fu complex. The spectra were offset for clarity. | ||
The FTIR spectra of MeOH, DHF, and MeOH–DHF are shown in Fig. 4. In the infrared absorption spectra, the 3488 and 3563 cm−1 bands are the OH-stretching fundamental transition of the MeOH–DHF complex with calculated OH-stretching mode frequency 3643 cm−1 and 3768 cm−1 (Table 4). This assignment is underscored by the distinct intensity evolution of these bands when varying the relative DHF concentration. Stronger OH-stretching vibration bands of the MeOH–DHF complex are observed in higher DHF/MeOH mixing ratio. The complex spectra obtained from the matrix isolation experiment belong to the more stable O–H⋯O hydrogen-bonded MeOH–DHF (b) complex according to the geometric and thermodynamic parameters discussed above. Similar measurements for the MeOH–Th and MeOH–Py complex were also carried out and the results are shown in Fig. 5 and S2,† respectively. The feature observed at 3666 cm−1 is due to MeOH monomer. The features observed around 3530 cm−1 are due to MeOH dimer, which agrees well with the literature value.55 The fundamental OH-stretching frequencies of MeOH–Th complex are recorded at 3614 cm−1, 3636 cm−1 in the infrared absorption spectra (calculated value 3776 cm−1 (Table 4)). It is hard to assign the spectra to the exact MeOH–Th conformer, because all hydrogen bond characteristic features we obtained for different MeOH–Th conformers are too close. It is likely that the two MeOH–Th conformers co-exist in the atmosphere. The integration regions for MeOH–Py are 3300–3700 cm−1. As shown in Fig. S2,† MeOH–Py (a) and Py–MeOH (b) were all detected in our matrix isolation experiments. The 3581 cm−1, 3583 cm−1 bands which have a small red shifts (ΔOH) are assigned to the MeOH–Py (a). It is plausible to assign the 3371 cm−1 band to the Py–MeOH (b), which is significantly lower in energy than the MeOH–Py (a) complex and has a larger red shifts (ΔOH). The calculated fundamental OH-stretching frequencies for the MeOH–Py complexes are 3740 cm−1, and 3536 cm−1. And this assignment follows the expected trend with the calculated results of the red shifts (ΔOH) in MeOH–Py complexes (Table 4).
 | ||
| Fig. 4 Infrared spectra of MeOH, DHF, and the co-deposition of them at 14 K. (a) DHF/Ar = 1/700; (b) MeOH/Ar = 1/700; (c) MeOH/DHF/Ar = 1/1/700; (d) MeOH/DHF/Ar = 1/5/700. Bands marked with asterisk are new features from MeOH–DHF complex. The spectra were offset for clarity. | ||
| Conformer | Calculated | Observed | ||||
|---|---|---|---|---|---|---|
| OH |
ΔOHa |
f | f/fMeOH | OHc |
ΔOHa |
|
| a ΔOH = MeOH − complex.b In the Py–MeOH (b) complex, MeOH acts as hydrogen bond acceptor. ΔNH = pyrrole − complex. The calculated NH-stretching transition wavenumber of pyrrole is 3671 cm−1.c The observed NH-stretching transition wavenumber of pyrrole is 3520 cm−1. |
||||||
| MeOH | 3828 | — | 5.8 × 10−6 | — | 3666 | — |
| MeOH–Fu (a) | 3770 | 58 | 4.2 × 10−5 | 7.3 | 3635, 3616 | 31–50 |
| MeOH–Fu (b) | 3772 | 56 | 6.2 × 10−5 | 10.6 | ||
| MeOH–DHF (a) | 3766 | 61 | 4.4 × 10−5 | 7.5 | 3488, 3563 | 103–178 |
| MeOH–DHF (b) | 3638 | 190 | 1.2 × 10−4 | 20.6 | ||
| MeOH–Py (a) | 3738 | 90 | 5.2 × 10−5 | 9.0 | 3581, 3583 | 85–87 |
| Py–MeOH (b)b | 3537 | 134 | 1.1 × 10−4 | 18.8 | 3371 | 149 |
| MeOH–Th (a) | 3775 | 53 | 4.1 × 10−5 | 7.0 | 3636, 3614 | 30–60 |
| MeOH–Th (b) | 3775 | 53 | 4.0 × 10−5 | 6.9 | ||
 | ||
| Fig. 5 Infrared spectra of MeOH, thiophene, and the co-deposition of them at 14 K. (a) Th/Ar = 1/700; (b) MeOH/Ar = 1/700; (c) MeOH/Th/Ar = 1/1/700; (d) MeOH/Th/Ar = 1/5/700. Bands marked with asterisk are new features from MeOH–Th complex. The spectra were offset for clarity. | ||
The OH-stretching bands of MeOH–Fu and MeOH–Th complexes show similar profile (Fig. S1†), which demonstrates that formation of hydrogen bond in the MeOH–Fu complex is similar to the one in the MeOH–Th complex. The OH-stretching transition frequency of MeOH–Fu (3616, 3635 cm−1) is similar to the one in the MeOH–Th complex (3614, 3636 cm−1). This confirms that these two MeOH-complexes are formed in a similar way, and S is an acceptor atom similar in strength to O. The previous experimental studies on methionine–tyrosine hydrogen bonded complexes also indicate that O–H⋯S interaction is perhaps as strong as O–H⋯O interaction.56 However, the OH-stretching transition frequency of MeOH–Py (3581, 3583 cm−1) is smaller than those of MeOH–Fu and MeOH–Th complexes. Comparing the fundamental OH-stretching transition frequencies of the two different MeOH–Py complexes, we found that our measured OH-stretching frequency of MeOH–Py (a) (3581, 3583 cm−1) is much larger than that of Py–MeOH (b) (3371 cm−1), and therefore, the Py–MeOH (b) complex is significantly more stable than the MeOH–Py (a) complex. Similar findings were also observed from quantum chemical calculations performed in this study. In addition, the OH-stretching transition frequency of MeOH–DHF (3488, 3563 cm−1) is smaller than that of MeOH–Fu complex (3653, 3616 cm−1). Accordingly, DHF forms stronger O–H⋯O hydrogen bonded complexes than furan.
The red shift value (ΔOH) of the OH-stretching transitions (ΔOH = MeOH − complex) correlates with the hydrogen bond strength. The smaller shift in the complex points out a weaker hydrogen bond formation. The observed bands origin for the complexes are red shifted with respect to their respective monomers. The observed and calculated fundamental OH-stretching transition frequency and the red shifts (ΔOH) of the fundamental OH-stretching in the complexes are summarized in Table 4. The maximum red shifts (ΔOH) are observed for O–H⋯O hydrogen bonded MeOH–DHF complex (103, 178 cm−1) followed by N–H⋯O hydrogen bonded Py–MeOH (b) (149 cm−1), O–H⋯π hydrogen bonded MeOH–Py (a) (85, 87 cm−1), O–H⋯O hydrogen bonded MeOH–Fu complex (31, 50 cm−1) and O–H⋯S hydrogen bonded MeOH–Th complex (30, 60 cm−1). The magnitudes of the red shifts (ΔOH) observed in MeOH–Fu complex are similar to those observed in MeOH–Th complexes, but they are all smaller than those of MeOH–DHF and MeOH–Py (a). Of all the MeOH complexes studied here, Py–MeOH (b) has larger red shifts (ΔOH) of the NH-stretching fundamental transition than MeOH–Py (a), which would suggest that N–H⋯O hydrogen bonded Py–MeOH (b) is more stable than O–H⋯π hydrogen bonded MeOH–Py (a). Hence, the hydrogen bond strength for MeOH-complexes can be sorted as follows: DHF forms stronger O–H⋯Y (YO, S) hydrogen bond than furan and thiophene, which is attributed to the nonaromatic properties of DHF. In addition, the O–H⋯S hydrogen bond strength of MeOH–Th is almost similar to O–H⋯O of the MeOH–Fu complex. These observed trends are in excellent agreement with the former measurement for other analogous hydrogen bonded complexes, which depicted that MeOH–TMA (ΔOH = 333 cm−1) is more stable than MeOH–DMS (ΔOH = 113 cm−1) and MeOH–DME (ΔOH = 103 cm−1).24,55,57 Experimental studies of MeOH-complex under different temperature conditions have drawn the same conclusion as well. The spectra of the mixture recorded at 14 K and 25 K are shown in Fig. S1.† The intensity of the OH-stretching frequency of the MeOH monomer decreases slightly with increasing temperature. Similar trend is also found in the bands of MeOH dimer. In contrast, the features of the relevant bands of the MeOH-complex formation are more obvious when the annealing temperature increases from 14 K to 25 K. This phenomenon also provides evidence to the formation of four different MeOH-complexes. Furthermore, we can also conclude that increasing temperature favors the complexes formation. A recent study about the thermodynamic properties of gas phase hydrogen bonded complexes has also confirmed that the OH-stretching vibration in the MeOH–DMA complex is temperature dependent.58 More detailed information of the co-deposition of MeOH and pyrrole are shown in Fig. S2.† The trend observed in the intermolecular stretching frequencies and relative red shifts of OH-stretching in the FTIR spectra are consistent with the binding energy values calculated with DFT methods.
3.5 Calculated OH-stretching transitions and intensities
Vibrational frequency calculations were performed on the optimized geometries to help assigning the wavenumbers observed in the matrix isolation experiment. The calculated OH-stretching frequencies are listed in Table 4. The experimental and calculated red shifts (ΔOH) of OH-stretching fundamental transition are also included. The experimental observations of OH-stretching fundamental vibrations wavenumbers are smaller than the calculated frequencies with respect to the monomer. The calculated red shifts of MeOH-complexes follow the same trends as the experimental observations. As given in Table 4, the calculated red shift (ΔOH) with the π-type hydrogen bond is in the range 53–90 cm−1, which is larger than that in previous studies which described that OH-stretching red shifts is about 45 cm−1 for weak intermolecular O–H⋯π hydrogen bond in MeOH–ethene complex.7 The maximum calculated red shifts (ΔOH) is the O–H⋯O hydrogen bonded MeOH–DHF complex (190 cm−1) followed by Py–MeOH (b) (134 cm−1), MeOH–Py (a) (90 cm−1), O–H⋯π hydrogen bonded MeOH–DHF (a) (61 cm−1), MeOH–Fu (a) (58 cm−1), MeOH–Fu (b) (56 cm−1) and MeOH–Th complex (53 cm−1). The results agree well with the experimental red shifts, and thus indicate that the hydrogen bond strength of O–H⋯O in MeOH–Fu complexes is similar to O–H⋯S hydrogen bond in the MeOH–Th complexes. In O–H⋯O hydrogen bonded complexes, MeOH–DHF is more stable than MeOH–Fu complex because oxygen atom in furan is affected by the delocalization of π-electrons in the furan ring, thus forming the weaker O–H⋯O hydrogen bond.
The oscillator strength (f) and the relative intensities (f/fMeOH) of the OH-stretching fundamental transition for the complexes are also listed in Table 4. The oscillator strengths of OH-stretching vibrations in MeOH complexes were over seven times stronger than MeOH monomer as a result of hydrogen bond formation. In addition, we found that the calculated oscillator strengths of the two different conformers of a same complex are quite different except for the MeOH–Th complex. The oscillator strengths of O–H⋯π hydrogen bonded conformers are smaller than O–H⋯O hydrogen bonded conformers, which means that the conformer (b) of the MeOH–Fu and MeOH–Th complexes is more stable than the conformer (a). This trend is also suitable for MeOH–Py complexes, as the strength of Py–MeOH (b) conformer is almost twice that of MeOH–Py (a) conformer. Nevertheless, there is no significant difference in oscillator strength between the O–H⋯π (4.1 × 10−5) and O–H⋯S (4.0 × 10−5) bonded conformers of the MeOH–Th complex. As the values are too close to each other, it is hard to tell which bond is stronger based on the relative intensities.
4. Conclusions
A competition between O–H⋯O/S and O–H⋯π binding sites exists in MeOH complexes when MeOH dock onto the heterocyclic organic compounds, such as furan, DHF, pyrrole and thiophene. To compare the competition between these two different bonding sites, we have investigated the hydrogen bond MeOH complexes using matrix isolation FTIR spectroscopy and theoretical calculations. Obviously, two different configurations (X–H⋯Y and O–H⋯π hydrogen bonded conformers) in each MeOH-complex were obtained. Generally, the structures with a hydrogen bond to the heteroatom are more stable than those bonding to the π systems. AIM results clearly predict the nature of interaction in MeOH complexes, and indicate the presence of the hydrogen bond between MeOH and the heterocyclic organic compounds. The measured OH-stretching transitions of the complexes were assigned as from the O–H⋯Y (YO, S) bonded structures, and the N–H⋯O bonded Py–MeOH complexes. The observed red shifts (ΔOH) of the OH-stretching fundamental transition are consistent with the theoretical computational results, which confirms that the complex spectra of MeOH–Fu and MeOH–DHF complexes obtained from the matrix isolation experiment belong to the O–H⋯O hydrogen bonded complexes. However, the O–H⋯π bonded MeOH–Th (a) conformer may co-exist with the O–H⋯S bonded MeOH–Th (b) conformer since all the values obtained from theoretical calculations for different MeOH–Th conformers are very close to each other. In addition, comparison of the red shifts (ΔOH) of the OH-stretching fundamental transition of the complexes shows that MeOH–DHF is more stable than MeOH–Fu complex, while the hydrogen bond strength of O–H⋯O in MeOH–Fu complexes is similar to O–H⋯S hydrogen bond in the MeOH–Th complexes. The results of this study help to understand the nature of hydrogen bonds in MeOH-complexes and their implication in new particle formation.
Acknowledgements
This work was supported by National Natural Science Foundation of China (21577080, 91644214, 21407095) and Shandong Provincial Natural Science Foundation, China (ZR2014BQ013).Notes and references
- F. Bianchi, J. Trostl, H. Junninen, C. Frege, S. Henne, C. R. Hoyle, U. Molteni, E. Herrmann, A. Adamov, N. Bukowiecki, X. Chen, J. Duplissy, M. Gysel, M. Hutterli, J. Kangasluoma, J. Kontkanen, A. Kuerten, H. E. Manninen, S. Muench, O. Perakyla, T. Petaja, L. Rondo, C. Williamson, E. Weingartner, J. Curtius, D. R. Worsnop, M. Kulmala, J. Dommen and U. Baltensperger, Science, 2016, 352, 1109–1112 CrossRef CAS PubMed.
- R. Zhang, Science, 2010, 328, 1366–1367 CrossRef CAS PubMed.
- J. Zhao, A. Khalizov, R. Zhang and R. McGraw, J. Phys. Chem. A, 2009, 113, 680–689 CrossRef CAS PubMed.
- P. Paasonen, T. Olenius, O. Kupiainen, T. Kurten, T. Petaja, W. Birmili, A. Hamed, M. Hu, L. G. Huey, C. Plass-Duelmer, J. N. Smith, A. Wiedensohler, V. Loukonen, M. J. McGrath, I. K. Ortega, A. Laaksonen, H. Vehkamaki, V. M. Kerminen and M. Kulmala, Atmos. Chem. Phys., 2012, 12, 9113–9133 CAS.
- M. Voehringer, E. B. Hansmann, H. Hernandez, J. S. Francisco, J. Troe and B. Abel, Science, 2007, 315, 497–501 CrossRef PubMed.
- B. Long, X. F. Tan, C. R. Chang, W. X. Zhao, Z. W. Long, D. S. Ren and W. J. Zhang, J. Phys. Chem. A, 2013, 117, 5106–5116 CrossRef CAS PubMed.
- M. Heger, R. A. Mata and M. A. Suhm, Chem. Sci., 2015, 6, 3738–3745 RSC.
- S. Tang, H. Zhao and L. Du, RSC Adv., 2016, 6, 91233–91242 RSC.
- B. J. van der Veken, W. A. Herrebout, R. Szostak, D. N. Shchepkin, Z. Havlas and P. Hobza, J. Am. Chem. Soc., 2001, 123, 12290–12293 CrossRef CAS PubMed.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2015, 119, 8497–8502 CrossRef CAS PubMed.
- A. A. Stolov, W. A. Herrebout and B. J. van der Veken, J. Am. Chem. Soc., 1998, 120, 7310–7319 CrossRef CAS.
- F. Kollipost, J. Andersen, D. W. Mahler, J. Heimdal, M. Heger, M. A. Suhm and R. W. Larsen, J. Chem. Phys., 2014, 141, 174314 CrossRef CAS PubMed.
- K. E. Otto, S. Hesse, T. N. Wassermann, C. A. Rice, M. A. Suhm, T. Stafforst and U. Diederichsen, Phys. Chem. Chem. Phys., 2011, 13, 14119–14130 RSC.
- A. Sharma, I. Reva, R. Fausto, S. Hesse, Z. Xue, M. A. Suhm, S. K. Nayak, R. Sathishkumar, R. Pal and T. N. G. Row, J. Am. Chem. Soc., 2011, 133, 20194–20207 CrossRef CAS PubMed.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2012, 116, 4089–4096 CrossRef CAS PubMed.
- M. Rozenberg and A. Loewenschuss, J. Phys. Chem. A, 2009, 113, 4963–4971 CrossRef CAS PubMed.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2014, 118, 1004–1011 CrossRef CAS PubMed.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2015, 119, 2271–2280 CrossRef CAS PubMed.
- J. P. Perchard, F. Romain and Y. Bouteiller, Chem. Phys., 2008, 343, 35–46 CrossRef CAS.
- M. Muzomwe, B. Boeckx, G. Maes and O. E. Kasende, S. Afr. J. Chem., 2011, 64, 23–33 CAS.
- P. R. Joshi, N. Ramanathan, K. Sundararajan and K. Sankaran, J. Phys. Chem. A, 2015, 119, 3440–3451 CrossRef CAS PubMed.
- A. Mellouki, T. J. Wallington and J. Chen, Chem. Rev., 2015, 115, 3984–4014 CrossRef CAS PubMed.
- G. Legreid, J. B. Loeoev, J. Staehelin, C. Hueglin, M. Hill, B. Buchmann, A. S. H. Prevot and S. Reimann, Atmos. Environ., 2007, 41, 8409–8423 CrossRef CAS.
- L. Du, K. Mackeprang and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2013, 15, 10194–10206 RSC.
- H. C. Gottschalk, J. Altnoeder, M. Heger and M. A. Suhm, Angew. Chem., Int. Ed., 2016, 55, 1921–1924 CrossRef CAS PubMed.
- D. L. Howard and H. G. Kjaergaard, J. Phys. Chem. A, 2006, 110, 9597–9601 CrossRef CAS PubMed.
- F. Kollipost, K. Papendorf, Y. F. Lee, Y. P. Lee and M. A. Suhm, Phys. Chem. Chem. Phys., 2014, 16, 15948–15956 RSC.
- T. G. Karl, T. J. Christian, R. J. Yokelson, P. Artaxo, W. M. Hao and A. Guenther, Atmos. Chem. Phys., 2007, 7, 5883–5897 CrossRef CAS.
- T. B. Nguyen, A. Laskin, J. Laskin and S. A. Nizkorodov, Faraday Discuss., 2013, 165, 473–494 RSC.
- S. Kumar, V. Pande and A. Das, J. Phys. Chem. A, 2012, 116, 1368–1374 CrossRef CAS PubMed.
- P. M. Lemieux, C. C. Lutes and D. A. Santoianni, Prog. Energy Combust. Sci., 2004, 30, 1–32 CrossRef CAS.
- F. Villanueva, B. Cabanas, E. Monedero, S. Salgado, I. Bejan and P. Martin, Atmos. Environ., 2009, 43, 2804–2813 CrossRef CAS.
- I. Kind, T. Berndt, O. Boge and W. Rolle, Chem. Phys. Lett., 1996, 256, 679–683 CrossRef CAS.
- T. J. Christian, B. Kleiss, R. J. Yokelson, R. Holzinger, P. J. Crutzen, W. M. Hao, B. H. Saharjo and D. E. Ward, J. Geophys. Res., 2003, 108, 1–13 Search PubMed.
- T. J. Christian, B. Kleiss, R. J. Yokelson, R. Holzinger, P. J. Crutzen, W. M. Hao, T. Shirai and D. R. Blake, J. Geophys. Res., 2004, 109, 1–12 CrossRef.
- B. Cabanas, M. T. Baeza, P. Martin, S. Salgado, F. Villanueva, E. Monedero and Y. D. De Mera, Int. J. Chem. Kinet., 2006, 38, 570–576 CrossRef CAS.
- H. Zhao, J. Chang and L. Du, Comput. Theor. Chem., 2016, 1084, 126–132 CrossRef CAS.
- H. Zhao, S. Tang, S. Li, L. Ding and L. Du, Struct. Chem., 2016, 27, 1241–1253 CrossRef CAS.
- L. Du, J. R. Lane and H. G. Kjaergaard, J. Chem. Phys., 2012, 136, 184305 CrossRef PubMed.
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Dkhissi, L. Adamowicz and G. Maes, J. Phys. Chem. A, 2000, 104, 2112–2119 CrossRef CAS.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244–9253 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- V. Vaida and J. E. Headrick, J. Phys. Chem. A, 2000, 104, 5401–5412 CrossRef CAS.
- F. Tran and T. A. Wesolowski, Int. J. Quantum Chem., 2005, 101, 854–859 CrossRef CAS.
- R. G. A. Bone and R. F. W. Bader, J. Phys. Chem., 1996, 100, 10892–10911 CrossRef CAS.
- P. L. A. Popelier and R. F. W. Bader, Chem. Phys. Lett., 1992, 189, 542–548 CrossRef CAS.
- H. Sasaki, S. Daicho, Y. Yamada and Y. Nibu, J. Phys. Chem. A, 2013, 117, 3183–3189 CrossRef CAS PubMed.
- M. Heger, J. Altnoeder, A. Poblotzki and M. A. Suhm, Phys. Chem. Chem. Phys., 2015, 17, 13045–13052 RSC.
- H. Sasaki, S. Daicho, Y. Yamada and Y. Nibu, J. Phys. Chem. A, 2013, 117, 3183–3189 CrossRef CAS PubMed.
- D. L. Howard and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2008, 10, 4113–4118 RSC.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- H. S. Biswal, P. R. Shirhatti and S. Wategaonkar, J. Phys. Chem. A, 2009, 113, 5633–5643 CrossRef CAS PubMed.
- A. S. Hansen, L. Du and H. G. Kjaergaard, J. Phys. Chem. Lett., 2014, 5, 4225–4231 CrossRef CAS PubMed.
- H. S. Biswal, S. Chakraborty and S. Wategaonkar, J. Chem. Phys., 2008, 129, 184311 CrossRef PubMed.
- B. Michielsen, C. Verlackt, B. J. van der Veken and W. A. Herrebout, J. Mol. Struct., 2012, 1023, 90–95 CrossRef CAS.
- A. S. Hansen, Z. Maroun, K. Mackeprang, B. N. Frandsen and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2016, 18, 23831–23839 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26076d |
| This journal is © The Royal Society of Chemistry 2017 |