
Driving and photo-regulation of myosin–actin motors at molecular and macroscopic levels by photo-responsive high energy molecules†
 ab
Nobuyuki
Tamaoki
*ab
ab
Nobuyuki
Tamaoki
*ab
Abstract
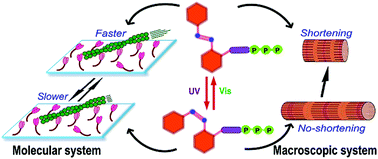
We employed an azobenzene based non-nucleoside triphosphate, AzoTP, in a myosin–actin motile system and demonstrated its efficiency as an energy molecule to drive and photo-regulate the myosin–actin motile function at the macroscopic level along with an in vitro motility assay. The AzoTP in its trans state induced shortening of a glycerinated muscle fibre whilst a cis isomer had no significant effect. Direct photoirradiation of a cis-AzoTP infused muscle fibre induced shortening triggered by a locally photo-generated trans-AzoTP in the muscle fibre. Furthermore, we designed and synthesized three new derivatives of AzoTPs that served as substrates for myosin by driving and photo-regulating the myosin–actin motile function at the molecular as well as the macroscopic level with varied efficiencies.
 Please wait while we load your content...
Please wait while we load your content...

