
Hydroxyapatite: catalyst for a one-pot pentose formation†
 *ab
*ab
Abstract
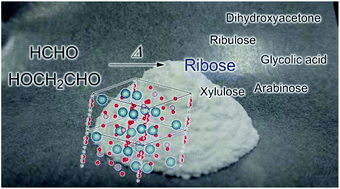
One of the possible synthetic routes to pentoses is the formose reaction pathway from C1 and C2 carbon sources, but preferential ribose generation in a one-pot reaction without any control of conditions has not been reported. We have tested a one-pot pentose formation and analyzed the products and mechanism in the reaction, using 1H-NMR and mass spectrometry. Hydroxyapatite (HAp), which consists of phosphate and calcium ions, worked continuously for cross-aldol reactions and Lobry de Bruyn–van Ekenstein transformations to yield ribose from formaldehyde and glycolaldehyde. The continuous reaction proceeds in one pot in hot water only in the presence of a HAp catalyst, without any fine pH control or any complicated condition control at each reaction step. Ribose production by HAp may be a reason why a pentose backbone was incorporated into nucleic acids in the prebiotic world.
 Please wait while we load your content...
Please wait while we load your content...

