
Application of a mild hydrothermal method to the synthesis of mixed transition-metal(ii)/uranium(iv) fluorides†
Abstract
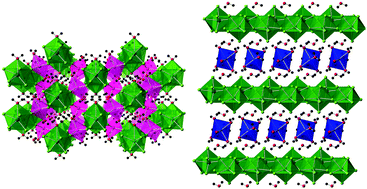
Single crystals of five transition metal uranium fluorides were obtained via the use of a mild hydrothermal route. Uranyl acetate was used as both the uranium source and the reducing agent for an in situ reduction of U(VI) to U(IV). The synthesized materials are present as both two- and three-dimensional structures and contain uranium in 9-fold coordination environments. Magnetic susceptibility measurements indicate that the reported materials remain paramagnetic down to 2 K, with no evidence for the existence of long-range magnetic ordering. Thermogravimetric analysis studies of the reported materials are also presented.
- This article is part of the themed collection: In honour of Mercouri G. Kanatzidis for his contributions to Inorganic Chemistry for over 30 years
 Please wait while we load your content...
Please wait while we load your content...

