
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Positive zeta potential of nanodiamonds†
Laia
Ginés
 *a,
Soumen
Mandal
a,
Ashek-I-Ahmed
b,
Chia-Liang
Cheng
b,
Maabur
Sow
c and
Oliver A.
Williams
a
*a,
Soumen
Mandal
a,
Ashek-I-Ahmed
b,
Chia-Liang
Cheng
b,
Maabur
Sow
c and
Oliver A.
Williams
a
aSchool of Physics and Astronomy, Cardiff University, UK. E-mail: GinesL@cardiff.ac.uk
bDepartment of Physics, National Dong Hwa University, Hualien 97401, Taiwan
cEPSRC Centre for Doctoral Training in Diamond Science and Technology, Warwick University, UK
First published on 10th August 2017
Abstract
In this paper, the origin of positive zeta potential exhibited by nanodiamond particles is explained. Positive zeta potentials in nano-structured carbons can be explained by the presence of graphitic planes at the surface, which leave oxygen-free Lewis sites and so promotes the suppression of acidic functional groups. Electron Microscopy and Raman Spectroscopy have been used to show that positive zeta potential of nanodiamond is only exhibited in the presence of sp2 carbon at the surface.
Introduction
When a solid surface comes into contact with an aqueous solution, a surface charge is created at the solid surface. This charge originates from either acid–base reactions between surface functional groups and the aqueous solution, or the adsorption of water ions. For this reason the majority of these interactions result in an overall net negative charge due to the de-protonation of surface oxides, carboxyl/hydroxyls etc. that are prevalent at most solid surfaces. Positive charges are less common and usually due to the protonation of amine surface groups, or in the case of hydrophobic surfaces, the ordering of water ions at the interface.The origin of the positive zeta potential (and thus surface charge) on diamond nanoparticles has been controversial, resulting in multiple models to explain its origin.1–4 However, diamond remains a model system where the surface groups can easily be controlled and are stable across a wide pH range.
Nanodiamond's surface groups strongly depends on their production methods and non-diamond areas (sp2) are commonly present in the surface, particularly in detonation diamond (DND),1,5 but also in bigger size particles. For most applications,6 a homogenized diamond surface is desirable and many studies have been performed to control the sp3/sp2 ratio, attempting to remove all the sp2 carbon for subsequent diamond's surface functionalization.7
On the other hand, the presence of sp2 carbon can lead to the diamond surface's reconstruction. The sp2 carbon in the form of graphitic-like carbon or amorphous carbon is highly reactive and can promote the diamond nanoparticles’ surface functionalization.8
In this work it is shown that the positive zeta potential in diamond cannot be generated without the presence of sp2 bonding. The unusual positive zeta potential origin is shown to be related to the protonation of the basal planes of graphite terminating the diamond nanoparticle surface. This has profound implications for many applications of nanodiamond, especially biological ones where reactive sp2 species are highly undesirable.9–11
Experimental section
Material preparation
Commercial nanodiamond powders sourced by Microdiamant Switzerland (MSY 0-0.05, monocrystalline diamond powder produced by high pressure-high temperature) were used in this study. Six different powders’ treatments were compared as well as untreated diamond powder.An air treatment was performed by heating the diamond particles in a furnace under air atmosphere at 480 °C for 5 hours. For the molecular hydrogen gas treatment, the diamond nanoparticles were annealed in hydrogen gas (hydrogen gas was flowed at 100 sccm) at 500 °C and at 10 mbar for 5 hours.1 For the vacuum annealing treatment, the diamond particles were heated in vacuum (1 × 10−3 mbar) at 1000 °C for 2 hours. The same vacuum annealing treatment was performed to another set of nanodiamond powder, but after the 2 hours of vacuum annealing treatment, the particles were taken outside the vacuum chamber and were placed inside again to perform a molecular hydrogen treatment under the same conditions as explained before. The same treatment was carried out (vacuum annealing followed by hydrogen annealing) but both the vacuum annealing an hydrogen annealing were performed inside the vacuum chamber, in situ, cooling down the samples from 1000 °C to 500 °C after the vacuum annealing. In the last treatment, the diamond particles were first annealed in air (480 °C, for 5 hours) and then the vacuum annealing followed by hydrogen treatment was conducted.
Different aqueous colloids were prepared from the above-mentioned treated powders by dispersing 0.1 g of powder in 200 ml of deionized water. The colloids were dispersed via high power ultrasound (Sonics Vibra-cell VCX 500) at 200 W (3 s on 2 s off duty cycle) for five hours and the colloids temperature was kept below 20 °C. All the solutions were decanted after been settled for 24 hours, to remove any large sonotrode contaminants. The solutions were centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 10 °C in a Sigma 3-30 KS centrifuge.1
000g at 10 °C in a Sigma 3-30 KS centrifuge.1
Material characterization
Dynamic light scattering (DLS) and Nanoparticle tracking analysis (NTA) measurements were performed to confirm the particles’ size distribution. A Malvern Zetasizer Nano ZS equipped with a 633 nm laser in backscattering configuration (173°) and a Malvern Nanosight LM10 equipped with a 635 nm laser were used respectively. Zeta potential measurements and pH titration measurements were made in the Malvern Zetasizer Nano ZS, using the MPT-2 autotitration accessory. 0.1 M HCL and 0.1 M of NaOH were used as acid and base titrants. Particle size distributions are the average of 100 × 30 s scans and zeta potential of 3 × 100 scans. The colloids were de-gassed by nitrogen flow.Reverse titrations were performed by making aqueous colloids of mass/volume concentration 1% and diluting by 1:10 seven times until a constant pH was reached. The pH was measured by a Mettler Toledo. The colloids were de-gassed by nitrogen flow as was the de-ionised water used for dilution.
Raman measurements were recorded in an inVia Renishaw confocal Raman microscope equipped with a 532 nm laser. All the measurements were acquired using the same parameters: 10 seconds acquisition time and 50 accumulations.
To study the surface groups present on nanodiamonds’ surfaces FTIR measurements were performed. For the FTIR measurements all the treated powders were prepared as previously described and were transferred to a UHV chamber.12 FTIR measurements were conducted in situ and chamber base pressure was maintained around 5 × 10−7 Torr. The samples were annealed using electrically controlled heating device under UHV at 550 °C to remove loosely bounded hydrocarbons and the spectra were collected using a Bomem MB154 FTIR with MCT-liquid nitrogen cooled detector. The resolution was set to be 4 cm−1, and 400 scans were used. To avoid IR background signal from the elevated temperatures, annealed samples were cooled down to room temperature for the measurement.
Particle surface morphology and structure were examined by High Resolution Transmission Electron Microscopy (HRTEM).
High resolution TEM images were performed in a FEI Titan.3 The microscope was operated at low acceleration voltage (80 kV) to minimize beam damage and prevent undesired diamond nanoparticles’ surface graphitization or sample contamination. As is well known, the electron beam can induce the formation of a graphitic-like outer shell,13 and can also promote volatile carbon species present in the sample to be re-absorbed into other areas of the sample.14,15
TEM specimens were prepared by dispersing the powders in ethanol via ultra-sonication and the solutions were drop cast onto a perforated-carbon TEM grid.
Results and discussion
Three different samples were imaged in HRTEM to study the particle surface morphology. The first one consists of the untreated diamond powder, shown in Fig. 1a and b. Most of the untreated particles imaged presented a crystalline diamond core, however some of them showed an amorphous outer shell surrounding the diamond core.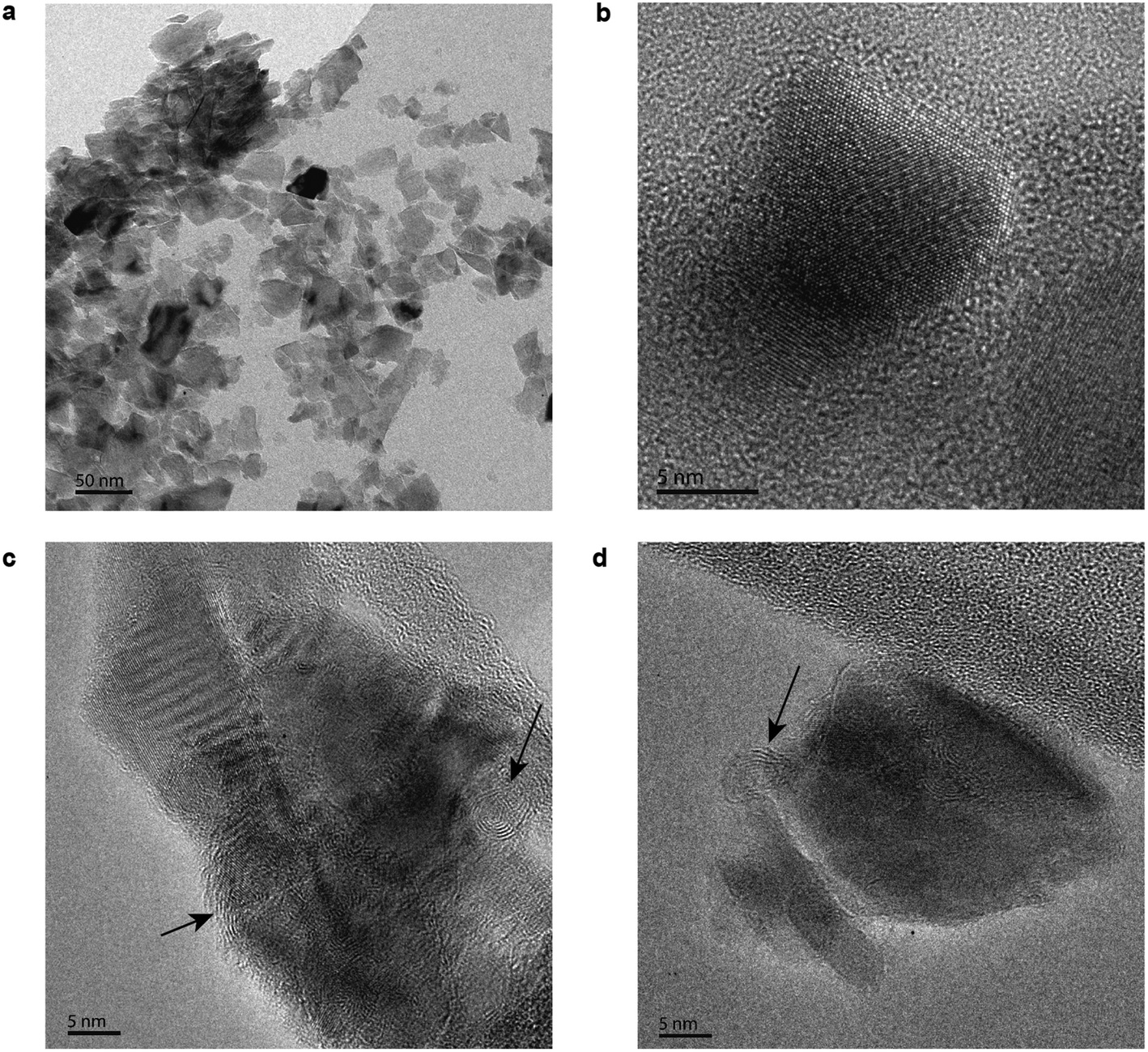 | ||
| Fig. 1 Structural characterization of 50 nm diamond nanoparticles. Unfiltered high resolution transmission electron microscope images. (a) Overview bright field image of the as received 50 nm particles. (b) Detailed HRTEM image of the as received 50 nm particles. (c) HRTEM image of the diamond nanoparticles after the vacuum annealing treatment. The arrows indicate the graphite planes formed. (d) HRTEM image of the hydrogenated particles. | ||
To confirm the influence of the sp2 in the diamond nanoparticles’ positive zeta potential and low temperature hydrogenation, we intentionally created sp2 bonding. We promoted surface graphitization by annealing the diamond nanoparticles in vacuum at 1000 °C.16 In Fig. 1c and d, particles after the vacuum annealing show a well-defined diamond core and an outer shell in which graphitization (marked by arrows) can be clearly observed, with spacing between the graphitic layers of 0.34–0.35 nm.
The surface graphitization can be further studied by Raman measurements. The Raman spectra of the variously treated nanodiamond are represented in Fig. 2a. All spectra show a clear first-order diamond peak at 1332 cm−1, broader and less intense for the powders that have been vacuum annealed. For samples not previously vacuum annealed, Fig. 2a(i)–(iii), a broad band at 1550 cm−1 confirms the presence of sp2 sites. This band, known as G-band, is due to the bond stretching of sp2 atoms in both rings and chains.17,18 Different Raman characteristic peaks are present in the samples that have been previously vacuum annealed, shown in Fig. 2a(iv)–(vi). The diamond peak is still present although it is less intense and broader compared to its G-band than in the previous treated samples. This is because nanodiamond can undergo a phase transition from diamond to graphite under high temperature vacuum annealing processes.19 Three different peaks arise. The first and broad peak appears between 1100 cm−1 and 1150 cm−1, and has been related to sp2 structures known as trans-polyacetylene structures, related to hydrogen presence.20,21 Although molecular hydrogen was not introduced in the chamber while annealing the sample in Fig. 2a(iv), hydrogen was present in the vacuum annealed sample as molecular hydrogen can be desorbed from the vacuum chamber walls.22 The second peak, around 1350 cm−1, corresponds to the well-known D-band, and is due to breathing motion of sp2 rings.18 The third emergent peak is located at 1582 cm−1 and is characteristic of graphitic-like materials, clearly indicating that graphitic structures have been formed after the annealing at 1000 °C.23
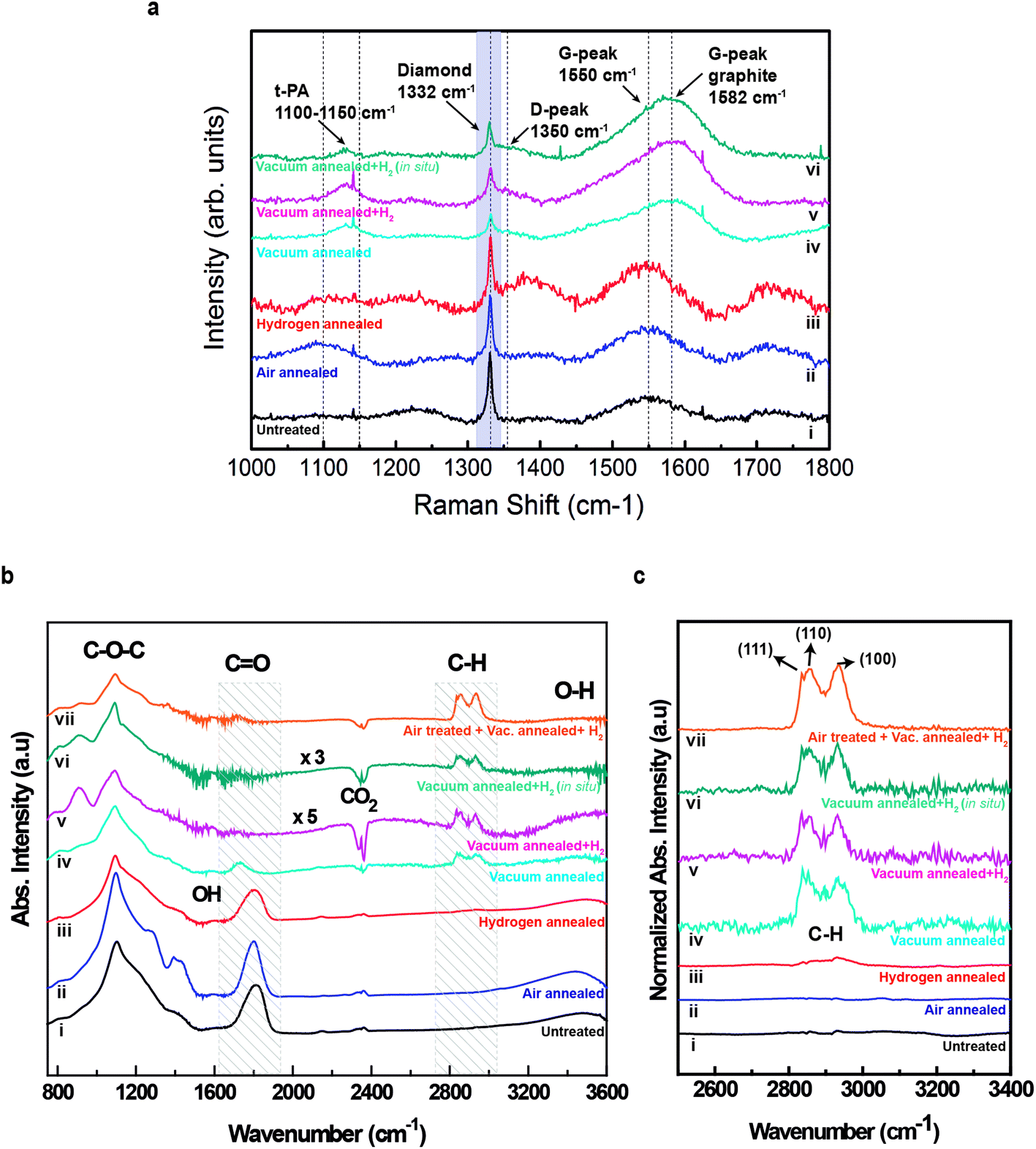 | ||
| Fig. 2 Raman and FTIR spectra of surface treated and untreated nanodiamond particles. (a) Raman spectra of (i) untreated 50 nm nanodiamond particles (ii) air annealed at 480 °C (iii) molecular hydrogen treated at 500 °C (iv) vacuum annealed at 1000 °C (v) sample (iv) treated with molecular hydrogen at 500 °C (vi) sample (iv) treated in situ with molecular hydrogen at 500 °C. (b) FTIR spectra; (i)–(vi) samples are treated in same condition as Raman, (vii) sample (ii) annealed in vacuum at 1000 °C and then treated in situ with molecular hydrogen at 500 °C. (c) Detailed feature of the C–H for the spectrum drawn on b. | ||
Apart from the graphitic structures formed on the nanodiamond's surface, the presence of different functional groups can influence the zeta potential, as well as the acidic or basic character of nanodiamond aqueous solutions. Fourier Transform Infrared Spectroscopy (FTIR) measurements were performed (see Fig. S2 in ESI† for details) to determine these surface complexes, and the spectra of the variously treated nanodiamonds are shown in Fig. 2b and c. It should be noted that these measurements were performed in high vacuum in order to remove any influence of organic contaminants.
As can be observed, there is a significant difference between the nanodiamonds annealed in vacuum at 1000 °C, Fig. 2b(iv)–(vii), and those that were not annealed, Fig. 2b(i)–(iii). For all the samples, the first band region with noticeable peaks is between 1000 cm−1 and 1300 cm−1. This broad band is generally associated with C–O–C stretching vibrations of cyclic ether24 and exits frequently on diamond surface but not in large quantity. The C–O–C peak can be changed through oxygenation processes (air annealing treatments) at different temperatures resulting in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O products on diamond surface with desorption of either CO2 or CO.25 Moreover, two distinguishable broad peaks are observed in the region between 1250 cm−1 and 1500 cm−1. The peak at 1265 cm−1 is attributed to CO bending vibration, and the peak between 1380 cm−1 and 1440 cm−1 corresponds to the asymmetric –CH bending vibration. These two peaks are due to the presence of the ester group (CH3–COOR). The CO intensity is significantly reduced in the samples annealed in vacuum and thus followed by hydrogen treatment represented in Fig. 2b(v)–(vii). This peak's intensity reduction indicates the –CO desorption/substitution by H during ND annealing at elevated temperature.26 A small intensity peak appears at 1620 cm−1. This peak is associated with –OH bending vibration due to the presence of water outside environment or due to the carboxyl group (–COOH) on ND surface. The corresponding –OH stretching mode is a wide band observed between 3200 cm−1–3600 cm−1.
O products on diamond surface with desorption of either CO2 or CO.25 Moreover, two distinguishable broad peaks are observed in the region between 1250 cm−1 and 1500 cm−1. The peak at 1265 cm−1 is attributed to CO bending vibration, and the peak between 1380 cm−1 and 1440 cm−1 corresponds to the asymmetric –CH bending vibration. These two peaks are due to the presence of the ester group (CH3–COOR). The CO intensity is significantly reduced in the samples annealed in vacuum and thus followed by hydrogen treatment represented in Fig. 2b(v)–(vii). This peak's intensity reduction indicates the –CO desorption/substitution by H during ND annealing at elevated temperature.26 A small intensity peak appears at 1620 cm−1. This peak is associated with –OH bending vibration due to the presence of water outside environment or due to the carboxyl group (–COOH) on ND surface. The corresponding –OH stretching mode is a wide band observed between 3200 cm−1–3600 cm−1.
There is a remarkable feature difference between the samples in which a vacuum annealing treatment and hydrogen annealing treatment were performed (samples v to vii) and the samples without a vacuum annealed treatment (samples i to iii). This is observable in the band range 1600 cm−1 to 1800 cm−1. The broad peak centred at 1778 cm−1 corresponds to CO stretching modes, and demonstrates the presence of carboxyl groups. This peak intensity is dramatically reduced in the samples after the vacuum annealing/hydrogenation process.26,27
Another meaningful change is observed on the band ranging from ∼2800 cm−1–3000 cm−1 shown in detail in Fig. 2c. The hydrogenation of diamond nanoparticles surfaces is confirmed with the presence of the C–H stretching modes. Two clear peaks at 2860 cm−1 and at 2934 cm−1 are observed in the samples that have been previously vacuum annealed, Fig. 2c(iv)–(vii). In detonation diamond, these peaks appear at 2870 cm−1 and 2940 cm−1.28 The peak at 2860 cm−1 corresponds to C(110):H mixed with C(111):H, and the peak at 2934 cm−1 for C(100):H.12 Beside these peaks, a clear sharp peak of C(111):H, plane adjacent with C(110):H, is also present at 2835 cm−1 and it is relevant for large size ND. The mechanism responsible for nanodiamond hydrogenation is the catalytic reaction between carbon radicals and hydrogen molecules.12 C2 and C3 carbon radicals are available after desorption of intentionally created sp2 carbon through vacuum annealing at 1000 °C.
This result indicates the effect of sp2 carbon on hydrogen dissociation as –CH peaks are not formed on the sample shown in Fig. 2c(iii). It is noted that for the sample in Fig. 2c(iv) annealed at 1000 °C in vacuum without the external source of hydrogen in chamber, –CH is formed through the interaction of hydrogen desorbed from chamber wall at elevated temperature (>500 °C).
To investigate the origin of the positive/negative zeta potential, we prepared colloids out of the treated diamond powders as detailed in the Experimental section.
Fig. 3a shows the zeta potential versus pH for the different colloids. The solid–liquid interface behaviour is strongly dependant on the surface treatment, due to the presence of different functional groups on the diamond nanodiamond's surface.2,29 Negative zeta potential values are well understood. For example, in untreated powders, most of the diamond surface is covered by oxygen containing groups, such as carboxyl, carbonyl, alcohol and ethers, generated during nanodiamond production and acid purification. The negative zeta potential can be explained due to the dissociation of these carboxyl (COOH) and carbonyl (CO) groups in the diamond particles’ surface.8
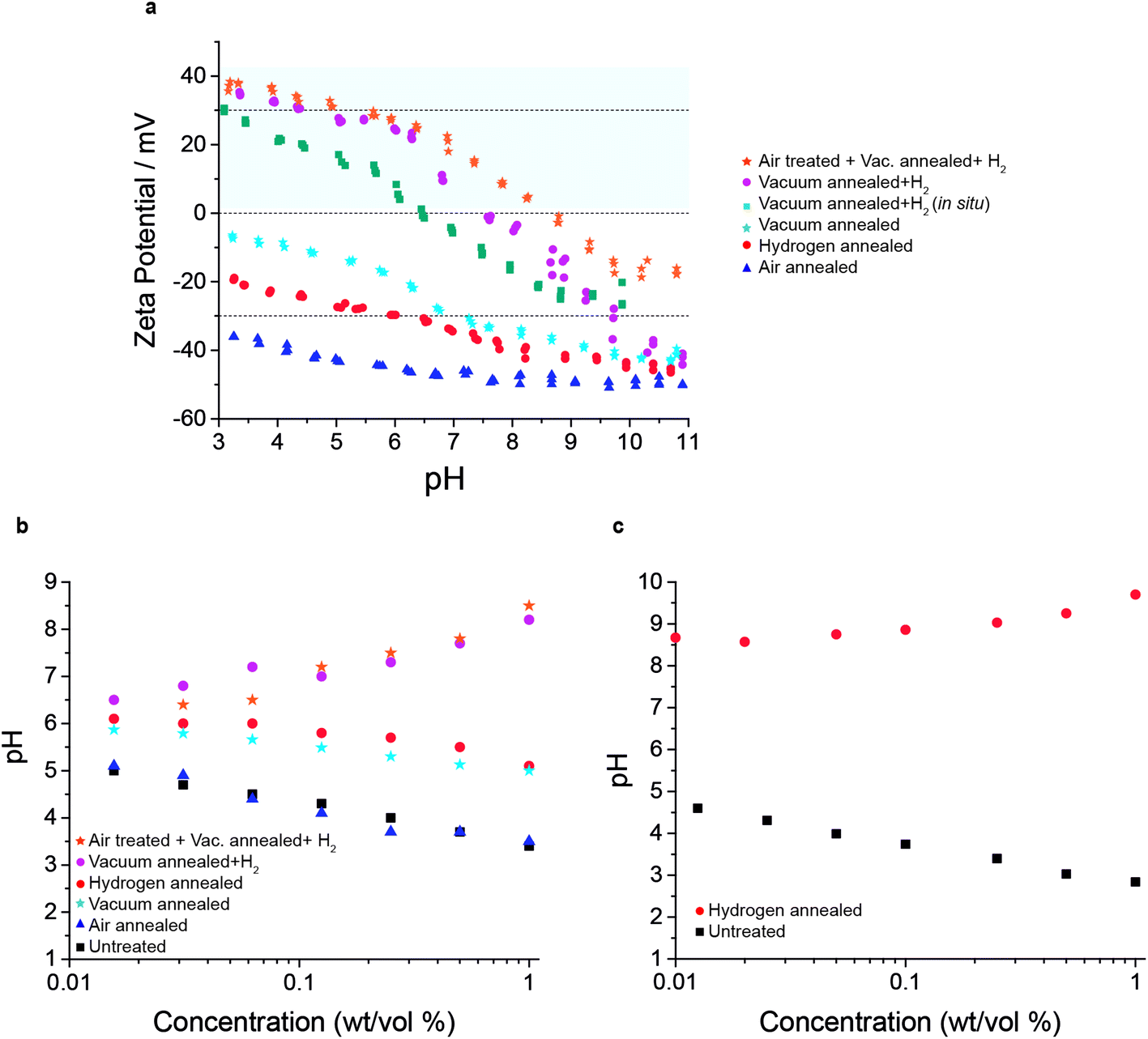 | ||
| Fig. 3 Zeta potential and back titration measurements of the colloids prepared from the annealed powders. (a) Zeta potential measurements over pH ranging from 3 to 11. (b) Back titration measurements of the 50 nm nanodiamond particles. (c) Back titration measurements of detonation nanodiamond (5 nm particle size). | ||
In this work, negative zeta potential values were also measured in the colloids prepared from powders previously annealed in air, in molecular hydrogen, and after a vacuum annealing at 1000 °C. The colloid obtained from air treated powder shows the most negative zeta potential, with values ranging from −35 mV at pH 3 to −55 mV at pH 11, being stable in the whole range of pH (colloids stability is defined as absolute zeta potential values greater than 30 mV).
Air annealing treatments have been thoroughly used not only to remove the non-diamond content in the particles’ surface, but also to oxidize diamond particles’ surfaces.7,8 This treatment allows the creation of the aforementioned oxygen containing groups. The dissociation of these COOH and CO groups present on the nanodiamond's surface lead to negative zeta potential values, which become less negative at low pH due to a higher hydrogen ion concentration at acidic pH values.
A similar trend is observed for the hydrogen annealing treatment (with no previous vacuum annealing). As confirmed in the FTIR spectra, oxygen groups (CO mainly) are still present after the hydrogen treatments, resulting in negative zeta potential values. Nevertheless, it has to be taken into account that previous works on detonation nanodiamond (5 nm particle size) show a positive zeta potential after the same molecular hydrogen treatment, but was proved to be ineffective for larger particles.1,30 This is due to the large quantity of non-diamond content (sp2) present on the detonation nanodiamond surface which cannot be completely removed, and was explained in terms of the basicity of carbon surfaces.31
Back titrations of the various surface treatments are shown in Fig. 3b. It is clear that the air annealed sample shows the strongest acidity vs. concentration, which is easily explained by its low concentration of sp2 and high concentration of COOH groups. These COOH groups become de-protonated making the solution more acidic with increasing concentration of particles as well as generating a negative zeta potential as seen in Fig. 3a. The concentration of these COOH groups is somewhat depleted by vacuum annealing or annealing in molecular hydrogen as evidenced by their reduced acidity vs. particle concentration and reduced zeta potential magnitude vs. pH. However, upon vacuum annealing followed by hydrogen annealing the samples exhibit clear basicity, increasing with concentration. This basicity is even more pronounced with the detonation nanodiamond (5 nm) particles.
The origin of the basicity of nanodiamond surfaces and the associated positive zeta potential has been controversial. Several models have been proposed to explain it but generally assume a pristine (sp2 free) diamond surface.4,32 This is directly contradicted by Streaming potential measurements performed by Härtl et al. that demonstrate the zeta potential of both hydrogen and oxygen terminated bulk diamond is negative over the whole pH range.33 These previous nanodiamond works do not show near-infrared Raman analysis to confirm the phase purity of the surface, and use hydrogen plasmas to terminate the surface which can easily graphitise nanodiamond particles due to poor thermal contact with substrate.
In this work it has been shown that the positive zeta potential and basicity is only exhibited with finite concentrations of sp2 at the surface. Boehm et al. correlate the basicity in carbon black and carbon-based materials with the absence of oxygen containing groups, driven off by hydrogen.34 Oxygen-containing surface groups structures, such as pyrones, have been suggested as the origin of carbon's basic properties.31,35,36 The strength37 or weakness35,38 of pyrone-type structures as basic sites is controversial, but a relatively high amount of these structures are necessary to explain carbon basicity, which are not seen in FTIR. The positive zeta potential can also be associated with the protonation of amino groups in acidic environments.39 But amino groups are also not present on these nanodiamonds. Petit et al.32 attribute positive zeta potentials in graphitised detonation nanodiamond (5 nm particle size) to O2 molecule adsorption on basal planes, but this would not explain the basicity which is simultaneously observed. In the case of this work, all titrations were performed with deoxygenated water.
One of the most generally accepted explanation regarding the carbon basicity comes from the electron–donor–acceptor (EDA) complex. In this complex, π electron-rich regions located in the basal planes, can interact with oxonium (H3O+) ions.40,41 In other words, oxygen-free carbon sites can absorb H3O+ ions to provide a diamond's surface with positive charge and so basic properties.
Another possibility is related to oxygen-free Lewis base sites on the basal planes within the graphene layers.42 Both models are represented in Fig. S4 (see ESI†). After annealing the powders in vacuum at high temperature (1000 °C), graphite is created forming an outer-shell of the diamond core particles, and leaving oxygen-free Lewis base sites. As the surface graphite is highly reactive, it can reabsorb oxygen on exposure to air, and hence exhibit a negative zeta potential. However, if samples after the vacuum annealing are terminated with hydrogen, active sites are no longer present for the oxygen absorption.42 This leaves particles with little or no COOH on the surface with graphitic planes exposed to the aqueous solution. These planes can become protonated by H+ from the solution, which would simultaneously generate a positive zeta potential whilst rendering the solution basic.42 This would only work with samples with little oxygen and some sp2 on the surface, as evidenced by the vacuum annealed/hydrogenated samples.
This is further evidenced by the enhanced basicity seen on detonation nanodiamond particles which have a much higher concentration of sp2 at the surface, as seen in Fig. 3c. Far stronger basicity is seen, which correlates with the higher concentration of basal planes for protons to diffuse into. These detonation nanodiamond particles also exhibit a strong positive zeta potential as previously reported.1
Conclusions
In summary, the positive zeta potential and associated basicity is only exhibited on nanodiamond with non-zero concentrations of sp2 bonding, specifically graphene like shells. These shells can become protonated, generating a net positive charge and zeta potential. At the same time the extraction of protons from the water results in pronounced basicity. This is significant as many applications in areas such as biology cannot tolerate sp2 bonding at the surface and thus mechanism for generation of the positive zeta potential such as surface derivatisation is required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Cardiff University authors would like to thank the Royal Society International Exchanges Scheme (IE131713) and EU FP7 FET Open “Wavelength tunable Advanced Single Photon Sources”. CLCheng appreciates the financial support of this research by the Ministry of Science and Technology (MOST) of Taiwan for research grant: MOST-103-2112-M-259-001-MY3 and Taiwan-UK exchange program under Grant No. MOST-103-2911-I-259-501. M. Sow acknowledges the EPSRC Centre for Doctoral Training in Diamond Science and Technology (EP/L015315/1). The TEM experiments have been conducted in the Laboratorio de Microscopias Avanzadas (LMA) at Instituto de Nanociencia de Aragon (INA) of the Universidad de Zaragoza, Spain. Authors acknowledge the LMA-INA for offering access to their instruments and expertise. L. G. acknowledges Dr César Magén from LMA-INA for his help with the TEM experiments.References
- O. A. Williams, J. Hees, C. Dieker, W. Jäger, L. Kirste and C. E. Nebel, ACS Nano, 2010, 4, 4824–4830 CrossRef CAS PubMed.
- N. Gibson, O. Shenderova, T. J. M. Luo, S. Moseenkov, V. Bondar, A. Puzyr, K. Purtov, Z. Fitzgerald and D. W. Brenner, Diamond Relat. Mater., 2009, 18, 620–626 CrossRef CAS.
- H. A. Girard, T. Petit, S. Perruchas, T. Gacoin, C. Gesset, J. C. Arnault and P. Bergonzo, Phys. Chem. Chem. Phys., 2011, 13, 11517–11523 RSC.
- V. Chakrapani, J. C. Angus, A. B. Anderson, S. D. Wolter, B. R. Stoner and G. U. Sumanasekera, Science, 2007, 318, 1424–1430 CrossRef CAS PubMed.
- A. Krueger, J. Mater. Chem., 2008, 18, 1485 RSC.
- B. R. Smith, D. Gruber and T. Plakhotnik, Diamond Relat. Mater., 2010, 19, 314–318 CrossRef CAS.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, Synthesis, 2006, 11635–11642 CAS.
- A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890–906 CrossRef CAS.
- E. Koike and T. Kobayashi, Chemosphere, 2006, 65, 946–951 CrossRef CAS PubMed.
- P. G. Barlow, A. Clouter-Baker, K. Donaldson, J. Maccallum and V. Stone, Part. Fibre Toxicol., 2005, 2, 11 CrossRef PubMed.
- D. M. Brown, V. Stone, P. Findlay, W. MacNee and K. Donaldson, Occup. Environ. Med., 2000, 57, 685–691 CrossRef CAS PubMed.
- A. Ahmed, S. Mandal, L. Gines, O. A. Williams and C. Cheng, Carbon, 2016, 110, 438–442 CrossRef CAS.
- M. Ozawa, H. Goto, M. Kusunoki and E. Osawa, J. Phys. Chem. B, 2002, 106, 7135–7138 CrossRef CAS.
- S. Turner, O. I. Lebedev, O. Shenderova, I. I. Vlasov, J. Verbeeck and G. Van Tendeloo, Adv. Funct. Mater., 2009, 19, 2116–2124 CrossRef CAS.
- K. Iakoubovskii, K. Mitsuishi and K. Furuya, Nanotechnology, 2008, 19, 155705 CrossRef CAS PubMed.
- I. P. Chang, K. C. Hwang, J. A. A. Ho, C. C. Lin, R. J. R. Hwu and J. C. Horng, Langmuir, 2010, 26, 3685–3689 CrossRef CAS PubMed.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2001, 64, 75414 CrossRef.
- J. Chen, S. Z. Deng, J. Chen, Z. X. Yu and N. S. Xu, Appl. Phys. Lett., 1999, 74, 3651–6353 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2001, 63, 121405 CrossRef.
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc., A, 2004, 362, 2477–2512 CrossRef CAS PubMed.
- Y. Hirohata, J. Vac. Sci. Technol., A, 1993, 11, 2637 CAS.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc., A, 2004, 362, 2271–2288 CrossRef CAS PubMed.
- T. Jiang and S. Jib, J. Chem. Soc., Faraday Trans., 1996, 92, 3401–3406 RSC.
- T. Ando, K. Yamamoto, M. Ishii, M. Kamo and Y. Sato, J. Chem. Soc., Faraday Trans., 1993, 89, 3635 RSC.
- T. Ando, M. Ishii, M. Kamo and Y. Sato, J. Chem. Soc., Faraday Trans., 1993, 89, 1783–1789 RSC.
- A. Dandekar, R. T. K. Baker and M. A. Vannice, Carbon, 1998, 36, 1821–1831 CrossRef CAS.
- C. L. Cheng, C. F. Chen, W. C. Shaio, D. S. Tsai and K. H. Chen, Diamond Relat. Mater., 2005, 14, 1455–1462 CrossRef CAS.
- M. Ozawa, M. Inaguma, M. Takahashi, F. Kataoka, A. Krüger and E. Osawa, Adv. Mater., 2007, 19, 1201–1206 CrossRef CAS.
- J. Hees, A. Kriele and O. A. Williams, Chem. Phys. Lett., 2011, 509, 12–15 CrossRef CAS.
- M. A. Montes-Moran, D. Suarez, J. A. Menendez and E. Fuente, Carbon, 2004, 42, 1219–1224 CrossRef CAS.
- T. Petit, H. A. Girard, A. Trouvé, I. Batonneau-Gener, P. Bergonzo and J.-C. Arnault, Nanoscale, 2013, 5, 8958–8962 RSC.
- A. Härtl, J. A. Garrido, S. Nowy, R. Zimmermann, C. Werner, D. Horinek, R. Netz and M. Stutzmann, J. Am. Chem. Soc., 2007, 129, 1287–1292 CrossRef PubMed.
- H. P. Boehm, Carbon, 1994, 32, 759–769 CrossRef CAS.
- H. P. Boehm, Carbon, 2002, 40, 145–149 CrossRef CAS.
- H. B. Man, B. Saha, D. Ho and G. C. Schatz, J. Phys. Chem. C, 2013, 117, 17256–17267 Search PubMed.
- D. Suarez, J. A. Menendez, E. Fuente and M. A. Montes-Moran, Langmuir, 1999, 15, 3897–3904 CrossRef CAS.
- A. Contescu, M. Vass, C. Contescu, K. Putyera and J. A. Schwarz, Carbon, 1998, 36, 247–258 CrossRef CAS.
- X. Xu, Z. Yu, Y. Zhu and B. Wang, Diamond Relat. Mater., 2005, 14, 206–212 CrossRef CAS.
- C. A. Leon y Leon, J. M. Solar, V. Calemma and L. R. Radovic, Carbon, 1992, 30, 797–811 CrossRef CAS.
- M. A. Montes-Moran, J. A. Menendez, E. Fuente and D. Suarez, J. Phys. Chem., 1998, 102, 5595–5601 CrossRef CAS.
- J. A. Menéndez, J. Phillips, B. Xia and L. R. Radovic, Langmuir, 1996, 12, 4404–4410 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: DLS, NTA and FTIR data. See DOI: 10.1039/c7nr03200e |
| This journal is © The Royal Society of Chemistry 2017 |