
An ionic liquid promoted approach to bitriazolyl compounds as succinate–ubiquinone oxidoreductase inhibitors
Rui
Zhang
,
Qiong-You
Wu
*,
Jun
Tao
,
Jin-Huan
Pan
and
Guang-Fu
Yang
*
Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry Central China Normal University, Wuhan 430079, P. R. China. E-mail: gfyang@mail.ccnu.edu.cn; qywu@mail.ccnu.edu.cn; Fax: +86-27-67867141; Tel: +86-27-67867800 Tel: +86-27-67867706
First published on 21st November 2016
Abstract
The respiratory chain succinate–ubiquinone oxidoreductase (SQR or complex II) is a promising target for fungicide discovery. As a continuation of our research work on the development of new fungicides, a series of bitriazolyl compounds were designed and synthesized in excellent yields by an ionic liquid promoted 1,3-dipolar Huisgen cycloaddition reaction of azides and akynes. These newly synthesized compounds were characterized by 1H NMR, 13C NMR and HR-MS spectroscopy. The in vitro assay indicated that several compounds displayed good inhibitory effects against porcine succinate–cyctochrome reductase (SCR) with IC50 values ranging from 2.89 to 61.19 μM. Compound 1b with an IC50 value of 2.89 μM, comparable to the commercial control penthiopyrad, was identified as the most promising inhibitor. Further evaluation of the representative compounds against respective SQR and cyt bc1 indicated that their inhibitory potency against SQR was much higher than that against cyt bc1, suggesting that SQR might be a potential target of these inhibitors. Furthermore, molecular docking studies suggested that strong hydrogen bonding and π–π stacking interactions might be responsible for a higher SQR inhibitory effect of compound 1b as compared to that of compounds 1d and 2b. Consequently, bitriazolyl compounds, a totally new skeleton that is distinct from the existing commercial SQR-inhibiting fungicides, were discovered, which could potentially be a new lead for further development of SQR inhibitors.
1. Introduction
The succinate–ubiquinone oxidoreductase (SQR, complex II) complex is one of the five consecutive mitochondria respiratory chain super-complex components, which catalyzes the oxidation of succinate into fumarate and the reduction of ubiquinone into ubiquionol.1–7 It is also the only membrane protein that is involved in the electron transfer of the mitochondria respiratory chain as well as the tricarboxylic acid cycle. Over the past several decades, the SQR complex has drawn attention amongst pesticide researchers, due to its unique biochemical characteristics and biomedical significance.8–13 In line with this, agricultural fungicides targeting SQR have achieved great success since the introduction of the first commercial SQR fungicide, carboxin, in 1966.14 So far, approximately 18 commercial SQR fungicides have been developed to prevent a broad range of fungal plant diseases.15 Structurally, all of these commercial fungicides belong to the class of carboxamide compounds, in which two aromatic components are connected by the carboxamide bond. Consequently, cross-resistance may be a potential risk of these fungicides since they share a highly similar structural moiety. Hence, identification of novel lead structures addressing the crop protection industry's needs is in high demand.The triazole unit and its analogs are considerably important in medicinal and agrochemical research because of their unique structure and properties. A notable example is ribavirine,16 a synthetic 1,2,4-triazole containing nucleoside mimic showing broad-spectrum activity against many DNA and RNA viruses (Scheme 1). Another representative example is the first-generation triazole antifungal medication fluconazole,17 which bears two 1,2,4-triazole residues and has been used to treat a variety of fungal infections, especially Candida infections. Additionally, 1,2,3-triazoles were identified as bioactive molecules, and earned a prominent place in medicinal chemistry because of their therapeutic properties, such as anticancer,18 antimicrobial,19 antioxidant,20 anti-HIV,21 and anti-inflammatory activities.22 The integration of the 1,2,4-triazole unit and the 1,2,3-triazole moiety into one backbone has been demonstrated to be an efficient method to construct new compounds with a broad spectrum and biological significance.23–25 In our ongoing project, which focuses on the discovery of novel compounds to combat agricultural fungal diseases,26–28 we are interested in developing bitriazolyl compounds. Using ionic liquid promoted Huisgen 1,3-dipolar cycloaddition, we developed an efficient procedure to synthesize bitriazolyl compounds starting with azido-triazole and alkynes. In a screening of succinate–ubiquinone oxidoreductase inhibitors, we discovered that several compounds show a promising SQR inhibitory potency. This suggested that bitriazolyl compounds, a totally new skeleton that is different from the existing SQR inhibitors, might have considerable potential to control fungal diseases in the field of agrochemical research. Herein, we report on the synthesis and characterization of these bitriazolyl compounds, as well as on the evaluation of their SQR inhibitory activities. Furthermore, molecular docking studies are also included to provide an insight into the molecular basis of the inhibition by various inhibitors.
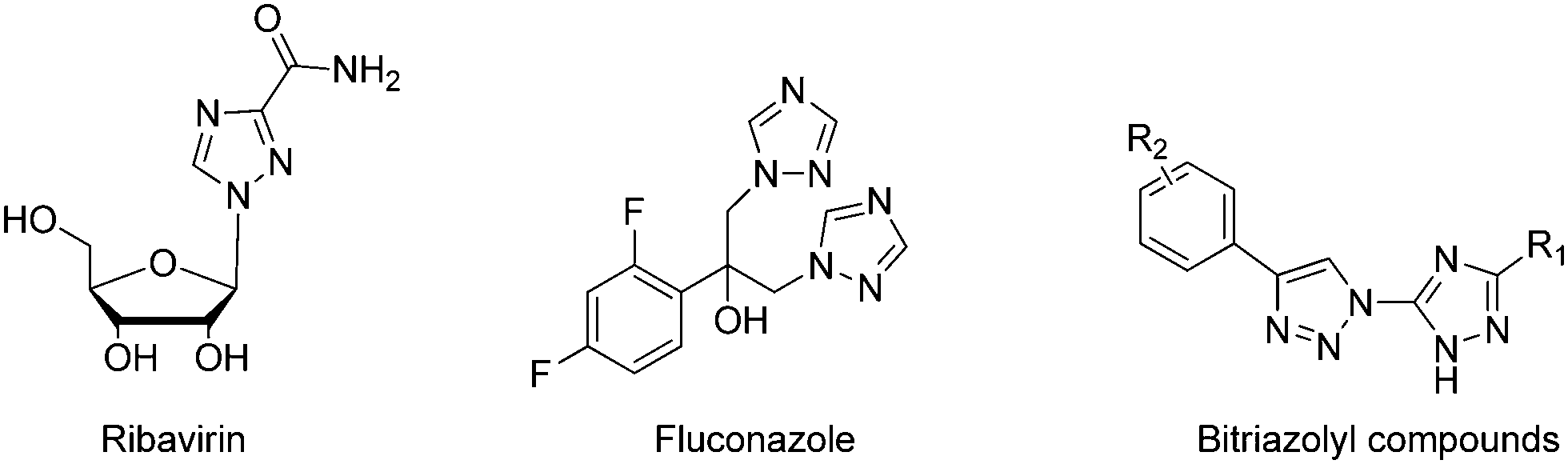 | ||
| Scheme 1 The structures of ribavirin, fluconazole and designed bitriazolyl compounds. | ||
2. Experimental
2.1 General techniques
All chemical reagents were commercially available and treated with standard methods before use. Solvents were dried and redistilled before use. Chromatography employed 200–300 mesh silica gel from Qingdao Makall Group Co., Ltd, Qingdao, China. 5-Azido-1,2,4-triazole was prepared using a literature reported method.291H NMR spectra were recorded on a VARIAN Mercury-Plus 600 or 400 spectrometer in CDCl3 or DMSO-d6 with TMS as the internal reference, 13C NMR spectra were recorded in CDCl3 or DMSO-d6 on a VARIAN Mercury-Plus 600 (150 MHz) or a 400 (100 MHz) spectrometer, and chemical shifts (δ) are given in ppm relative to the center line of a triplet at 77.0 ppm of CDCl3. The following abbreviations are used to designate multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad. HR-MS was conducted on an Agilent 6520 Accurate-Mass Q-TOF instrument or a Bruker Daltonics microTOF-QII instrument.2.2 General procedure for the preparation of the designed compounds
Azides 3 (0.6 mmol) and alkynes 4 (0.72 mmol) were dissolved in an ionic liquid (1 mL). Sodium ascorbate (0.09 mmol, freshly prepared solution in water) was added, followed by CuSO4·5H2O (0.09 mmol, freshly prepared solution in water). The yellowish mixture was stirred at 90 °C. The progress of the reaction was monitored by TLC, and when it was complete, the reactant was extracted with ether (3 × 10 mL) and the combined solvents were evaporated to give the product.2.3 Enzyme assay
The succinate–cytochrome c reductase (SCR, mixture of respiratory complex II and bc1 complex) from porcine heart was prepared as reported previously.30 The activity of SCR was measured by monitoring the increase of cytochrome c at 550 nm, by using an extinction coefficient of 18.5 mM−1 cm−1. The succinate–ubiquinone reductase (complex II) activity was measured by monitoring the decrease of 2,6-dichlorophenolindophenol (DCIP) at 600 nm, by using an extinction coefficient of 21 mM−1 cm−1. The reaction mixture was scaled down to 1.8 mL with final concentrations of PBS (pH 7.4), 100 mM; EDTA, 0.3 mM; succinate, 20 mM; oxidized cytochrome c, 60 μM (or DCIP, 53 μM); and appropriate amounts of enzyme to start the reaction.312.4 Computational simulations
The software of SYBYL 2.0 was used to construct the three-dimensional chemical structures of compounds ZR766, ZR768 and ZR783. Then, the optimized geometries were used as the starting structures for the following docking study. The structures of complex II (1ZOY) and the ligand were prepared with AutoDock Tools4.2 and docking calculations were performed with the program of AutoDock4.2. A total of 256 runs were performed for each molecule. Most of the parameters for the docking calculation were set to the default values recommended by the program.3. Results and discussion
3.1 Chemistry
The most popular method for the construction of a 1,2,3-triazole framework is the 1,3-dipolar Huisgen cycloaddition reaction of azides and akynes. However, the early Huisgen cycloaddition reaction requires a strong electron-withdrawing substituent on the alkynes or a strong electron-donating substituent on the azides.32 Additionally the reaction is often conducted at a high temperature for a long period, and usually leads to a mixture of 1,4-disubstituted and 1,5-disubstituted-1,2,3-triazoles.33 Therefore, considerable efforts have been made by several research groups to develop a convenient, regioselective controllable approach to construct triazoles. Noteworthy achievements are the copper and ruthenium catalyzed azide alkyne cycloaddition reaction developed by Sharpless and Meldal allowing the regioselective formation of 1,4- and 1,5-regioisomer, respectively.34–36 Thereafter, a number of methodologies were investigated to improve the reaction conditions including metal-free methods,37 ultrasound promoted reactions,38 microwave assisted click-chemistry cycloaddition39 and ionic liquid accelerated reactions.40 The use of an ionic liquid in the copper catalyzed azido alkyne cycloaddition appeared to be a reasonable solution to overcome a few obstacles, for instance, instability or tendency to oxidation of the metal.41,42 Furthermore it also accelerated some less active precursor involved reaction affording the product in a simplified and easy isolation manner. Thus, we envisioned to synthesize the designed bitriazolyl compounds via a Huisgen reaction using an ionic liquid to facilitate the transformation of the less active azido-triazole precursor.As a starting point for the development of our methodology, we first examined the template reaction of 5-azido-3-methylsulfanyl-1H-[1,2,4]triazole and phenylacetylene (1.2 equiv.) under various reaction conditions in an ionic liquid (1 mL). As shown in Table 1, no reaction occurred without any catalyst at room temperature, and only trace amounts of the product were detected when the reaction was heated up to a temperature of 90 °C for a long time (Table 1, entry 1 and 2). The starting materials were recovered in the yield of up to 90% in these cases. The poor reactivity might be attributed to the highly electro-deficient feature of the triazole ring, which did not favor the Huisgen reaction. Previously it was demonstrated that Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition of azides and alkynes afforded superior regioselectivity and high transformation under mild conditions. Therefore, the Cu(I)-catalyst was introduced to the reaction, and positive results were observed. However, CuCN and CuCl only gave the yield of 29% and 50%, respectively. In addition, two isomers, 1,4-disubstituted triazole and 1,5-disubstituted triazole, were detected concomitantly. Fortunately, when CuBr was chosen as a catalyst, 1,4-disubstitued-1,2,3-triazole was obtained regioselectively with a yield of 65%. Encouraged by this positive result, we tested some other Cu(I) sources. CuI could yield comparatively to the same level as CuBr in a relatively short reaction time. Furthermore, some Cu(II) salts, such as CuSO4·5H2O, Cu(OAc)2 in the presence of reducing reagents such as ascorbic acid or sodium ascorbate, were also evaluated for the catalytic efficiency. We noticed that the yield was improved dramatically by means of the Cu(I)-catalysts generated in situ. Consequently 1,4-disubstituted-1,2,3-triazoel was obtained as the sole product, no 1,5-disubstituted triazole isomer was observed. The products were easily separated by several rounds of extractions, with no need for further chromatographic purification. In comparison, when the reactions were conducted under common Huisgen cycloaddition reaction conditions as described in the literature, the results are far from satisfactory. For instance, we examined the cycloaddition reaction using toluene as a solvent, the target product was isolated only in a 26% yield even after prolonging the reaction time to 24 h under the optimized catalytic system (entry 17). The poor yield can be ascribed to the limitation of the solubility of CuSO4·5H2O in toluene which retards the in situ generation of the Cu(I) species. When the reaction was performed in a mixture of CH3CN/H2O or dioxane/H2O, it was found that the starting material could not be consumed even after 24 h (entry 18 and 19). A moderate yield was achieved when the reaction medium was changed to a mixed solvent THF/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (entry 16). The special effect of this co-solvent system on the reaction might be attributable to the enhanced hydrophobic interactions occurring between the substrates during the activation process, which favors the reaction.43,44 Apparently, the results obtained in the ionic liquid are far superior to that in the organic solvent. Furthermore, other ionic liquids with 1,3-diakylimidazolium cations and PF6, NTf2 or Br anions were also investigated. These ionic liquids led to a relatively low yield of the product regardless of the Cu(I) catalyst sources being the CuBr/CuI or Cu(I)-catalyst generated in situ by reduction of the Cu(II) salts like CuSO4·5H2O and Cu(OAc)2 with sodium ascorbate. The physicochemical properties of ionic liquids affect the ability of the anion and cation to interact with dissolved species, including catalysts.45 The strong hydrophobic ionic liquids [bmim][PF6] and [bmim][NTf2] are better than hydrophilic ionic liquid [bmim][Br] in terms of the yield. The ionic liquid [bmim][BF4], whose hydrophobicity positioned between the above mentioned ionic liquids [bmim][NTf2] and [bmim][Br], provided the best result in these experiments. Therefore, the optimum reaction conditions were identified as azido-triazole (1.0 mmol), alkynes (1.2 mmol) with CuSO4·5H2O/VcNa as a catalyst (0.15 mmol) in an ionic liquid ([bmim][BF4], 1 mL) at 90 °C. Under the optimized reaction conditions, a number of substrates were investigated (Table 2). All the reactions resulted in the formation of 1,4-disubstituted-1,2,3-triazoles in excellent yields at 90 °C within 1.5 h. No significant steric or electronic effects on the outcome of the reaction were observed, and the crude products required no purification.
:2 (entry 16). The special effect of this co-solvent system on the reaction might be attributable to the enhanced hydrophobic interactions occurring between the substrates during the activation process, which favors the reaction.43,44 Apparently, the results obtained in the ionic liquid are far superior to that in the organic solvent. Furthermore, other ionic liquids with 1,3-diakylimidazolium cations and PF6, NTf2 or Br anions were also investigated. These ionic liquids led to a relatively low yield of the product regardless of the Cu(I) catalyst sources being the CuBr/CuI or Cu(I)-catalyst generated in situ by reduction of the Cu(II) salts like CuSO4·5H2O and Cu(OAc)2 with sodium ascorbate. The physicochemical properties of ionic liquids affect the ability of the anion and cation to interact with dissolved species, including catalysts.45 The strong hydrophobic ionic liquids [bmim][PF6] and [bmim][NTf2] are better than hydrophilic ionic liquid [bmim][Br] in terms of the yield. The ionic liquid [bmim][BF4], whose hydrophobicity positioned between the above mentioned ionic liquids [bmim][NTf2] and [bmim][Br], provided the best result in these experiments. Therefore, the optimum reaction conditions were identified as azido-triazole (1.0 mmol), alkynes (1.2 mmol) with CuSO4·5H2O/VcNa as a catalyst (0.15 mmol) in an ionic liquid ([bmim][BF4], 1 mL) at 90 °C. Under the optimized reaction conditions, a number of substrates were investigated (Table 2). All the reactions resulted in the formation of 1,4-disubstituted-1,2,3-triazoles in excellent yields at 90 °C within 1.5 h. No significant steric or electronic effects on the outcome of the reaction were observed, and the crude products required no purification.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ionic liquid/solvent | Cata. | T (°C) | Time (h) | Yield (%) |
| a bmim: 1-butyl-3-methylimidazolium. b Starting material can not be consumed. | |||||
| 1 | [bmim][BF4]a | — | 20 | 72 | — |
| 2 | [bmim][BF4] | — | 90 | 72 | 5 |
| 3 | [bmim][BF4] | CuCN | 90 | 48 | 29 |
| 4 | [bmim][BF4] | CuCl | 90 | 24 | 50 |
| 5 | [bmim][BF4] | CuBr | 90 | 7 | 65 |
| 6 | [bmim][BF4] | CuI | 90 | 2 | 60 |
| 7 | [bmim][BF4] | Cu(OAc)2/VCNa | 90 | 1.2 | 85 |
| 8 | [bmim][BF4] | CuSO4·5H2O/VCNa | 90 | 1.5 | 95 |
| 9 | [bmim][BF4] | Cu(OAc)2/VC | 90 | 2 | 89 |
| 10 | [bmim][PF6] | CuSO4·5H2O/VCNa | 90 | 6 | 83 |
| 11 | [bmim][PF6] | CuBr | 90 | 24 | 45 |
| 12 | [bmim][PF6] | CuI | 90 | 24 | 56 |
| 13 | [bmim][PF6] | CuSO4·5H2O/VC | 90 | 6 | 73 |
| 14 | [bmim][NTf2] | CuSO4·5H2O/VCNa | 90 | 1 | 60 |
| 15 | [bmim]Br | CuSO4·5H2O/VCNa | 90 | 3 | 20 |
| 16 | THF/H2O (1:2) |
CuSO4·5H2O/VCNa | 90 | 2.5 | 81 |
| 17 | Toluene | CuSO4·5H2O/VCNa | 90 | 24 | 26 |
| 18 | CH3CN/H2O (1:2) |
CuSO4·5H2O/VCNa | 90 | >36 | —b |
| 19 | Dioxane/H2O (1:2) |
CuSO4·5H2O/VCNa | 90 | >24 | —b |
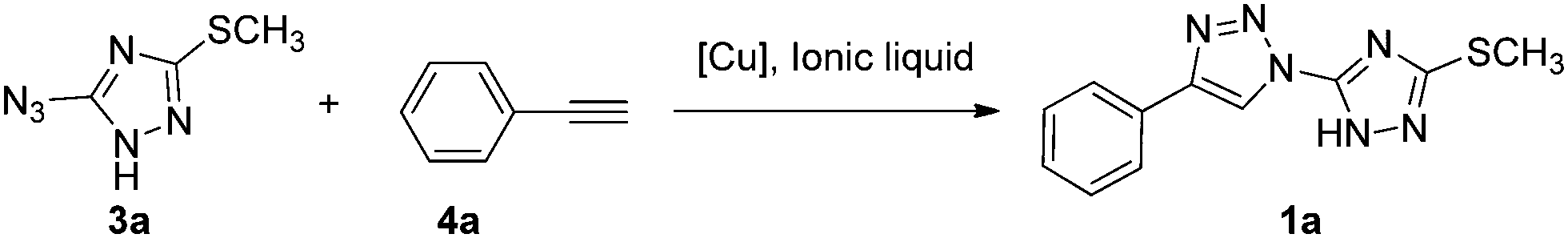
|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | Yield (%) | Inhibition ratio (%) | IC50 (μM) |
| a Not test. | |||||
| 1a | SCH3 | — | 90 | 42 | —a |
| 1b | SCH3 | 2-Cl | 93 | 77 | 2.89 ± 1.25 |
| 1c | SCH3 | 3-Cl | 93 | 67 | 20.72 ± 1.16 |
| 1d | SCH3 | 4-Cl | 94 | 9 | — |
| 1e | SCH3 | 2-CH3 | 88 | 55 | 18.01 ± 1.47 |
| 1f | SCH3 | 3-CH3 | 89 | 52 | — |
| 1g | SCH3 | 4-CH3 | 90 | 16 | — |
| 1h | SCH3 | 3-CH3O | 85 | 29 | — |
| 1i | SCH3 | 4-CH3O | 86 | <10 | — |
| 1j | SCH3 | 3-F | 92 | 38 | — |
| 1k | SCH3 | 4-F | 93 | 37 | — |
| 1l | SCH3 | 4-CH3(CH2)2 | 87 | 54 | 33.00 ± 1.09 |
| 2a | COOCH3 | — | 91 | 14 | — |
| 2b | COOCH3 | 2-Cl | 92 | 48 | — |
| 2c | COOCH3 | 3-Cl | 95 | 60 | — |
| 2d | COOCH3 | 4-Cl | 96 | 46 | — |
| 2e | COOCH3 | 2-CH3 | 89 | 51 | 61.19 ± 1.17 |
| 2f | COOCH3 | 3-CH3 | 89 | 44 | — |
| 2g | COOCH3 | 4-CH3 | 88 | 34 | — |
| 2h | COOCH3 | 3-CH3O | 90 | <10 | — |
| 2i | COOCH3 | 4-CH3O | 86 | 20 | — |
| 2j | COOCH3 | 3-F | 94 | 58 | — |
| 2k | COOCH3 | 4-F | 94 | 41 | — |
| 2l | COOCH3 | 4-CH3(CH2)2 | 88 | 81 | 13.10 ± 1.20 |
| Penthiopyrad | 1.32 ± 0.011 | ||||
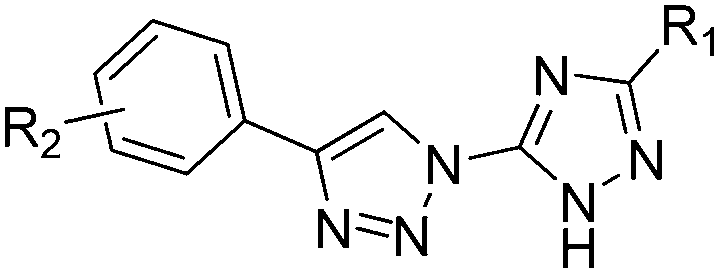
3.2 Inhibition activities of compounds against porcine succinate–cytochrome c reductase (SCR), succinate–ubiquinone oxidoreductase (SQR) and ubihydroquinone–cytochrome (cyt) c oxidoreductase (cyt bc1)
The in vitro activities of the prepared compounds were assayed against porcine succinate–cytochrome reductase (SCR), which composed of respiratory complex II (SQR) and complex III (bc1 complex). The complex II (SQR) firstly passes electrons from succinate to ubiquinone, and then the cytochrome bc1 complex passes electrons from reduced ubiquinone to cytochrome c. The activity of complex II in SCR was selectively determined using succinate and dichlorophenolindophenol (DCIP) as substrates, and the sole activity of the cytochrome bc1 complex in SCR was determined using decylubiquinol (DBH2) and cytochrome c as substrates, while the overall activity of SCR (both complex II and bc1 complex) was determined using succinate and cytochrome c as substrates.The inhibition results against SCR from porcine heart mitochondria are listed in Table 2. The IC50 values of compounds with inhibitory activities higher than 50% were further determined. Obviously, the activities varied largely depending on the substitution pattern of the bitriazoles. Although a number of synthesized compounds did not show significant inhibitory effects, other compounds with excellent SCR inhibition activity were discovered. It may be difficult to draw a clear structure–activity relationship due to the limited data obtained, some interesting conclusions can still be drawn according the biological results. First, it is clear that both the R1 group at the 1,2,4-triazole ring and the R2 group at the phenyl ring are crucial to maintain a strong SCR inhibitory activity. If R1 is fixed as a methylthio group, the substituent on the phenyl ring will have a conspicuous effect on their activities. When there is no further substituent on the phenyl ring, the resulting compound 1a with a simple phenyl ring connected to the 1,2,3-triazole has no activity. Substituting a chlorine atom in the ortho-position of the phenyl ring led to the most promising compound 1b with an IC50 value of 2.89 μM. Moving the chlorine atom to the meta-position resulted in a dramatic decrease of the activity, while placing the chlorine atom in the para-position, the resulting compound 1d lost its activity in terms of its inhibition ratio. Other substituents investigated including electron-donating groups such as methyl or methoxyl, and electron-withdrawing group such as fluorine all showed a limited activity, regardless of placing them on the meta- or the para-position. However, the n-propyl group, when it was assembled in the para-position of the phenyl ring, produced a moderate inhibitory activity (1l, IC50 = 33 μM). Additionally, changing the methylthio group to the methoxycarbonyl, the corresponding analogues 2 displayed elusive structure–activity relationship with regard to the effect of the R2 group on their activity. Unlike the series 1 compounds, which exhibited a promising activity when the chlorine atom was incorporated at the o- or the m-position, the chlorine containing analogues 2b, 2c and 2d with the chlorine atom at the o-, m- and p-position, respectively, all did not elicit inhibitory potential. Interestingly, the p-propyl substituted compound 2l showed slightly stronger activity than the counterpart analogue 1l.
Because SCR is composed of the respiratory complex II (SQR) and complex III (bc1 complex), it is very interesting to further determine which one, SQR or bc1, is responsible for the inhibition activity against the SCR system. Thus, the highly active compounds 1b, 1c and 1e with respective IC50 values of 2.89 μM, 20.72 μM and 18.01 μM were selected and assayed against SQR and bc1 alone (Table 3). In comparison, compounds 1b, 1c and 1e showed slightly higher activity against SQR than that against SCR, but much higher inhibitory potency as compared to that of the bc1 complex in terms of their inhibition ratios. These results suggest that the inhibition of succinate–cyctochrome reductase (SCR) is mainly ascribed to the interruption of the electron transfer from succinate to ubiquinone in the mitochondrial matrix. Thus, it can be deduced that SQR might be the potential target of these inhibitors.
| Compound | IC50 (μM), SCR | I% (50 μM), SCR | I% (50 μM), SQR | I% (50 μM), cyt bc1 | Selectivity |
|---|---|---|---|---|---|
| 1b | 2.89 ± 1.25 | 77 | 81 | 37 | SQR |
| 1c | 20.72 ± 1.16 | 67 | 78 | 30 | SQR |
| 1e | 18.01 ± 1.47 | 55 | 71 | 15 | SQR |
3.3 Computational simulations
To gain further insights into the structure–activity relationship, three representative compounds 1b, 1d and 2b were selected to perform the computational simulation. Compound 1b with a chlorine atom at the ortho-position of the phenyl ring and an additional methylthio group on the 1,2,4-triazole ring was identified as the most promising inhibitor with an IC50 value of 2.89 μM. Changing the position of the chlorine substituent from the o- to p-position led to the formation of compound 1d, which showed a much weaker activity as compared to compound 1b. In comparison, replacement of the methylthio group in compound 1b with methoxycarbonyl produced compound 2b and resulted in a significant reduction in the inhibitory activity. The binding model of these three compounds with a porcine heart QCR enzyme was illustrated in Fig. 1. From the simulated binding model of 1b in Fig. 1A, three characteristic interactions between the inhibitor and the enzyme can be observed. The 2-chlorophenyl ring underwent a π–π interaction with W35 and the two nitrogen atoms of the 1,2,3-triazole ring formed hydrogen bonds with the residues W173 and Y91, respectively. Furthermore, the 1,2,4-triazole ring underwent a π-ionic interaction with the residue R46. Therefore, the strong QCR inhibitory capacity may be attributed to these robust interactions. In contrast, the binding model of 1d, in which the chlorine atom was changed to the p-position, lost one of the two hydrogen bonds which formed in compound 1b due to the conformational deflection, and consequently led to a dramatic reduction of the inhibitory effect. Compound 2b, in which the methylthio group was replaced with methoxycarbonyl, adopted a totally different binding model from that of compound 1b and a conformational change occurred in the simulated binding model as illustrated in Fig. 1C. The 1,2,4-triazole moiety of 2b, instead of the 2-chlorophenyl ring in 1b, underwent a weak hydrophobic interaction with W35. Two important interactions, the hydrogen-bonding interaction between the nitrogen atom of the 1,2,3-triazole and W173 and a π-ion interaction between the 1,2,4-triazole ring and R46 in compound 1b, are lost in this binding model. This may account for why 2b showed a much weaker potency than 1b.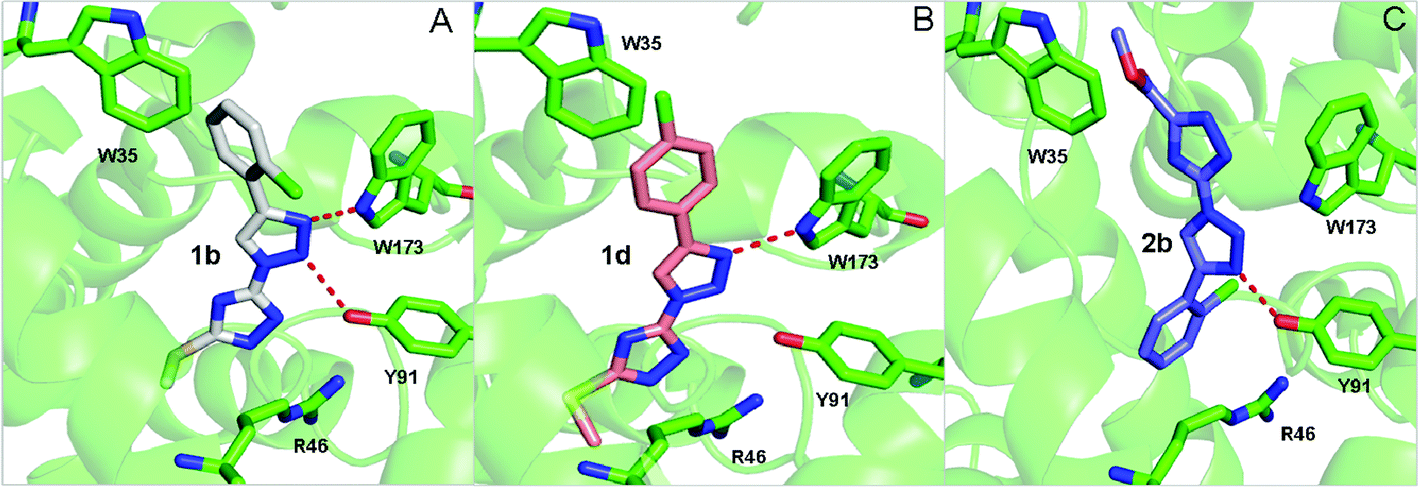 | ||
| Fig. 1 The binding model of compounds 1b, 1d and 2b with porcine QCR. | ||
4. Conclusion
In summary, a convenient and efficient methodology was developed to synthesize a series of bitriazolyl compounds via an ionic liquid promoted 1,3-dipolar Huisgen cycloaddition reaction of azides and akynes. This approach afforded a small library of bitriazolyl compounds in excellent yields with a simplified workup procedure, without requirement for tedious chromatographic purification. The results from an in vitro bioassay indicated that these newly prepared compounds exhibited varied inhibition against porcine succinate–cyctochrome reductase (SCR) depending on the substitution pattern of the triazole moiety. Compound 1b with an IC50 value of 2.89 μM was identified as the most promising inhibitor. Further evaluation against respective SQR and cyt bc1 indicated that the representative compounds had a much higher inhibitory potency against SQR than that of the bc1 complex in terms of their inhibition ratios, implying that SQR might be the potential target of these inhibitors. Thus, bitriazolyl compounds, a totally novel skeleton that is different from existing commercial SQR-inhibiting fungicides, can pave the way for development of novel SQR inhibitors.Acknowledgements
This research was supported by the National Key Technologies R&D Program (2014BAD23B01), the National Natural Science Foundation of China (No. 21272091, 21332004, 21472063) and self-determined research funds of CCNU from the colleges' basic research and operation of MOE (CCNU15A02014). We thank Dr Behrooz Moosavi for his careful reading of this manuscript.References
- D. Xia, L. Esser, L. Yu and C.-A. Yu, Photosynth. Res., 2007, 92, 17 CrossRef CAS PubMed.
- H. Kim, D. Xia, C. A. Yu, J. Z. Xia, A. M. Kachurin, L. Zhang, L. Yu and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8026 CrossRef CAS.
- C. Hunte, S. Solmaz, H. Palsdottir and T. Wenz, Results Probl. Cell Differ., 2008, 45, 253 CAS.
- E. A. Berry, M. Guergova-Kuras, L. Huang and A. R. Crofts, Annu. Rev. Biochem., 2000, 69, 1005 CrossRef CAS PubMed.
- F. Sun, X. Huo, Y. Zhai, A. Wang, J. Xu, D. Su, M. Bartlam and Z. Rao, Cell, 2005, 121, 1043 CrossRef CAS PubMed.
- V. Yankovskaya, R. Horsefield, S. Tornroth, C. Luna-Chavez, H. Miyoshi, C. Leger, B. Byrne, G. Cecchini and S. Iwata, Science, 2003, 299, 700 CrossRef CAS PubMed.
- E. Maklashina and G. Cecchini, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 1877 CrossRef CAS PubMed.
- S. J. Ralph, R. Moreno-Sánchez, J. Neuzil and S. Rodríguez-Enríquez, Pharm. Res., 2011, 28, 2695 CrossRef CAS PubMed.
- H. Sierotzki and G. A. Scalliet, Phytopathology, 2013, 103, 880 CrossRef CAS PubMed.
- S. Harada, D. K. Inaoka, J. Ohmori and K. Kita, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 658 CrossRef CAS PubMed.
- L. S. Huang, T. M. Borders, J. T. Shen, C. J. Wang and E. A. Berry, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, D61, 380 CAS.
- L. S. Huang, J. T. Shen, A. C. Wang and E. A. Berry, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1073 CrossRef CAS PubMed.
- L. S. Huang, G. Sun, D. Cobessi, A. C. Wang, J. T. Shen, E. Y. Tung, V. E. Anderson and E. A. Berry, J. Biol. Chem., 2006, 281, 5965 CrossRef CAS PubMed.
- P. Gunasekaran and P. Tauro, J. Biosci., 1982, 4, 219 CrossRef CAS.
- Mode of Action of Fungicides. FRAC Classification on Mode of Action, Crop life, 2015, http://www.frac.info.
- R. W. Sidwell, J. H. Huffman, G. P. Khare, L. B. Allen, J. T. Witkowski and R. K. Robins, Science, 1972, 177, 705 CAS.
- Y. A. Al-Soud, M. N. Al-Dweri and N. A. Al-Masoudi, Farmaco, 2004, 59, 775 CrossRef CAS PubMed.
- J. C. Cindy, M. R. Gregory, M. Janina, F. B. Laurent, K. Kasiram, M. Julia, A. C. Susan, V. Daniela, T. S. Claudiu and P. Sally-Ann, J. Med. Chem., 2013, 56, 9623 CrossRef PubMed.
- B. S. Holla, M. Mahalinga, M. S. Karthikeyan, B. Poojary, P. M. Akberali and N. S. Kumari, Eur. J. Med. Chem., 2005, 40, 1173 CrossRef CAS PubMed.
- M. F. Mady, G. E. A. Awad and K. B. Jørgensen, Eur. J. Med. Chem., 2014, 84, 433 CrossRef CAS PubMed.
- S. Velazquez, R. Alvarez, C. Perez, F. Gago, E. De Clercq, J. Balzarini and M. J. Camarasa, Antiviral Chem. Chemother., 1998, 9, 481 CrossRef CAS.
- P. S. Rao, C. Kurumurthy, B. Veeraswamy, G. S. Kumar, Y. Poornachandra, C. G. Kumar, V. S. Babu, S. Kotamraju and B. Narsaiah, Eur. J. Med. Chem., 2014, 80, 184 CrossRef PubMed.
- Y. Xia, Z. J. Fan, J. H. Yao, Q. Liao, W. Li, F. Q. Qu and L. Peng, Bioorg. Med. Chem. Lett., 2006, 16, 2693 CrossRef CAS PubMed.
- W. Li, Y. Xia, Z. J. Fan, F. Q. Qu, Q. Y. Wu and L. Peng, Tetrahedron Lett., 2008, 49, 2804 CrossRef CAS.
- M. H. Wang, R. Z. Zhu, Z. J. Fan, Y. F. Fu, L. Feng, J. H. Yao, A. Maggiani, Y. Xia, F. Q. Qu and L. Peng, Bioorg. Med. Chem. Lett., 2011, 21, 354 CrossRef CAS PubMed.
- H. Cheng, Y. Q. Shen, X. Y. Pan, Y. P. Hou, Q. Y. Wu and G. F. Yang, New J. Chem., 2015, 39, 7281 RSC.
- L. Lin, N. Mulholland, Q. Y. Wu, D. Beattie, S. W. Huang, D. Irwin, J. Clough, Y. C. Gu and G. F. Yang, J. Agric. Food Chem., 2012, 60, 4480 CrossRef CAS PubMed.
- L. Lin, N. Mulholland, S. W. Huang, D. Beattie, D. Irwin, Y. C. Gu, J. Clough, Q. Y. Wu and G. F. Yang, Chem. Biol. Drug Des., 2012, 80, 682 CAS.
- Y. Xia, F. Q. Qu, W. Li, Q. Y. Wu and L. Peng, Heterocycles, 2005, 65, 345 CrossRef CAS.
- L. Yu and C. A. Yu, J. Biol. Chem., 1982, 257, 2016 CAS.
- T. E. King, Methods Enzymol., 1967, 10, 216 CAS.
- A. R. Katritzky, Y. Zhang and S. K. Singh, Heterocycles, 2003, 60, 1225 CrossRef CAS.
- K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed.
- L. Zhang, X. Chen, P. Xue, H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef CAS PubMed.
- D. B. Ramachary, Chem. – Eur. J., 2013, 19, 13175 CrossRef CAS PubMed.
- P. Cintas, A. Barge, S. Tagliapietra, L. Boffa and G. Cravotto, Nat. Protoc., 2010, 5, 607 CrossRef CAS PubMed.
- D. Ashok, D. M. Gandhi, G. Srinivas and A. Vikas Kumar, Med. Chem. Res., 2014, 23, 3005 CrossRef CAS.
- A. A. Zarei, H. Fariba and K. Mosadegh, Synth. Commun., 2013, 43, 2100 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 5, 3508 CrossRef PubMed.
- D. V. Merbergen, J. W. Wijnen and J. B. F. N. Engberts, J. Org. Chem., 1998, 63, 8801 CrossRef.
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed., 1993, 32, 1545 CrossRef.
- A. Marra, A. Vecchi, C. Chiappe, B. Melai and A. Dondoni, J. Org. Chem., 2008, 73, 2458 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |