
Evaluating the friction of rotary joints in molecular machines
Tad
Hogg
 *a,
Matthew S.
Moses
*b and
Damian G.
Allis
*c
*a,
Matthew S.
Moses
*b and
Damian G.
Allis
*c
aInstitute for Molecular Manufacturing, Palo Alto, CA, USA. E-mail: tad@imm.org
bIndependent Consultant, Lafayette, CO, USA. E-mail: matt.moses@jhu.edu
cDepartment of Chemistry, Syracuse University, Syracuse, NY, USA. E-mail: dgallis@syr.edu
First published on 16th May 2017
Abstract
A computationally-efficient method for evaluating friction in molecular rotary bearings is presented. This method estimates drag from fluctuations in molecular dynamics simulations via the fluctuation–dissipation theorem. This is effective even for simulation times short compared to a bearing's energy damping time and for rotation speeds comparable to or below typical thermal values. We apply this method to two molecular rotary bearings of similar size at 300 K: previously studied nested (9,9)/(14,14) double-walled carbon nanotubes and a hypothetical rotary joint consisting of single acetylenic bonds in a rigid diamondoid housing. The acetylenic joint has a rotational frictional drag coefficient of 2 × 10−35 kg m2 s−1. The friction for the nested nanotubes is 120 times larger, comparable to values reported by previous studies. This fluctuation-based method could evaluate dissipation in a variety of molecular systems with similarly rigid and symmetric bearings.
Design, System, ApplicationReducing friction is important when designing molecular machines. One approach uses computer simulations to estimate friction in various machines and identify the best candidates to synthesize. These simulations start a proposed machine in motion and measure how rapidly it slows. Unfortunately, accurate measurements can require many long simulations because of thermal fluctuations in the speed. However, friction ultimately arises from those fluctuations. So instead of measuring friction directly, a more efficient method infers the friction from the fluctuations. We illustrate this method for two molecular rotary bearings: nested nanotubes and a new bearing based on rotation around single carbon–carbon bonds. With no sliding surfaces, the single-bond bearing has about 100 times less friction than nested nanotubes. Thus this bearing could greatly reduce energy dissipation in future molecular machines. |
1 Introduction
Bonds between carbon atoms are among the strongest chemical bonds that are free to rotate, subject only to the steric and electrostatic interactions between bound substituents. Hydrocarbons and organic molecules often have small potential energy barriers to this rotation, allowing a molecule to freely change configuration as constituent atoms rotate.1,2 The primary advantages of these molecular rotary joints, apart from their incredibly small size, are very low friction and zero wear. This phenomenon has enabled low-dissipation rotary motion in a number of recent nanoscale structures. For example, molecular machines with rotating or sliding joints have been created by various techniques, including traditional chemical synthesis,2–12 manipulation of carbon nanotubes,13,14 manipulation of graphene,15 and the re-purposing of biological motors.16 In theory, molecular machines using carbon bonds include planetary gearboxes,17,18 flywheels for energy storage,19 and mechanical computers.20 Such nanomachines will require large-scale atomically precise manufacturing, which is a challenging problem that is as yet unsolved in the general case.A key performance measure for molecular machines is their energy dissipation. Unfortunately, experimental measurements are often difficult to obtain, and impossible for molecular machines that cannot yet be fabricated. Computational studies are a reasonable alternative for evaluating and optimizing designs for various applications. Conceptually, estimating dissipation for a molecular machine is straightforward: simulate the machine's behavior over enough time for energy introduced into the machine's operational degrees of freedom to irreversibly transfer to many other degrees of freedom. Unfortunately, such computational studies are challenging for machines with many atoms and when the machine's operation is only weakly coupled to other atoms, leading to low friction and long damping times. Simulating such machines long enough to observe this damping may not be feasible, even for machines operating in vacuum, where the absence of gas or solvent molecules greatly reduces the computational cost. Moreover, even if simulating a single design is feasible, identifying low-power designs may require examining many machine variants, leading to a computationally costly overall design task.
One approach to reduce the computation time is to simulate the machine at speeds well above the typical thermal speed of the machine's motion. In this case, thermal fluctuations are relatively small, allowing definite estimates of damping directly from the change in speed, even over times short compared to the damping time. For example, this approach has been applied to determine friction in nested nanotubes21 However, this method may not apply to large molecular machines intended to operate near or below thermal speeds. This is because the machine may behave very differently at low and high speeds. For instance, machines at high speeds could involve forces strong enough to significantly distort components from their low-speed geometries or even break chemical bonds. More generally, high-speed operation may excite high-frequency vibrational modes that would not be relevant at intended operational speeds, thereby significantly overestimating the damping the machine would experience at its intended operating speed.
To address this challenge, this paper examines the effectiveness of an alternate method:22 using fluctuations in the simulated behavior to infer friction via the fluctuation–dissipation theorem. In contrast to measuring the damping directly from the decreasing energy during the simulation, fluctuations are evident even in simulations over times much shorter than the machine's damping time and when operating at speeds comparable to or less than thermal speeds.
This paper evaluates this fluctuation-based method for two examples. The first, rotating nested nanotubes, has been studied extensively and allows comparison with direct estimation of dissipation from the decrease of the energy. In this case, the relatively small number of atoms and high dissipation make both the direct and fluctuation-based methods feasible with modest computational cost.
The second example is a hypothetical molecular rotor using acetylenic bonds. These bonds give exceptionally low rotary friction, and thereby would require simulating the machines over long times to reliably measure dissipation directly. Moreover, including enough atoms around those bonds to capture interactions with the rotor's housing results in a significantly larger number of atoms than used for the nanotube example. Thus this rotor is an example where the fluctuation-based method could provide a significant benefit.
Specifically, after discussing related studies, section 3 describes the fluctuation-based method. We then present the two examples. The concluding section discusses future directions for applying the method and its possible limitations. The appendix provides details of the simulations.
2 Related work
Several studies have used molecular dynamics (MD) simulations to investigate friction in nanomachines with sliding or rotating components.In ref. 21, four different MD simulation configurations using the LAMMPS code and AIREBO force field were used to obtain consistent friction estimates for a rotating nested nanotube bearing. Other dissipation estimates for rotating nested nanotube bearings have been determined using intralayer interactions based on the Brenner potential23 with interlayer interactions based on the Kolmogorov–Crespi registry-dependent potential,24 and with the COMPASS force field.25 Dissipation estimates for linear sliding nested nanotube bearings have been determined using a custom force field and custom numerical simulator in ref. 26 and 27, and a custom force field based on ref. 28 and the r-RESPA integrator in ref. 29.
A MD model of experimentally realized graphene-on-graphene sliding is presented in ref. 15. The self-retracting motion of sheared graphene sheets is studied with an in-house MD integrator in ref. 30. Energy dissipation during high speed sliding of graphene sheets is investigated with GROMACS31 and the DREIDING force field32 in ref. 33.
In addition to the relatively simple cases of nested nanotubes and graphene–graphene sliding, MD studies have evaluated more complex nanomachines. One example is a study of meshing gears made from functionalized carbon nanotubes34 using the Brenner potential.23 In ref. 18 a complex molecular planetary gear mechanism is analyzed using the UFF forcefield and Gasteiger partial charges. In ref. 35 a custom integrator is used with a custom force field based on CHARMM and AMBER force fields, with partial charges determined by ab initio calculations, to model the behavior of the experimentally realized “nanocars”3 rolling on a gold surface.
An experimental and MD study measured the energy required to force rotation of a sterically-congested molecular bond in a surface-bound molecule.36 The work to rotate one bond was 5 × 10−20 J. The energy barrier involved in rotating the bond in this molecule is much larger than that of the rotary joint presented in this paper. All of this energy is not necessarily dissipated since much could be recovered by a properly designed mechanism. Nevertheless, this study provides an upper bound on the energy dissipated, and also indicates the work that would need to be supplied and, ideally, recovered for a single rotation.
3 Estimating drag from fluctuations
A physical system used as a machine distinguishes a few operational dynamic properties from other degrees of freedom of the machine and its environment. Conservative forces, derived from potential energy gradients, may impede the machine's motion. But they do not by themselves account for dissipation: energies associated with conservative forces can, in principle, be recovered during cyclic operation. On the other hand, friction is a dissipative force arising from random thermal interactions among the atoms of the machine and its environment. These interactions dissipate organized energy in the machine's motion into heat, and appear as a damping force. The distinction between conservative and dissipative forces is important for evaluating dissipation.213.1 Stochastic model of rotational damping
This paper considers rotary bearings which, ideally, have a single operational degree of freedom: rotation about a fixed axis. A molecular rotary joint can approximate this behavior,2 with additional degrees of freedom providing rapidly fluctuating torques on the rotation, i.e., a thermal bath. These torques produce a drag on the rotor.We model the effect of these torques with a phenomenological Langevin equation37,38 for the rotor's angular momentum L:2
 | (1) |
The first term on the right-hand side is a frictional damping torque, with characteristic damping time 1/γ. The second term describes conservative torques, arising from a potential energy V(θ) depending on the rotor's orientation angle θ. For the machines considered in this paper, torques arising from the potential are relatively small. In the final term, W is a standard Wiener stochastic process37 modeling the fluctuating torques. Operationally, σdW denotes independent normally-distributed random values with zero mean and standard deviation  over a time interval δt. The rotor's angular position changes as dθ/dt = ω, where ω = L/J is the angular velocity and J is the rotor's moment of inertia. Eqn (1) describes a stochastic process37 and numerical integration37,39 allows sampling solutions to this equation. This stochastic process is the rotational analog of a similar model applied to friction associated with linear motion.22,40
over a time interval δt. The rotor's angular position changes as dθ/dt = ω, where ω = L/J is the angular velocity and J is the rotor's moment of inertia. Eqn (1) describes a stochastic process37 and numerical integration37,39 allows sampling solutions to this equation. This stochastic process is the rotational analog of a similar model applied to friction associated with linear motion.22,40
The damping term in eqn (1) corresponds to a drag torque τ = γL = γJω. This drag is commonly expressed in terms of the rotational frictional drag coefficient, krd, defined by the relation τ ≡ krdω, so that krd = γJ. This drag dissipates energy at the rate Pdissipated = krdω2. The energy dissipated due to this friction when rotating by angle ϕ at a uniform speed is τϕ. If this rotation takes place over time t, then ω = ϕ/t and the dissipation is
| Edissipated = Pdissipatedt = krdωϕ = krdϕ2/t | (2) |
When operating at temperature T, the fluctuation–dissipation theorem2,37,38 relates the damping and fluctuation parameters, γ and σ. The physical mechanism underlying this relation is that damping arises from continual interactions with the environment. Random variations in these interactions provide fluctuating torques that, on average, damp the rotor's motion. However, instead of coming to rest, these torques make the rotor continue to move with a fluctuating angular momentum that is zero on average but has a nonzero variance denoted by L2th, where Lth is called the thermal angular momentum. The stochastic process of eqn (1) has equilibrium variance σ2/(2γ).37 Thus L2th = σ2/(2γ). The thermal angular momentum is related to temperature through the equipartition theorem,41 which states the average rotational kinetic energy equals kBT/2, where kB is Boltzmann's constant. Since the rotational kinetic energy of a rotor with angular momentum L is L2/(2J), the average kinetic energy in equilibrium is L2th/(2J). Thus L2th/(2J) = kBT/2, so  . Combining the expressions for Lth from the variance of eqn (1) and equipartition gives
. Combining the expressions for Lth from the variance of eqn (1) and equipartition gives
| σ2 = 2kBTJγ | (3) |
3.2 Range of model validity
Eqn (1) is an approximate model of a molecular rotor. Using this model to estimate drag requires determining when the model is an adequate approximation. In particular, the model assumes drag is linearly proportional to angular momentum and the motions of the other degrees of freedom of the machine provide uncorrelated random torques on the rotor's angular momentum.In general, the drag can have a nonlinear dependence on the speed. Such nonlinearity can be significant at high speeds, e.g., close to the speed of sound in the structure or involving forces large enough to break bonds. On the other hand, at sufficiently low speeds, the linear term dominates in a Taylor expansion of the drag as a function of speed. As one comparison, the speed of sound in diamond is about 104 m s−1, whereas the examples in this paper involve rotation of carbon molecules with radius r ≈ 1 nm at up to angular velocities of ω = 100 rad ns−1, with corresponding speed rω ≈ 102 m s−1.
The second assumption, uncorrelated random torques from other degrees of freedom, fails at sufficiently short time scales. For example, brief tilts of the rotor axis or oscillating normal modes of the housing could introduce short correlations in torques applied to the rotor. Eqn (1) assumes that these correlation times are short compared to times relevant for machine operation, which include the damping time. This requires a significant separation in time scales between the rotary degree of freedom and other motions. Molecular machines can achieve this separation with strongly bonded atoms throughout the structure except the parts intended to rotate, which instead have relatively weak interactions.
Thus we expect the stochastic model to apply to rotors operating at sufficiently low speeds and for sufficiently long times. We identify an appropriate time scale by comparing the standard deviation of changes in the rotor's angular momentum over various time scales δt. Specifically, consider ΔL(t) = L(t + δt) – L(t), the change in angular momentum starting at a time t. From a sample of the angular momenta, we compute s, the standard deviation of the changes ΔL at various times t in this sample, as a function of δt. Provided δt is short compared to the damping time 1/γ and the potential has only a small effect, the fluctuation term of eqn (1) dominates the changes, so  . In this case, the ratio
. In this case, the ratio  from the simulations should be nearly independent of δt. We evaluate this behavior with a “fluctuation plot” of this ratio vs. δt. The range of times δt achieving this independence indicates the time scale at which the molecular machine behaves approximately as a thermally-driven rotor.
from the simulations should be nearly independent of δt. We evaluate this behavior with a “fluctuation plot” of this ratio vs. δt. The range of times δt achieving this independence indicates the time scale at which the molecular machine behaves approximately as a thermally-driven rotor.
Another check on the model validity is comparing the power spectrum of the angular momentum with that of the stochastic process of eqn (1) using parameters estimated from simulations of the rotor behavior. Frequencies with a close match indicate corresponding times over which the model is a good approximation. In our cases, we find the time scales determined from power spectra are similar to those from fluctuation plots.
3.3 Molecular dynamics simulations
We evaluated rotational drag with molecular dynamics simulations. These simulations have two stages: warmup and spin-down.The warmup stage starts by adjusting the initial geometry to an energy minimum without constraints on any part of the structure. Next, we impose boundary conditions, i.e., fix the positions of a specific subset of the atoms. This simulation starts the system at zero temperature and couples it to a temperature bath at 300 K. We run the warmup simulation long enough bring the atoms to this temperature.
The spin-down stage starts with a warmed up configuration and adds velocities to the atoms of the rotary component to give it a specified initial angular velocity ω0. We then simulate the system in isolation, i.e., without coupling to a thermal bath. This is appropriate for the machines considered here, operating in vacuum, because the system is not in intimate contact with a thermal reservoir.
We repeat this procedure, with different random seeds, to obtain multiple samples of the behavior. Appendix A describes the simulation procedure in more detail.
3.4 Estimating drag from fluctuations in angular momentum
The simplest approach to estimating the damping γ is to average angular momenta over multiple simulations, since eqn (1) implies the average decays exponentially: 〈L〉 ∼ exp(−γt). However, as discussed in section 1, this direct approach may not be feasible for molecular machines containing many atoms and with low dissipation.On the other hand, fluctuations in the angular momentum are readily apparent in simulations over relatively short times. Thus we can use short simulations to estimate the fluctuation parameter σ and then determine the damping γ viaeqn (3). We do so by finding the value of σ that maximizes the likelihood of the observed changes in angular momentum over a fixed increment time δt according to eqn (1) and subject to the constraint of eqn (3). The second derivative of the log-likelihood at the maximum gives approximate confidence intervals for the estimated parameters.
In this fluctuation-based method, the choice of δt is somewhat arbitrary within a broad range. On the one hand, δt must be large enough that eqn (1) is a good approximation for the rotor, as discussed in section 3.2. On the other hand, δt must be short compared to the overall simulation time to have enough samples for estimation. As illustrated with the examples discussed below, this approach can estimate damping with simulation times short compared to the rotor's damping time even when operating at speeds comparable to thermal motion, so fluctuations are relatively large. For such situations, the fluctuation-based approach is computationally less costly than running simulations long enough to directly estimating damping with similar confidence.
In general, eqn (1) does not have a simple solution. In such cases, evaluating the likelihood requires sampling the stochastic process or numerically solving its Fokker–Planck equation.37 However, if the variation in the rotation potential V is small compared to thermal energies, the potential can be neglected, which greatly simplifies the estimation. In particular, when V is independent of the rotation angle, eqn (1) is an Ornstein–Uhlenbeck (O–U) process.42 In this case, the likelihood is readily computed. The examples considered in this paper have small potentials, allowing us to apply this simplification.
4 Friction of rotating nested nanotubes
The nested nanotube is a prototypical nanomechanical assembly. It has been demonstrated experimentally13 and its structural and operational behavior has been extensively discussed. Because of this importance, many molecular modeling studies have been performed on single- and multi-walled nanotubes, providing a considerable body of theory against which to compare our proposed technique for evaluating friction. This includes prior studies on the rotational friction of nested nanotubes.21 For this reason, we use nested nanotubes as a test case for our method.We applied the fluctuation-based method for evaluating friction to rotating nested nanotubes shown in Fig. 1. In this model system, a (9,9) single-wall carbon nanotube (SWCNT) rotates inside a (14,14) SWNCT. This example focuses on the rotary bearing itself, and ignores any damping that might arise from the housing to which the bearings would be connected in a molecular machine.
 | ||
| Fig. 1 Nested nanotubes. The inner tube is 6 nm long and the outer tube is 5 nm long. These consist of 882 and 1120 carbon atoms, respectively. | ||
The relatively small number of atoms in the nanotubes and their short damping time allow feasible simulations to show significant damping, even at relatively low rotation speeds, comparable to thermal speeds. This nanotube system thus provides a test case to compare damping estimated directly from the decrease in angular momentum with that estimated from fluctuations over a time scale much shorter than the damping time. Moreover, these simulations allow direct comparison with the extensive prior study of these nanotubes with simulations using other boundary conditions.21Table 1 describes the inner nanotube, and Appendix A.1 describes our simulations.
 and ωth = Lth/J, respectively, where kB is Boltzmann's constant
and ωth = Lth/J, respectively, where kB is Boltzmann's constant
| Parameter | Value | |
|---|---|---|
| Temperature | T | 300 K |
| Radius | r | 0.6 nm |
| Mass | m | 1.76 × 10−23 kg |
| Moment of inertia | J | 6.56 × 10−42 kg m2 |
| Thermal angular momentum | L th | 1.65 × 10−31 kg m2 s−1 |
| Thermal angular velocity | ω th | 25.1 rad ns−1 |
4.1 Nanotube behavior
Fig. 2 shows how the angular momentum of the inner nanotube changes during the simulations, starting with angular velocity ω0 = 50 rad ns−1. The initial angular velocity is smaller, and closer to typical thermal velocities, than prior studies of this system.21 Hence thermal fluctuations are relatively large, making it somewhat difficult to identify the decrease in average angular momentum even with the simulation time comparable to the damping time. This identification is even more difficult for the 0.1 ns simulation time that we use for the fluctuation-based method. | ||
| Fig. 2 The angular momentum of the inner nanotube about its axis vs. time for each of the spin-down simulations, sampled at 0.01 ns intervals. Successive samples are connected with straight lines, which do not show the high-frequency fluctuations between samples. The vertical dashed line indicates the extent of the truncated simulations used to estimate drag based on fluctuations. | ||
To identify the time scale over which the nanotubes match the stochastic model of eqn (1), Fig. 3 shows a fluctuation plot, described in section 3.2. The curves are roughly the same for δt above 0.01 ns. This indicates eqn (1) approximates the nanotube motion for times longer than this. In particular, short simulations, of just 0.1 ns, should be more than enough to estimate damping using the fluctuation-based method. These nested nanotubes have negligible potential barriers to rotation,26 so the potential is not relevant for friction estimation.
 | ||
Fig. 3 Standard deviation of changes in nanotube angular momentum in time δt divided by  , for each of the nanotube simulations (upper, colored curves). The solid horizontal black line shows the value of this ratio expected from the stochastic process model of the nanotubes. Times are shown on a logarithmic scale. , for each of the nanotube simulations (upper, colored curves). The solid horizontal black line shows the value of this ratio expected from the stochastic process model of the nanotubes. Times are shown on a logarithmic scale. | ||
4.2 Nanotube drag
Table 2 compares the drag estimates from two methods: 1) a direct estimation by fitting an exponential decay e−γt to the average angular momentum of the simulations, and 2) the fluctuation-based method applied to the first 0.1 ns of each simulation. The damping estimates are similar, showing the fluctuation-based method provides a useful estimate using simulations much shorter than the damping time, which is 1/γ ≈ 2.4 ns in this case. The corresponding drag is krd = 2.9 (±1.5) × 10−33 kg m2 s−1. On the other hand, applying the direct method to the first 0.1 ns of the simulations gives a poor estimate.| Method | Simulation time | Damping constant γ |
|---|---|---|
| Direct | 1 ns | 0.40 (±0.3)/ns |
| Direct | 0.1 ns | 1.5 (±1.1)/ns |
| Fluctuation | 0.1 ns | 0.45 (±0.2)/ns |
This estimate for nanotube drag is consistent with that of a previous study21 based on simulations at higher speeds, above 25 GHz. As described in Appendix A.1, this study gives krd = 3.4 (±0.8) × 10−33 kg m2 s−1. This consistency indicates the linear fit of frictional force vs. speed of ref. 21 extends to the lower speed used in our simulation, i.e., 8 GHz.
We find similar behaviors when operating the nanotubes at higher speed and lower temperature, as described in Appendix A.2.
5 Friction of an Acetylenic single-bond rotor
Bearings consisting of single bonds are molecular rotary joints that avoid sliding surfaces. This contrasts with molecular rotors such as nested nanotubes, in which rotor and housing are not bonded and involve sliding friction from many atoms.This section describes and evaluates such a design. The rotary joints consist of covalent acetylenic bonds between rotor and housing. Their linear geometry makes acetylenic bonds ideal as low barrier linkages in rigid organic frameworks. This property has been exploited in nanoscale mechanical design, where several designs of the Nanocar3 employed acetylenic linkages as axles between fullerene wheels and a planar chassis. The structure includes enough of a housing around the rotor to include long range interactions between the rotor and the rest of a molecular machine using the rotary joint. As with the nanotube bearing, we consider operation in vacuum at room temperature.
To evaluate rotor behavior, we used two computational techniques. First, we applied density functional theory (DFT) calculations to the rotor and part of the surrounding structure under symmetry constraints to determine how the rotor interacts with the housing. This included the predicted rotational barrier due to the shortest range rotor-housing interactions, and the vibrational mode energies associated with the rotor motion. Second, we used the DFT results to generate missing AMBER force field terms and RESP partial charges of the rotor and surrounding structure for the molecular dynamics simulations, which then were used to sample the rotor's motion within the full structure. We used these samples to estimate the drag. Appendix A.3 describes our simulations.
5.1 Rotor and housing
Fig. 4 shows our rotary system: a molecule supporting a mobile rotor with two coaxial molecular rotary joints attached to a stationary housing. The rotor is made of hexagonal diamond and suspended on two molecular rotary joints connected to a housing made of faceted cubic diamond. The rotor is bonded to the housing at each end by a so-called C2 spacer, which resembles the organic molecule 2-butyne. Table 3 describes the rotor. While it is not yet possible to synthesize this rotary joint molecule, Appendix B describes why it would be chemically stable. | ||
Fig. 4 A rotor bonded to a housing. This molecule contains 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 345 atoms, 7693 carbon (gray) and 2652 hydrogen (white), and occupies a volume of about 4 nm × 7 nm × 7 nm. The rotor consists of 200 carbon atoms and 150 hydrogens on its surface. The housing has 7493 carbon atoms and 2502 hydrogens. The rotor is shown in the minimum energy “staggered” configuration, which defines the origin of the rotation angle: θ = 0. 345 atoms, 7693 carbon (gray) and 2652 hydrogen (white), and occupies a volume of about 4 nm × 7 nm × 7 nm. The rotor consists of 200 carbon atoms and 150 hydrogens on its surface. The housing has 7493 carbon atoms and 2502 hydrogens. The rotor is shown in the minimum energy “staggered” configuration, which defines the origin of the rotation angle: θ = 0. | ||
 and ωth = Lth/J, respectively, where kB is Boltzmann's constant
and ωth = Lth/J, respectively, where kB is Boltzmann's constant
| Parameter | Value | |
|---|---|---|
| Temperature | T | 300 K |
| Mass | m | 4.24 × 10−24 kg |
| Moment of inertia | J | 1.76 × 10−42 kg m2 |
| Amplitude of potential | V 0 | 4.15 × 10−22 J |
| Thermal angular momentum | L th | 8.54 × 10−32 kg m2 s−1 |
| Thermal angular velocity | ω th | 48.5 rad ns−1 |
We designed this specific molecule as likely to give a rotary joint with low dissipation. Specifically, this molecule:
• provides a complete rotor and housing that includes all thermal motion in and around the rotor, and long-range interactions between the rotor and its housing
• isolates the rotor motion from the housing vibrations by use of a stiff material (diamond)
• has a low rotational energy barrier
• has closely matched rotor length and housing spacing so there is little strain (compression or stretching) between the C2 spacers and either the rotor or the housing.
This molecule has two properties that simplify computational studies. First, the high symmetry of the rotor and nearby parts of the housing reduces the computational cost of determining its properties, including geometries corresponding to extrema of the rotational potential, normal modes of vibration, and partial charges on the atoms. Second, the molecule consists of just three atom types: sp3 (tetrahedral) carbon, terminal C–H bond hydrogen, and sp (acetylenic) carbon. These standard atom types readily transfer to any molecular force field for additional testing and validation.
5.2 Forces and rotation potential
Evaluating the rotor's behavior requires accurate forces between the rotor and the housing. A significant portion of these forces arises from partial charges in the rotor and housing, which we evaluated using the RESP partial charge model described in Appendix C.3.We use these charges as part of the force field for our molecular dynamics simulations, both to sample rotor behavior for determining drag and to estimate the rotation potential V(θ). This potential is an aggregate property of the molecular dynamics force field. It quantifies the conservative torques on the rotor in the stochastic model given by eqn (1). As described in Appendix A.5, the potential energy as a function of rotation angle is well-approximated by
| V(θ) = V0(1 − cos(3θ)) | (4) |
This potential barrier, 2V0 = 8.3 × 10−22 J, is comparable to estimates of barriers for rotation about triple bonds.2 This barrier is about 0.2kBT, which is relatively small compared to thermal energy.
The potential applies a torque –dV/dθ to the rotor. The maximum torque is 3V0 = 1.2 pN nm. As an estimate of corresponding forces, if the maximum torque is mostly at the rotor's outer rim, with radius r = 0.9 nm (see Fig. 11 in Appendix B), the force is 3V0/r = 1.4 pN. If instead the torque mainly acts through the axle, of radius r = 0.05 nm, the force is 25 pN.
5.3 Rotor behavior
Fig. 5 shows the z component of the rotor angular momentum about its center of mass during five spin-down MD simulations, starting with angular velocity ω0 = 100 rad ns−1. During the 5 ns simulation time, the angular momentum does not decrease significantly, indicating the damping time is considerably larger than the simulation time. | ||
| Fig. 5 The z component of rotor angular momentum about the center of mass vs. time for each of the spin-down simulations, sampled at 0.1 ns intervals. Successive samples are connected with straight lines, which do not show the high-frequency fluctuations between samples. | ||
The atoms of the rotor change position slightly during the simulations. However, due to the strong bonding, these motions have only a small effect on the moment of inertia. In particular, the standard deviation of the moment of inertia about the rotor axis, J, is 6 × 10−45 kg m2, less than 1% of the mean value, given in Table 3.
To identify the time scale over which the rotor matches the stochastic model of eqn (1), Fig. 6 shows a fluctuation plot, described in section 3.2. The values are roughly the same for δt above 0.2 ns or so. For shorter times, the observed fluctuations are larger than the expected  behavior from eqn (1). This indicates the stochastic model does not correctly describe short-time motion of the rotor, below about 0.2 ns or above 5 GHz. This conclusion is similar to that from the peaks in the power spectrum shown in Fig. 7. These observations indicate eqn (1) provides a reasonable approximation of the rotor's motion for times longer than about 0.2 ns.
behavior from eqn (1). This indicates the stochastic model does not correctly describe short-time motion of the rotor, below about 0.2 ns or above 5 GHz. This conclusion is similar to that from the peaks in the power spectrum shown in Fig. 7. These observations indicate eqn (1) provides a reasonable approximation of the rotor's motion for times longer than about 0.2 ns.
 | ||
Fig. 6 Standard deviation of changes in rotor angular momentum in time δt divided by  , for each of the rotor simulations (upper, colored curves). The dashed and solid black curves show the value of this ratio expected from the stochastic process model of the rotor, eqn (1) discussed in section 5.5, with and without the potential, respectively. Times are shown on a logarithmic scale. , for each of the rotor simulations (upper, colored curves). The dashed and solid black curves show the value of this ratio expected from the stochastic process model of the rotor, eqn (1) discussed in section 5.5, with and without the potential, respectively. Times are shown on a logarithmic scale. | ||
 | ||
| Fig. 7 Average of the angular momentum power spectra from the spin-down simulations. The horizontal axis shows frequency on a logarithmic scale. Power values are in decibels, normalized so the total power equals the sum of the squared angular momenta from the simulations. For comparison, the dashed and solid black curves show the power spectrum of the stochastic model of the rotor, eqn (1), with and without the potential, respectively. | ||
The potential is relatively small compared to kBT. In addition, δt above 0.2 ns involves motion of more than a complete revolution of the rotor for the angular velocities in the simulation, which remain close to the initial 100 rad ns−1 because damping does not slow the rotor much during the 5 ns simulation time. Thus the small effects of the potential are further reduced by the rotor averaging over the potential during the time steps we consider.
5.4 Rotor drag
Table 4 gives the parameters maximizing the likelihood using increments with time step δt = 0.5 ns. Other choices of δt in the range 0.2–1.0 ns give the same values within the range of the confidence intervals. The estimated fluctuation magnitude, σ, is consistent with Fig. 6 for this range of δt.| Parameter | Value | |
|---|---|---|
| Damping constant | γ | 0.014 (±0.005)/ns |
| Damping time | 1/γ | 70 (±40) ns |
| Fluctuation magnitude | σ |

|
| Frictional drag coefficient | k rd | 2.4 (±0.9) × 10−35 kg m2 s−1 |
For example, with the angular velocity in our simulations, ω0 = 100 rad s−1, the time for a full rotation is t = 2π/ω0 = 0.06 ns. From eqn (2), the frictional energy dissipation during that rotation is 2πkrdω0 ≈ 1.5 × 10−23 J. By comparison, a full rotation of this molecular rotor moves through three potential wells, each of depth 2V0 (see Table 3). Thus climbing the potential barriers requires 6V0 ≈ 2.5 × 10−21 J. However, this work could, in principle, be recovered as the rotor moves into each potential well. The energy dissipated by friction is about 100 times smaller than the (possibly recoverable) energy required to climb the potential, which in turn is an order of magnitude smaller than the experimentally measured energy, 5 × 10−20 J, to force rotation of a surface-bound molecule.36
5.5 Stochastic process for rotor behavior
The stochastic process eqn (1) with parameters from Table 4 gives a model of the rotor as a thermally-fluctuating molecular machine with a single degree of freedom. As a consistency check on this model and its range of validity, we compare the behavior predicted by this stochastic process with that from the simulations.Fig. 6 shows one such comparison. The rotational potential accounts for some of the increase in fluctuations at short times. In particular, the rotor converts between kinetic and potential energy as it moves between the top and bottom of the potential wells. For our simulations, the angular velocity remains close to its initial value 100 rad ns−1. The potential, in eqn (4), is 3-fold symmetric, so rotation from one potential well to the next requires rotation through 2π/3 ≈ 2 rad, which occurs in 0.02 ns, corresponding to 50 GHz. This motion leads to the oscillating fluctuation size shown in the dashed curve of Fig. 6.
Another perspective on short-time rotor behavior is the average of the simulations' power spectra, shown in Fig. 7. By comparison, the power spectrum of an O–U random process is proportional to 1/(γ2 + (2πf)2).37 For frequencies large compared with γ, this power decreases as 1/f,2 and hence appears as a straight line on a log–log plot. Fig. 7 compares this behavior with that of the rotor simulation, showing close correspondence with the rotor's angular momentum up to about 10 GHz.
The behavior including the potential (dashed curve in Fig. 7) has the broad peak near 50 GHz, showing the potential ripple accounts for both the frequency (due to time it takes rotor to move over one potential barrier at 100 rad ns−1) and the magnitude of the peak.
Above about 50 GHz, both the fluctuation plot and power spectrum show deviations from the stochastic process model, even when including the potential. One source of these deviations is temporal correlations in torques applied to the rotor, e.g., due to movement of its axis of rotation. The oscillation frequencies of the structure's normal modes indicate the time scale of these motions. We evaluated the normal modes of the rotor-housing structure using GROMACS. The lowest mode is the rotor twisting about its axis, at about 7.24 GHz. This is close to the oscillation frequency of the potential minimum:  . This mode corresponds to the rotary motion given by the stochastic model. The next lowest modes involve the rotor tilting, at around 100 GHz. These modes characterize deviations from the stochastic model, and show the large separation in time scales between the rotary degree of freedom and other modes in the structure. This separation is the expected behavior for a rotary bearing consisting of strongly bonded structures and weak barriers to rotation.
. This mode corresponds to the rotary motion given by the stochastic model. The next lowest modes involve the rotor tilting, at around 100 GHz. These modes characterize deviations from the stochastic model, and show the large separation in time scales between the rotary degree of freedom and other modes in the structure. This separation is the expected behavior for a rotary bearing consisting of strongly bonded structures and weak barriers to rotation.
These comparisons between simulations and the stochastic process with the estimated parameters show the atoms in the rotor and housing behave approximately as a stochastic rigid rotor, with a single degree of freedom around the rotary joint. The atoms' other degrees of freedom act as a source of uncorrelated random fluctuations, i.e., a thermal bath.
6 Discussion
This paper applies a fluctuation-based method for evaluating friction in molecular machines to rotary bearings. The method performs well even when simulation times are short compared to the machine's damping time. Thus, the method can use much less computational time than a direct estimate from the average decrease in energy.The method was applied to nested nanotubes and a rotor covalently bonded to a housing by single acetylenic bonds. For the nested nanotubes, damping time is 1/γ = 2.4 ns and drag coefficient is krd = 2.9 × 10−33 kg m2 s−1 (section 4.2). The bonded rotor joint has 1/γ = 70 ns and krd = 2.4 × 10−35 kg m2 s−1 (section 5.4). From eqn (2), this means rotational friction dissipates 120 times less energy for the bonded rotor than for the nested nanotubes when they rotate at the same speed. Thus acetylenic rotary joints can have exceptionally low dissipation, which could make them useful bearings in future molecular machines.
Machine design often focuses on performance and ease of manufacture. For molecular machines that are not yet possible to fabricate, an important additional criterion is the computational cost to simulate their behavior. As an example of designs that simplify drag analysis, rotors with small potential barriers compared to thermal energy allow neglecting the potential in the stochastic model used with the fluctuation-based method. In addition, strong bonding among degrees of freedom other than those of the machine's operation (in this case the single rotational degree of freedom) allows treating the machine as a rigid body, with other degrees of freedom providing uncorrelated random fluctuations.
An open question is the range of conditions under which the fluctuation-based method performs well. For instance, potentials with larger barriers to rotation, less rigid molecular housings or less symmetric rotational geometry than the examples considered in this paper may significantly reduce the accuracy of modeling the rotor as a stochastic process with a single degree of freedom. Since neglecting the rotational potential simplifies the analysis, a specific issue for future work is determining design rules under which a molecular rotor is sufficiently well-approximated as having zero potential when evaluating its friction.
The fluctuation-based method discussed in this paper could evaluate drag in a range of operating conditions. Examples are how rotary friction depends on rotor size, rotation speed and temperature. This could require accounting for additional physical effects. For instance, when operating well below room temperature, the variation in the potential is significant compared to kBT, leading to more complicated behavior than seen in the examples of this paper.2,43 Moreover, at low temperatures, quantum effects alter how energy spreads among degrees of freedom and introduce additional dissipative mechanisms, as described in Appendix A.6.
The interactions between rotor and housing arise mainly from forces between the rotor and nearby atoms in the pyramid connectors. These interactions largely determine the potential barrier to rotation. Future work could more precisely quantify these short-range interactions and the extent to which they are responsible for the friction. Identifying the main contributions to friction will suggest how changes to the housing structure affect dissipation, e.g., by altering stresses on bonds near the rotary joint.
The fluctuation-based method can apply to many molecular machines in addition to the rotary bearings considered in this paper. In particular, a single bearing will only be a small part of a useful molecular machine using the bearings, such as a computer.20 Designing such machines, containing large numbers of atoms, will require efficient computational methods. Thus the fluctuation-based method could aid computational explorations of a wide variety of designs to identify low-power molecular machines. Fluctuations can estimate drag from simulations much shorter than damping time, which is particularly useful for low-friction molecular machines with many atoms. However, the simulation must still be long enough to average over dynamics of other modes of behavior than the degree(s) of freedom involved in the machine's intended operation. Otherwise fluctuations may not be independent, as assumed by the stochastic process model. More specifically, the method is useful when the correlation time of fluctuations is much smaller than the damping time of the machine's operation. Molecular machines made of strongly bonded structures can have such separation of time scales, as illustrated by the examples discussed in this paper. Thus the method described in this paper could be a useful tool for evaluating dissipation in such machines.
A Molecular dynamics simulations
Section 3.3 gives an overview of our molecular dynamics simulations, which used GROMACS (v. 5.0.4).31,44 The GROMACS simulations used double precision on a PC with an Intel Core i7-5960X 3.00 GHz processor and 48 GB RAM, running on 64-bit Ubuntu 14.04.4 LTS. GROMACS structure, topology, configuration files and supporting scripts are available on request.For both the nanotube and rotor simulations, the warmup stage starts with all atoms at zero velocity and couples them to a heat bath, i.e., the simulations used a canonical (NVT) ensemble. The spin-down simulations used a microcanonical (NVE) ensemble, i.e., without temperature control.
The warmup and spin-down simulations are performed in vacuum and use 1 fs time steps. In accordance with best-practices for molecular dynamics simulation in vacuum while reducing computational resource use, all GROMACS simulations were performed under periodic boundary conditions using 25 × 25 × 25 nm boxes with rlist, rcoulomb, and rvdw cut-offs of 10 nm, particle mesh Ewald (PME)45 coulomb type and vdwtype (with pme-order of 6), and the Verlet cutoff scheme under a grid-based neighbor list determination.
The box size used for periodic boundary conditions is larger than the sum of half the molecule's dimensions and the cut-off lengths. The volume of this box remains constant during the simulation. Since the molecule is in vacuum, the simulation has both constant volume for the full simulation volume and zero pressure on the molecule.
The warmup simulation uses the following parameters: 50000 steps at 1 fs per step (50 ps total simulation time), xyz periodic boundary conditions with Verlet cutoff, 10 nm cutoff distance (which exceeds the largest atom-to-atom distance in the structure, and is less than the distance between copies of the structure due to periodic boundary conditions), v-rescale temperature control with 1 ps time constant, and random seed set to 42, 43, 44, 45, or 46. These seeds give five different samples of the structure at 300 K. During the warmup simulations, the structures reach 300 K within about 10 ps, indicating the 50 ps warmup simulation time is more than sufficient to eliminate transients during warmup.
A.1 Molecular dynamics for nested nanotubes
For our evaluation of nanotube drag, we focus on the inner nanotube and hold fixed the atoms on one edge of the outer tube. After the warmup, velocities added to the atoms of the inner tube give it initial angular velocity ω0 = 50 rad ns−1, corresponding to 8 GHz rotation frequency. The subsequent spin-down simulations were for 1 ns. We performed five separate tests of the nanotubes. A 50 picosecond warmup nanotube simulation takes one minute to complete, while a five-nanosecond spin-down simulation takes about 20 minutes.Table 5 provides the force field parameters46 used for the nested nanotube bearing simulations. The bond stretch and bond angle terms are based on the AMBER force field.47 The Ryckaert–Bellemans dihedral terms are based on the bending rigidity of graphene,48 and the Lennard-Jones terms are based on ref. 27.
| Interaction type | Parameter | Value | Unit |
|---|---|---|---|
| Morse potential bond stretch | b | 0.142 | nm |
| Morse potential bond stretch | D | 722.7 | kJ mol−1 |
| Morse potential bond stretch | β | 18.33 | nm−1 |
| Harmonic angle potential | k | 527.2 | kJ mol−1 rad−2 |
| Harmonic angle potential | θ 0 | 120.0 | Degrees |
| Ryckaert–Bellemans dihedral | C1, C3, C4, C5 | 0.0 | kJ mol−1 |
| Ryckaert–Bellemans dihedral | C 0 | 17.843 | kJ mol−1 |
| Ryckaert–Bellemans dihedral | C 2 | −17.843 | kJ mol−1 |
| Lennard-Jones | ∈ | 0.28454 | kJ mol−1 |
| Lennard-Jones | σ | 0.341 | nm |
Our nanotube simulations are similar to the “coast-down” method of ref. 21. Minor differences arise from our use of GROMACS instead of LAMMPS, the force field of Table 5 instead of AIREBO, and having the outer tube anchored at one edge instead of freely counter-rotating.
Our friction estimate is consistent with the linear fit to normalized values given in ref. 21. This fit is F* = Av* where F* and v* are normalized friction force and sliding velocities [eqn (23) and (24)],21A = 4.62 × 10−4 and the fit is consistent with having zero friction at zero velocity.† Converting the friction force to a torque, and the sliding velocity to the corresponding angular velocity, the linear relation between F* and v* gives the rotational frictional drag coefficient described in section 3.1:
 | (5) |
A.2 Nanotube damping and operating conditions
Table 2 gives drag estimates for one operating condition for the nanotubes. This appendix describes the drag for other operating conditions to assess our method's generality.When the nanotube rotates at speeds well above thermal velocity, fluctuations are relatively unimportant. In that case, the decrease in angular momentum is readily apparent, as illustrated in Fig. 8 for nested nanotubes with initial angular velocity 500 rad ns−1. This initial speed is ten times the angular velocity considered in section 4, and is well above typical thermal speeds (see Table 1). Table 6 compares drag estimated with the direct and fluctuation-based methods for this case. Because thermal fluctuations are relatively small, the direct method gives good estimates even over short times. Both methods give comparable drag estimates, and are consistent with the values at the lower speed, shown in Table 2. This similarity is consistent with the linear relation between frictional torque and angular velocity found in other studies,21,27 as well as the linear form for the drag in eqn (1).
 | ||
| Fig. 8 The angular momentum of the inner nanotube about its axis vs. time for spin-down simulations with initial angular velocity ω0 = 500 rad ns−1, sampled at 0.01 ns intervals. Successive samples are connected with straight lines, which do not show the high-frequency fluctuations between samples. | ||
| Method | Simulation time | Damping constant γ |
|---|---|---|
| Direct | 0.5 ns | 0.38 (±0.03)/ns |
| Direct | 0.1 ns | 0.46 (±0.1)/ns |
| Fluctuation | 0.1 ns | 0.41 (±0.1)/ns |
Fluctuations and damping depend on temperature. As an example, Table 7 shows the damping estimates from the two methods at T = 80 K. We performed 5 simulations for each of the two initial angular velocities shown in the table. In this case, the thermal angular velocity is ωth = 13.0 rad ns−1 so even the smaller initial speed, ω0 = 50 rad ns−1, is well above the thermal speed. This means fluctuations are less important than at higher temperatures, and both the direct and fluctuation-based methods give similar results, as shown in Table 7. At 80 K, the damping time 1/γ is about 15 ns, so there is relatively little damping during our 0.5 ns simulations.
| Method | Initial angular velocity | Damping constant γ |
|---|---|---|
| Direct | 500 rad ns−1 | 0.067 (±0.01)/ns |
| Fluctuation | 500 rad ns−1 | 0.066 (±0.01)/ns |
| Direct | 50 rad ns−1 | 0.071 (±0.06)/ns |
| Fluctuation | 50 rad ns−1 | 0.068 (±0.03)/ns |
The fluctuation–dissipation theorem, eqn (3), relates the fluctuation (σ) and damping (γ) parameters to temperature (T). Specifically the ratio σ2/γ is proportional to T. However, this does not determine how the two parameters vary individually. Comparing the damping at 80 K and 300 K identifies how these parameters depend on temperature. Fitting the parameter estimates from our simulations at both temperatures to a power-law dependence, σ ∝ Tα and γ ∝ Tβ, gives α = 1.23 ± 0.08 and β = 1.47 ± 0.15. This is consistent with a study of nanotube friction over a wider range of temperatures that found β = 1.53 ± 0.04.27
A.3 Molecular dynamics for the bonded rotary joint
The boundary condition for the simulation holds fixed the atoms in the stem indicated in Fig. 4. After warmup, we set the rotor's initial angular velocity to ω0 = 100 rad ns−1, corresponding to rotation at 16 GHz. The subsequent spin-down simulations were for 5 ns. We performed five rotor simulations. A 50 picosecond warmup rotor simulation takes less than an hour to complete, while a five-nanosecond simulation of rotor spin-down takes about 44 hours.Structure optimizations, normal mode analyses, and molecular dynamics simulations used the AMBER99 (ref. 47) force field C–H and C–C bend, stretch, dihedral, and C6/C12 parameters serving as a basis for the calculations. Terms for the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (triple bond) and C–C# bonds were defined as given in ref. 50.
C (triple bond) and C–C# bonds were defined as given in ref. 50.
The full structure (rotor plus housing) is first energy-minimized without applying anchors to any part of the structure. Next, anchors are applied to 514 atoms in the stem of the housing, and the structure is heated to 300 K in a warmup simulation.
The output of each of the five warmup simulations is a molecular structure in G96 format which contains the position and velocity for each atom in the structure. The initial angular velocity of the rotor is set by using a Matlab/GNU Octave script to adjust the velocities of individual atoms. First the principal axes of the rotor are determined, and then the full system is rotated into a coordinate frame in which the z axis aligns with the rotor's main principal axis. The component of the initial angular momentum due to thermal excitation about the main principal axis Lwarmupz and initial polar moment of inertia about the main principal axis Jz are calculated. A correction velocity of
 | (6) |
The spin-down simulation uses the speed-adjusted G96 files as inputs and anchors the atoms in the stem. The simulation parameters are: 5000000 steps at 1 fs per step (5 ns total simulation time), xyz periodic boundary conditions with Verlet cutoff, 10 nm cutoff distance.
A.4 Energy and temperature drift for the bonded rotary joint
The classical Dulong-Petit51 law gives a fair approximation for the heat capacity of the molecular machine at 300 K, although the heat capacity of bulk diamond at this temperature is only one-fourth this value. The full system has N = 10345–514 = 9831 atoms that are free to move (514 atoms in the stem are frozen during the simulations). The total heat capacity of the system is then 3NkB = 4.1 × 10−19 J K−1. If all of the initial kinetic energy of the rotor is dissipated as heat, the expected temperature increase would be ΔT = (Jω02/2)/(3NkB) = 0.022 K for an initial angular velocity of ω0 = 100 rad ns−1. This upper bound on temperature increase is comparable to temperature drift due to numerical effects. Moreover, the five-nanosecond spin-down duration is too short to fully dissipate the rotor's kinetic energy, so the expected temperature increase is even smaller than this estimate. Thus there are no significant heating effects during the spin-down simulation.
Table 8 shows the energy drift for the 5 ns spin-down simulation with random seed = 42, calculated with the GROMACS utility g_energy_d.46 The other simulations have similar values. For the N = 9831 atoms free to move, 3NkBT = 1.2 × 10−16 J (or 7.4 × 104 kJ mol−1), about half the average energy in the table. This is due to GROMACS including an offset of about 9.46 × 104 kJ mol−1 from the minimum-potential configuration of the rotor.
| Quantity | Average | Standard deviation | Total drift | Units |
|---|---|---|---|---|
| Total energy | 1.7 × 105 | 12 | 3.3 | kJ mol−1 |
| Temperature | 303 | 1.8 | −0.01 | K |
During each of the five spin-down simulations, the total system energy remained nearly constant. Specifically, over the full five nanosecond spin-down, the largest increase in total system energy, as computed by the GROMACS analysis tool g_energy_d, of any of the simulations was 6.51 × 10−21 J (4 kJ mol−1). This drift is small enough to not effect the conclusions of this study.
A.5 Rotation potential for the bonded rotary joint
The 3-fold symmetry of the rotor leads to a corresponding symmetry in the rotation potential: V(θ + 120°) = V(θ). Using GROMACS, we measured the potential energy at various rotation angles in static configurations, i.e., at zero temperature. That is, for several angles θ between −60° and 60°, we evaluated the potential energy of each rotor atom according to the force field. The value of V(θ) is the sum of these individual potentials. Fig. 9 shows these measured values are very close to eqn (4). | ||
| Fig. 9 Potential energy of the rotor and housing structure as a function of rotor rotation angle within one of the three symmetric potential wells. The points are the values measured with GROMACS and the curve is eqn (4). The angle 0° is the configuration shown in Fig. 4. | ||
For the purposes of our analysis, the key quantity is the height of the potential barrier 2V0 compared to kBT. As a check on the size of the potential, we performed a spin test at zero temperature. This test started the rotor spinning and measured the change in angular momentum as the rotor moved from one potential well to the next. This case has no thermal motion so the rotor is affected only by the potential. The observed changes were consistent with eqn (4) and the value V0 in Table 3.
Fig. 10 shows the configuration of atoms at the rotary joint corresponding to the extremes of the potential. The staggered geometry is the minimum of the rotational potential, while the eclipsed geometry is the maximum. The staggered configuration corresponds to rotor rotation angles (see Fig. 4) of θ = 0, 120, 240 degrees. The eclipsed configuration corresponds to angles of θ = 60, 180, 300 degrees.
 | ||
| Fig. 10 Ball-and-stick and space-filling models of the molecular joint in two configurations: eclipsed and staggered on left and right, respectively. C2 spacers are indicated by the arrows. These geometries have molecular D3d and D3h symmetries,52 respectively. | ||
The potential arises mainly from non-bonding interactions between atoms of the rotor and the pyramid portions of the housing. The single C2 spacer is, then, a compromise between minimizing the rotational barrier and maintaining stiffness along the rotation axis.
A.6 Quantum effects
Molecular dynamics simulations treat atoms as classical particles with defined bonds and persistent connectivity. The rotary molecular machines considered in this paper do not form or break bonds, thereby maintaining atom connectivity throughout the simulation. This appendix considers the validity of the classical approximation to the atoms' motions.The thermal de Broglie wavelength of an object with mass m is  which is 2 × 10−3 nm for the rotor, much smaller than its size. Thus the rotor is well-approximated as a localized classical mass.
which is 2 × 10−3 nm for the rotor, much smaller than its size. Thus the rotor is well-approximated as a localized classical mass.
At the temperature considered here, typical angular momenta of the nanotube and rotor (see Tables 1 and 3) are about 1000 times larger than the quantum of angular momentum ℏ = 10−34 kg m2 s−1. Thus the classical approximation of continuous variation of angular momentum is reasonable. These observations indicate the rotor behaves as a classical object for operation at up to at least a few GHz.
Drag arises from spreading organized energy in rotor motion to the many other degrees of freedom of the molecular structure. Thus in addition to classical physics being sufficient to describe the rotor motion itself, molecular dynamics simulations assume classical behavior for other modes of motion. In particular, classical equipartition of energy among these modes only applies when thermal energy, kBT, is large compared to the quantum level spacing hf, where f is a mode's frequency.
The rotor speeds we consider (below 100 GHz) have kBT large compared to the quantum level spacing. Thus the classical behavior assumed in molecular dynamics simulations is a reasonable approximation for the rotor itself.
In addition to the rotary motion, the simulations include much higher frequency modes, particularly the hydrogen bond motions. Resolving these motions requires simulation time steps, 1 fs, much shorter than the time scales of the rotary behavior. For these high-speed motions, the quantum energy spacing is large compared to kBT. Thus these modes will have less energy than predicted by classical equipartition and GROMACS simulations. As a check on whether this suppression affects the rotor drag results, we repeated the simulations with the GROMAS H-bond constraint, which effectively keeps the hydrogen–carbon bonds in their ground state throughout the simulation. With this constraint, we find the same value for the rotor drag, within the reported confidence intervals, as in our original simulations. Nevertheless, this constraint significantly reduced the short-time fluctuations. With this reduction, the fluctuations closely match those expected from the stochastic process model (shown as the dashed curve in Fig. 6) for time differences as short as 1 ps. This correspondence suggests the large fluctuations at short times seen in Fig. 6 may be a simulation artifact, arising from ignoring quantum suppression of high frequency modes.
Quantum effects are more important at low temperature, where thermal energy is smaller. Furthermore, dissipative forces arising from quantum effects53 may be important at low temperatures.54
B Bonded rotary joint structure and stability
The housing is a faceted piece of crystalline diamond of nominal C2v symmetry52 at its extreme energy geometries, i.e., with the rotor adopting a staggered or eclipsed geometry with respect to the pyramids. The housing contains two mirror plane symmetries and one rotation axis along the mirror symmetry plane axis denoted in Fig. 13. Nanodiamonds with dimensions in the nanometer range are routinely synthesized by the detonation method.55 Detonation nanodiamonds typically have a highly ordered crystalline core covered by a layer of graphene-like patches or other functional groups that stabilize dangling bonds on the surface. This non-crystalline outerlayer is largely a result of the detonation nanodiamond formation and purification processes, and while it is sufficient for chemical stability, it is not necessary. DFT calculations have shown that nanometer-sized diamond structures are stable when the surface is passivated with hydrogen atoms.56,57 The hydrogen-passivated facets of the housing shown in Fig. 4 are predicted to be stable and free from surface reconstruction.The rotor itself is a hexagonally-shaped piece of hexagonal-lattice diamond, or lonsdaleite, as shown in Fig. 11. DFT calculations indicate that lonsdaleite would be stable,58 and it has been fabricated.59 As with the housing, the hydrogen-passivated facets of the rotor are predicted to be stable and free from surface reconstruction.
 | ||
| Fig. 11 The rotor consists of two mirrored and stacked {111} layers of hexagonal lattice diamond. The curved arrow indicates the direction of positive angular velocity: counterclockwise when viewed from above, i.e., from along the positive direction of the rotation axis. While the overall shape of the rotor is hexagonal, the atomic structure has threefold rather than sixfold symmetry, as can be seen by close inspection of the atomic structure along the rotor's edges. | ||
A ball-and-stick atomic model of the rotor and its two rotary joints is shown in Fig. 13. Each rotary joint is composed of four co-linear carbon atoms, similar to the organic molecule 2-butyne. The single bonds at the outside ends of the co-linear chain (e.g., bond C9442–C9443 and bond C9444–C9446) provide for the rotation. Such four-carbon chains have been shown experimentally to function as stable mechanical rotary joints.3 Of the four carbon atoms, the outside two (e.g., C9422 and C9446) are embedded within the framework of the rotating components – one within the housing and one in the rotor, while the inner two (e.g., C9443 and C9444) are free to rotate. We refer to the inner two carbon atoms as a “C2 spacer”.
Many proposed applications for molecular machines based on the rotary joint structure are intended to operate in vacuum (e.g. mechanical computation devices). Nevertheless, it is likely that the structure will be stable at room temperature in atmosphere. Several examples of atomically precise mechanical interactions are known to be stable in air, including the self-retraction of nested nanotubes,14 self-retraction of graphene flakes,60 and rotation of acetylenic axles.3 In addition, the hydrogen-passivated carbon {111} surface is chemically stable in air.61,62 These experimental results on similar systems strongly suggest that the rotary joint molecule would be stable under ambient conditions.
C Density functional theory methods
Density functional theory (DFT) calculations were used to determine partial charges on atoms around the rotary joint, and to check the rotational potential determined with GROMACS, as described in Appendix A.5.C.1 Rotor-pyramid assemblies
DFT is computationally demanding so was applied to only a subset of the full structure: the rotor and surrounding pyramids shown in Fig. 12, which we call a “rotor-pyramid assembly”. | ||
| Fig. 12 Rotor-pyramid assembly used for DFT calculations, shown as insets and in the context of the full structure. | ||
The rotor and housing structure conforms to C2v symmetry52 at certain rotor geometries. In the absence of the housing, the rotor-pyramid assembly has significantly higher symmetry at certain rotor geometries. Specifically, defined against the nearest-neighbor H⋯H interactions between the rotor and pyramids, two symmetry-constrained geometries bring the 12 total pairs of H⋯H interactions between rotor and pyramid eclipsed and staggered conformations with D3d or D3h symmetries, respectively. Fig. 10 shows these conformations. These symmetries allow calculations at significantly higher (i.e., more accurate) levels of theory than is possible for lower-symmetry structures.
C.2 DFT computations
Our DFT calculations were performed with Gaussian09, ver. D.01 (ref. 63) using program-option “tight” convergence criteria (force criterion RMS < 1.0 × 10−5, density matrix RMS < 1.0 × 10−8), “ultrafine” grid size (99 radial shells and 590 angular points per shell), and symmetry constraints as applicable to all structures. Optimized geometries for all structures and RESP charges are available on request.The B3LYP hybrid density functional64,65 was employed for all calculations, along with various calculations with the 6-31G(d,p),66 6-311G(2d,p), and 6-311G(2d,2p)67 Gaussian-type basis sets and D3-type Grimme Dispersion correction.68
RESP model69,70 charge calculations for the rotor-pyramid assemblies were produced at the B3LYP/6-31G(d,p) level of theory, employing the Merz–Singh–Koller scheme71,72 as a basis for Antechamber script input (program option “Pop=MK IOp(6/33=2,6/41=10,6/42=17)”).
C.3 Partial charges
We determined partial charges on atoms near the rotary joint by way of the AMBER Antechamber program.73Fig. 13 shows the resulting restrained electrostatic potential (RESP) charges. Values for these charges are comparable to published values used for similar molecular joints, e.g.ref. 35.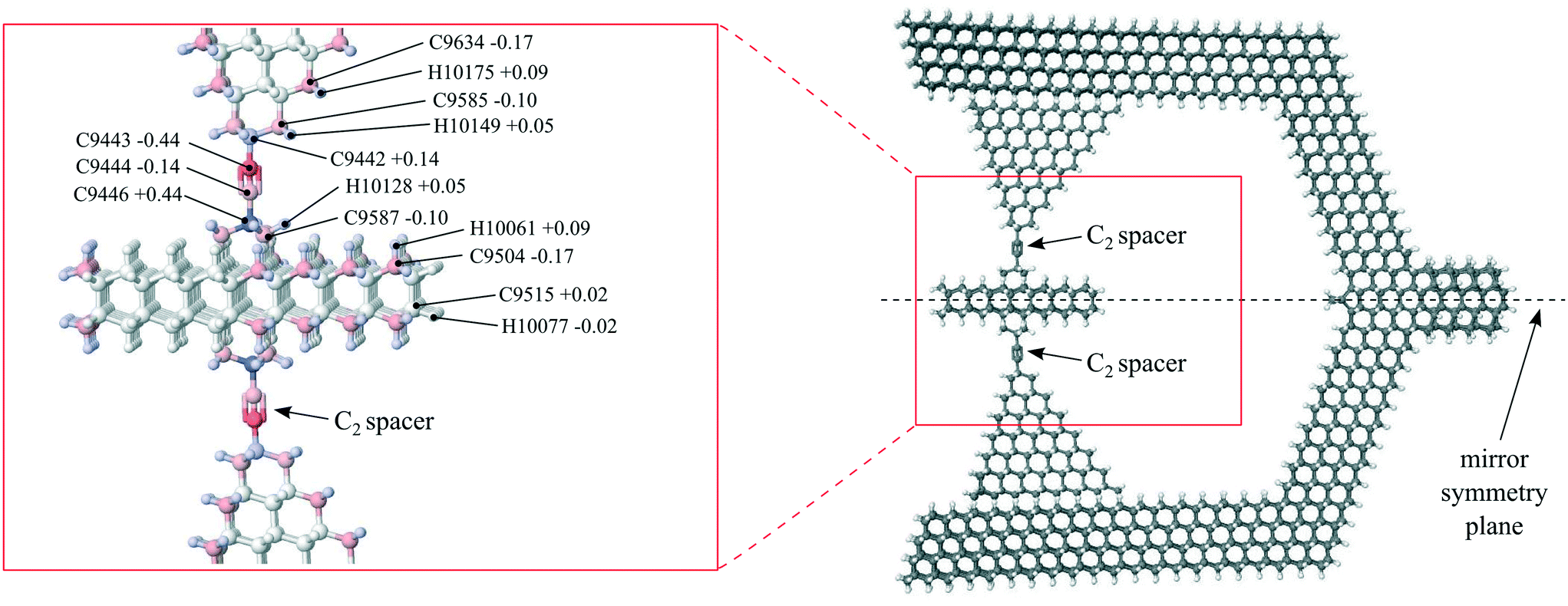 | ||
| Fig. 13 Rotor, rotary joints and symmetry plane. Colors indicate restrained electrostatic potential (RESP) charges on atoms in and around the molecular rotary joint. Charge ranges from −0.44 to +0.44 electron charges (red and blue, respectively). Some atoms in the structure are denoted by type (C or H) and index number, along with their partial charges. These index numbers are those used in the GROMACS structure files, described in Appendix A.3. | ||
C.4 Rotational potential barrier of rotor-pyramid systems
The key property of the rotational potential used for our drag estimation is that the barrier, 2V0, is relatively small compared to kBT as we found to be the case with the GROMACS evaluation of the potential given in Appendix 5. As a check on this relationship, we also evaluated the barrier with the more accurate DFT method.To reduce the computational cost of the DFT calculations, we restricted the evaluation to rotor-pyramid assemblies shown in Fig. 14. These assemblies have few enough atoms to allow feasible DFT calculations, while including most of the interactions between the rotor and the housing. We further reduced the cost by only evaluating the highly symmetric eclipsed and staggered conformations of the assemblies described in Appendix C.1. These conformations correspond to the extrema of the potential evaluated with GROMACS in Appendix A.5. Evaluating just these two cases is sufficient to compute the potential barrier, i.e., as the difference in energy between the eclipsed and staggered geometries of the joint.
 | ||
| Fig. 14 Groups of atoms around a rotary joint used for DFT evaluation of rotational potential: Pyr1 (adamantane), Pyr2 (pentamantane), Pyr3 (undecamantane). The bottom layer of atoms is the top of the rotor, and the upper layers are portions of the pyramid. | ||
Table 9 shows the values for the barrier, i.e., 2V0, for assemblies with various numbers of atoms in the pyramid. The calculated barrier between eclipsed and staggered geometries is well below kBT = 2.5 kJ mol−1 at the temperature we consider. The rotational barrier does not change noticeably when including more atoms from the pyramid in the calculation, indicating that the barrier is a local phenomenon primarily due to interactions among atoms close to the rotary joint. The small size of these computed energy barriers (0.05 kJ mol−1) are in excellent agreement with experimental measurements and theoretical calculations for the rotational barrier around the acetylene linkage in the symmetric 2-butyne molecule.74 Diphenylacetylene, a structurally similar molecule with closer H⋯H interactions than diadamantylacetylene and a preference for maintaining planarity, has a measured rotational barrier of 2.5 kJ mol−1 (calculated to be 3.3 kJ mol−1 at the B3LYP/6-311+G(d,p) level of theory [Table 2]).74 At the operational temperature we consider, the barrier for the C2 spacer motif is expected to be inconsequential to the operation of the full assembly.
| PyrN-C2–Rot–C2-PyrN | ||||
|---|---|---|---|---|
| DFT theory level | C10–C2–C10 | Pyr1 | Pyr2 | Pyr3 |
| B3LYP-GD3/6-311G(2d,2p) | 0.022 | — | — | — |
| B3LYP-GD3/6-31G(d,p) | 0.030 | 0.063 | −0.006 | 0.096 |
| B3LYP/6-31G(d,p) | 0.003 | 0.122 | 0.057 | 0.154 |
The barrier computed by GROMACS for the complete structure, 2V0 = 0.5 kJ mol−1 from Table 3, is larger than that determined by DFT for isolated rotor-pyramid structures (−0.006 to +0.154 kJ mol−1, see Table 9). Molecular mechanics force fields using RESP charges are of inherently lower accuracy than DFT methods.47 It is therefore not surprising that GROMACS overestimates the rotational barrier since the barrier predicted by DFT is extremely small. The larger barrier calculated by GROMACS may be due to interactions in the full structure modeled by GROMACS that are not present in the smaller structures modeled by DFT, such as strain induced by the housing or long range interactions between RESP charges. Regardless of the origin of the discrepancy, a barrier height of 0.5 kJ mol−1 is comparable to values reported for smaller molecules with similar rotational structure, e.g., 12 kJ mol−1 for internal rotation in ethane and 0.07 kJ mol−1 for 2-butyne.1,74
Acknowledgements
We thank Jeremy Barton, Robert A. Freitas Jr., Michael S. Marshall, Ralph C. Merkle, Thomas E. Moore and James Ryley for helpful discussions. We appreciate Eugene Cook's clarification of ref. 21.References
- K. S. Pitzer, Potential energies for rotation about single bonds, Discuss. Faraday Soc., 1951, 10, 66–73 RSC.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Artificial molecular rotors, Chem. Rev., 2005, 105, 1281–1376 CrossRef CAS PubMed.
- Y. Shirai, A. J. Osgood, Y. Zhao, K. F. Kelly and J. M. Tour, Directional control in thermally driven single-molecule nanocars, Nano Lett., 2005, 5, 2330–2334 CrossRef CAS PubMed.
- S. Ogi, T. Ikeda, R. Wakabayashi, S. Shinkai and M. Takeuchi, A bevel-gear-shaped rotor bearing a double-decker porphyrin complex, Chem. – Eur. J., 2010, 16, 8285–8290 CrossRef CAS PubMed.
- J. K. Gimzewski, C. Joachim, R. R. Schlittler, V. Langlais, H. Tang and I. Johannsen, Rotation of a single molecule within a supramolecular bearing, Science, 1998, 281, 531–533 CrossRef CAS PubMed.
- S. Pekker, et al. Rotor–stator molecular crystals of fullerenes with cubane, Nat. Mater., 2005, 4(10), 764–767 CrossRef CAS PubMed.
- U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S.-W. Hla, Controlled clockwise and anticlockwise rotational switching of a molecular motor, Nat. Nanotechnol., 2013, 8, 46–51 CrossRef CAS PubMed.
- Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun, C. Joachim, G. Rapenne and S.-W. Hla, Simultaneous and coordinated rotational switching of all molecular rotors in a network, Nat. Nanotechnol., 2016, 11, 706–712 CrossRef CAS PubMed.
- C. Manzano, W.-H. Soe, H. S. Wong, F. Ample, A. Gourdon, N. Chandrasekhar and C. Joachim, Step-by-step rotation of a molecule-gear mounted on an atomic-scale axis, Nat. Mater., 2009, 8, 576–579 CrossRef CAS PubMed.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Artificial molecular machines, Angew. Chem., Int. Ed., 2000, 39(19), 3348–3391 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Making molecular machines work, Nat. Nanotechnol., 2006, 1(1), 25–35 CrossRef CAS PubMed.
- B. Champin, P. Mobian and J.-P. Sauvage, Transition metal complexes as molecular machine prototypes, Chem. Soc. Rev., 2007, 36(2), 358–366 RSC.
- J. Cumings and A. Zettl, Low-friction nanoscale linear bearing realized from multiwall carbon nanotubes, Science, 2000, 289, 602–604 CrossRef CAS PubMed.
- R. Zhang, Z. Ning, Y. Zhang, Q. Zheng, Q. Chen, H. Xie, Q. Zhang, W. Qian and F. Wei, Superlubricity in centimetres-long double-walled carbon nanotubes under ambient conditions, Nat. Nanotechnol., 2013, 8, 912–916 CrossRef CAS PubMed.
- E. Koren, E. Lörtscher, C. Rawlings, A. W. Knoll and U. Duerig, Adhesion and friction in mesoscopic graphite contacts, Science, 2015, 348, 679–683 CrossRef CAS PubMed.
- D. V. Nicolau Jr., M. Lard, T. Korten, F. C. M. J. M. van Delft, M. Persson, E. Bengtsson, A. Månsson, S. Diez, H. Linke and D. V. Nicolau, Parallel computation with molecular-motor-propelled agents in nanofabricated networks, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2591–2596 CrossRef PubMed.
- K. E. Drexler, Nanosystems: Molecular Machinery, Manufacturing, and Computation, John Wiley & Sons, Inc., New York, 1992 Search PubMed.
- T. Cagin, A. Jaramillo-Botero, G. Gao and W. A. Goddard III, Molecular mechanics and molecular dynamics analysis of Drexler-Merkle gears and neon pump, Nanotechnology, 1998, 9(3), 143 CrossRef CAS.
- R. A. Freitas Jr., volume I: Basic Capabilities, Nanomedicine, Landes Bioscience, Georgetown, TX, 1999 Search PubMed.
- R. C. Merkle, R. A. Freitas Jr., T. Hogg, T. E. Moore, M. S. Moses and J. Ryley, Molecular mechanical computing systems. techreport 46, Institute for Molecular Manufacturing, Palo Alto, CA, 2016 Search PubMed.
- E. H. Cook, M. J. Buehler and Z. S. Spakovszky, Mechanism of friction in rotating carbon nanotube bearings, J. Mech. Phys. Solids, 2013, 61, 652–673 CrossRef CAS.
- E. S. Torres, S. Gonçalves, C. Scherer and M. Kiwi, Nanoscale sliding friction versus commensuration ratio: Molecular dynamics simulations, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 035434 CrossRef.
- D. W. Brenner, Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9458 CrossRef CAS.
- S. Zhang, W. K. Liu and R. S. Ruoff, Atomistic simulations of double-walled carbon nanotubes (DWCNTs) as rotational bearings, Nano Lett., 2004, 4(2), 293–297 CrossRef CAS.
- B. Kan, J. Ding, Z. Zhang, Y. Zhu and G. Cheng, Molecular dynamics simulations for rotational nanobearing formed by double walled carbon nanotube, J. Comput. Theor. Nanosci., 2014, 11(2), 324–330 CrossRef CAS.
- J. Servantie and P. Gaspard, Translational dynamics and friction in double-walled carbon nanotubes, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(12), 125428 CrossRef.
- J. Servantie and P. Gaspard, Rotational dynamics and friction in double-walled carbon nanotubes, Phys. Rev. Lett., 2006, 97, 186106 CrossRef CAS PubMed.
- Y. Quo, N. Karasawa and W. A. Goddard, Prediction of fullerene packing in C60 and C70 crystals, Nature, 1991, 351, 464–467 CrossRef.
- J. L. Rivera, C. McCabe and P. T. Cummings, The oscillatory damped behaviour of incommensurate double-walled carbon nanotubes, Nanotechnology, 2005, 16, 186 CrossRef CAS PubMed.
- A. M. Popov, I. V. Lebedeva, A. A. Knizhnik, Y. E. Lozovik and B. V. Potapkin, Molecular dynamics simulation of the self-retracting motion of a graphene flake, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84(24), 245437 CrossRef.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, Gromacs 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem. Theory Comput., 2008, 4(3), 435–447 CrossRef CAS PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, Dreiding: a generic force field for molecular simulations, J. Phys. Chem., 1990, 94(26), 8897–8909 CrossRef CAS.
- Y. Liu, F. Grey and Q. Zheng, The high-speed sliding friction of graphene and novel routes to persistent superlubricity, Sci. Rep., 2014, 4, 4875 CrossRef CAS PubMed.
- J. Han, A. Globus, R. Jaffe and G. Deardorff, Molecular dynamics simulations of carbon nanotube-based gears, Nanotechnology, 1997, 8, 95 CrossRef CAS.
- A. V. Akimov, A. V. Nemukhin, A. A. Moskovsky, A. B. Kolomeisky and J. M. Tour, Molecular dynamics of surface-moving thermally driven nanocars, J. Chem. Theory Comput., 2008, 4(4), 652–656 CrossRef CAS PubMed.
- Ch. Loppacher, M. Guggisberg, O. Pfeiffer, E. Meyer, M. Bammerlin, R. Lüthi, R. Schlittler, J. K. Gimzewski, H. Tang and C. Joachim, Direct determination of the energy required to operate a single molecule switch, Phys. Rev. Lett., 2003, 90, 066107 CrossRef PubMed.
- D. T. Gillespie, Markov Processes: An Introduction for Physical Scientists, Academic Press, Boston, 1992 Search PubMed.
- D. S. Lemons, An Introduction to Stochastic Processes in Physics, Johns Hopkins Univ. Press, Baltimore, 2002 Search PubMed.
- D. J. Higham, An algorithmic introduction to numerical simulation of stochastic differential equations, SIAM Rev., 2001, 43, 525–546 CrossRef.
- B. N. J. Persson and A. Nitzan, Linear sliding friction: on the origin of the microscopic friction for Xe on silver, Surf. Sci., 1996, 367, 261–275 CrossRef CAS.
- K. Huang, Statistical Mechanics, John Wiley & Sons, New York, 1963 Search PubMed.
- G. E. Uhlenbeck and L. S. Ornstein, On the theory of the Brownian motion, Phys. Rev., 1930, 36, 823–841 CrossRef CAS.
- J. Vacek and J. Michl, Molecular dynamics of a grid-mounted molecular dipolar rotor in a rotating electric field, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5481–5486 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: fast, flexible, and free, J. Comput. Chem., 2005, 26(16), 1701–1718 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- M. J. Abraham, D. van der Spoel, E. Lindahl and B. Hess, and the GROMACS Development Team, GROMACS User Manual version 5.0.4. www.gromacs.org, 2014 Search PubMed.
- J. Wang, P. Cieplak and P. A. Kollman, How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?, J. Comput. Chem., 2000, 21(12), 1049–1074 CrossRef CAS.
- Q. Lu, M. Arroyo and R. Huang, Elastic bending modulus of monolayer graphene, J. Phys. D: Appl. Phys., 2009, 42(10), 102002 CrossRef.
- E. H. Cook, Personal communication, 2016 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- N. W. Ashcroft and N. D. Mermin, Solid State Physics, Holt, Rinehart and Winston, New York, 1976 Search PubMed.
- K. J. Laidler and J. H. Meiser, Physical Chemistry, Benjamin Cummings Publishing, 1982 Search PubMed.
- A. I. Volokitin and B. N. J. Persson, Quantum friction, Phys. Rev. Lett., 2011, 106, 094502 CrossRef CAS PubMed.
- U. D. Jentschura, M. Janke and M. DeKieviet, Theory of noncontact friction for atom-surface interactions, Phys. Rev. A, 2016, 94, 022510 CrossRef.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, The properties and applications of nanodiamonds, Nat. Nanotechnol., 2011, 7, 11–23 CrossRef PubMed.
- A. S. Barnard, N. A. Marks, S. P. Russo and I. K. Snook, Hydrogen stabilization of (111) nanodiamond, Technical report, DTIC Document, 2003 Search PubMed.
- D. Tarasov, E. Izotova, D. Alisheva, N. Akberova and R. A. Freitas, Jr, Structural stability of clean and passivated nanodiamonds having ledge, step, or corner features, J. Comput. Theor. Nanosci., 2012, 9, 144–158 CrossRef CAS.
- P. Németh, L. A. J. Garvie, T. Aoki, N. Dubrovinskaia, L. Dubrovinsky and P. R. Buseck, Lonsdaleite is faulted and twinned cubic diamond and does not exist as a discrete material, Nat. Commun., 2014, 5, 5447 CrossRef PubMed.
- T. B. Shiell, D. G. McCulloch, J. E. Bradby, B. Haberl, R. Boehler and D. R. McKenzie, Nanocrystalline hexagonal diamond formed from glassy carbon, Sci. Rep., 2016, 6, 37232 CrossRef CAS PubMed.
- Q. Zheng, B. Jiang, S. Liu, Y. Weng, L. Lu, Q. Xue, J. Zhu, Q. Jiang, S. Wang and L. Peng, Self-retracting motion of graphite microflakes, Phys. Rev. Lett., 2008, 100(6), 067205 CrossRef PubMed.
- P. G. Lurie and J. M. Wilson, The diamond surface: I. the structure of the clean surface and the interaction with gases and metals, Surf. Sci., 1977, 65(2), 453–475 CrossRef CAS.
- O. A. Williams, J. Hees, C. Dieker, W. Jäger, L. Kirste and C. E. Nebel, Size-dependent reactivity of diamond nanoparticles, ACS Nano, 2010, 4(8), 4824–4830 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09 Revision E.01, Gaussian Inc. Wallingford CT, 2009 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28(3), 213–222 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. Stephen Binkley, Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets, J. Chem. Phys., 1984, 80(7), 3265–3269 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97(40), 10269–10280 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly and P. A. Kollmann, Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation, J. Am. Chem. Soc., 1993, 115(21), 9620–9631 CrossRef CAS.
- U. C. Singh and P. A. Kollman, An approach to computing electrostatic charges for molecules, J. Comput. Chem., 1984, 5(2), 129–145 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, Atomic charges derived from semiempirical methods, J. Comput. Chem., 1990, 11(4), 431–439 CrossRef CAS.
- D. A. Case, et al., AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- S. Toyota, Rotational isomerism involving acetylene carbon, Chem. Rev., 2010, 110(9), 5398–5424 CrossRef CAS PubMed.
Footnote |
| † The corrected linear relation between force and speed49 corresponds to [Fig. 9]21 rather than [eqn (28)].21 |
| This journal is © The Royal Society of Chemistry 2017 |