
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the boundaries of direct detection and characterization of labile isomers – a case study of copper(II)–dipeptide systems†
Eszter N.
Tóth
 ab,
Nóra V.
May
c,
Antal
Rockenbauer
de,
Gábor
Peintler
f and
Béla
Gyurcsik
*a
ab,
Nóra V.
May
c,
Antal
Rockenbauer
de,
Gábor
Peintler
f and
Béla
Gyurcsik
*a
aDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: gyurcsik@chem.u-szeged.hu; Fax: +36-62-544-340; Tel: +36-62-544-335
bPhD Program in Human Biology, School of Integrative and Global Majors, University of Tsukuba, Japan
cResearch Centre for Natural Sciences, Hungarian Academy of Sciences, 1117 Budapest, Magyar tudósok körútja 2, Hungary
dInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, 1117 Budapest, Magyar tudósok körútja 2, Hungary
eDepartment of Physics, Budapest University of Technology and Economics and MTA-BME Lendület Spintronics Research Group (PROSPIN), P.O. Box 91, H-1521 Budapest, Hungary
fDepartment of Physical Chemistry and Material Sciences, University of Szeged, Aradi Vértanuk tere 1, H-6720 Szeged, Hungary
First published on 22nd May 2017
Abstract
The investigation of the linkage isomers of biologically essential and kinetically labile metal complexes in aqueous solutions poses a challenge, as these microspecies cannot be separately studied. Therefore, derivatives are commonly used to initially determine the stability or spectral characteristics of at least one of the isomers. Here we directly detect the isomers, describe the metal ion coordination sphere, speciation and thermodynamic parameters by a synergistic application of temperature dependent EPR and CD spectroscopic measurements in copper(II)–dipeptide systems including His-Gly and His-Ala ligands. The ΔH = (−23 ± 4) kJ mol−1 value of the standard enthalpy change corresponding to the peptide-type to histamine-type isomerisation equilibrium of the [CuL]+ complex was corroborated by several techniques. The preferential coordination of the side-chains was observed at lower temperatures, whereas, metal-binding of the backbone atoms became favourable upon increasing temperature. This study exemplifies the necessity of using temperature dependent multiple methodologies for a reliable description of similar systems for upstream applications.
Introduction
Similarly to the understanding of basic biological and chemical processes, the design of a new drug or a catalyst also demands the characterization of isomeric species. Metal complexes exhibit structural (e.g. linkage, coordination isomers, or complexes of ligand isomers), and stereo (such as geometrical, optical and conformational) isomers. More sophisticated induced geometrical,1mer/fac,2,3 electronic,4 topological5–7 isomers or rotamers,8–10 as well as the controversial bond-stretch or distortion isomerism11–14 are also frequently discussed in the literature. The isomers exhibit altered physico-chemical properties that affect their reactivity and structure. The microenvironment around the metal ion affects the catalytic activity and selectivity of metalloenzymes or their small molecular model compounds. It determines their ability to recognize substrates, provide reactive sites by transactivation, electrostatically activate the substrate, generate nucleophile agents in hydrolytic reactions, or stabilize either the transition state or the products.15–26 The binding mode of metal ions greatly influences the conformation of ligands27,28 and vice versa. The change in the protein structure induced by metal binding may lead to functional29–35 or pathogenic36–44 complexes. The recognition of the importance of coordination isomers has opened a new era in metallopharmaceutical design.45,46 Drug delivery,47 bioprocessing,48–50 drug activity,51–53 drug selectivity26,54 and resistance55–59 can be optimized by selecting an isomeric drug or a prodrug with the best properties.All metal complexes formed with multidentate ligands may exhibit linkage isomers.60–63 By means of a “two-dimensional” approach to electron paramagnetic resonance (EPR) evaluation,64 we have previously suggested the presence of linkage isomers in a number of copper(II) complex systems.65–69 This method defines EPR intensity as a function of the magnetic field, pH and the concentration of the metal complex. It furnishes the pertinent formation constants in the mass-balance equations together with the individual EPR parameters for all species simultaneously. Linkage isomers have also been studied by UV-Vis absorption,62,70 circular dichroism (CD),71–73 nuclear magnetic resonance (NMR)74 spectroscopic investigations or applying multiple methods.75 These isomers of kinetically labile metal complexes cannot be studied separately. The spectrum decomposition to obtain individual thermodynamic and spectral data is practically impossible due to the intrinsic limitation by the constant concentration ratios of the microspecies during the pH-dependent series of measurements. The most commonly applied method to explore such systems is the traditional way of selecting appropriate derivatives (mutants) to obtain basis limiting parameters for one or more isomers.
On the other hand, the ratio of the isomeric species can be varied by e.g. a change of temperature76–78 or photoirradiation.79–81 The conformational changes of the heat shock proteins exemplify such temperature dependent isomeric switches with a biological significance.82,83 There is, however, still no comprehensive study on solution microequilibria of labile coordination linkage isomers proving their presence by temperature-dependent multiple methods.
As a simple case we selected the His-Gly![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu(II) = 1:1 system, where the predominant [CuL]+ complex at pH ∼ 5.0 was suggested to exhibit two protonation isomers: {NH2,Nim} histamine-type (isomer h) and {NH2,N−,COO−} peptide-type (isomer p) complexes (Fig. 1).84 In this work we made an attempt to directly distinguish these two complex isomers by temperature dependent series of measurements. Their thermodynamic and spectral properties were determined by EPR, conventional and synchrotron radiation (SR)CD spectroscopy and differential scanning calorimetry (DSC) in the systems containing copper(II) and His-Gly or His-Ala dipeptides.
:Cu(II) = 1:1 system, where the predominant [CuL]+ complex at pH ∼ 5.0 was suggested to exhibit two protonation isomers: {NH2,Nim} histamine-type (isomer h) and {NH2,N−,COO−} peptide-type (isomer p) complexes (Fig. 1).84 In this work we made an attempt to directly distinguish these two complex isomers by temperature dependent series of measurements. Their thermodynamic and spectral properties were determined by EPR, conventional and synchrotron radiation (SR)CD spectroscopy and differential scanning calorimetry (DSC) in the systems containing copper(II) and His-Gly or His-Ala dipeptides.
 | ||
| Fig. 1 Peptide-type (left) and histamine-type (right) coordination in the linkage isomers of [CuL]+ complex of His-Gly (L). | ||
Experimental
Materials
Sodium and copper(II) perchlorate were from Fluka and Reachim. The latter solution was standardized complexometrically. pH-Metric titrations were performed by NaOH (Fluka) standard solution. The dipeptides were Sigma-Aldrich products. All the materials were used without further purification.pH-Metric measurements
The protonation and coordination equilibria were investigated by potentiometric titrations in aqueous solution (I = 0.1 M NaClO4, T = 298.2 ± 0.1 K and 328.2 ± 0.1 K) under an argon atmosphere, using an automatic titration set including a PC controlled Dosimat 665 (Metrohm) autoburette and an Orion 710A precision digital pH-meter. The Metrohm Micro pH glass electrode (125 mm) was calibrated in aqueous solution via the modified Nernst equation:where JH and JOH are fitted parameters in acidic and alkaline media, respectively for taking into account the deviations from the Nernst equation due to the liquid junction potential and either the acidic or the alkaline errors of the glass electrode,85 pKw = 13.75 is the negative logarithm of the autoprotolysis constant of water at 298.2 K,86 while the corresponding value at 328.2 K is 13.10.87 The parameters were calculated by the non-linear least squares method. The complex formation was characterized by the general equilibrium process:

where L− denotes the non-protonated ligand molecule. The corresponding formation constants (β(CupLqHr(2p−q+r)+) ≡ βpqr) were calculated using the PSEQUAD computer program.88 The protonation constants were determined from three independent titrations (70–80 data points per titration) with the peptide concentration of (1.0–3.0) × 10−3 M. The complex formation constants were evaluated from three independent titrations (50–90 data points per titration) in the case of copper(II) containing systems, respectively. The applied ratio of the ligands and metal ions was 1
 :1 with the metal ion concentration of (1.0–3.0) × 10−3 M at the beginning of the titrations. The temperature range for the pH-metric titrations did not fully cover the range used for the EPR or CD experiments, 283.2–328.2 K for the former and 283.2–333.2 K for the latter. However, given the minimal change observed for the species distribution in the pH-potentiometric titrations over the 298.2–328.2 K range (Fig. S1†) it is safe to assume that there would be no significant change in the unexamined region.
:1 with the metal ion concentration of (1.0–3.0) × 10−3 M at the beginning of the titrations. The temperature range for the pH-metric titrations did not fully cover the range used for the EPR or CD experiments, 283.2–328.2 K for the former and 283.2–333.2 K for the latter. However, given the minimal change observed for the species distribution in the pH-potentiometric titrations over the 298.2–328.2 K range (Fig. S1†) it is safe to assume that there would be no significant change in the unexamined region.
Continuous wave-EPR measurements
EPR spectra were recorded with a Bruker EleXsys X-band spectrometer (microwave frequency 9.81 GHz, microwave power 10 mW, modulation amplitude 5 G, modulation frequency 100 kHz). The [CuL]+ complex of His-Gly and for comparison the [CuLH−1] complex of Gly-Gly were obtained as major species in equimolar aqueous solutions of the components at 5 mM copper(II) and ligand concentration, the pH of which were adjusted to 5.05 and 7.12, respectively (I = 0.1 M NaClO4). 100 μl solution of each complex was transferred to a quartz EPR tube and the spectra were recorded between 283.2–328.2 K in steps of 5.0 K and at 77 K, respectively. The temperature was adjusted by a liquid nitrogen based temperature controller system with an accuracy of ±0.1 K.Circular dichroism measurements
The Vis-CD spectra were recorded on a Jasco J-815 spectropolarimeter in the wavelength interval from 300 to 800 nm with 0.5 nm increments in a cell with 1 cm optical path length. The synchrotron radiation CD (SRCD) spectra of the free ligand and the metal complexes were recorded at the SRCD facility at the UV1 beamline89 on the storage ring ASTRID at the Institute for Storage Ring Facilities (ISA), University of Aarhus, Denmark. The light passed from the synchrotron through a CaF2 window into a CD instrument purged constantly with nitrogen. In the instrument an initial linear polarization is converted into alternating circularly polarized light with a CaF2 photo-elastic modulator (PEM, model I/CF50, Hinds, USA) operating at 50 kHz. The signal is detected with a photo-multiplier tube (PMT, 9402B, Electron Tubes, UK) using a lock-in amplifier. All spectra were recorded with 1 nm steps and a dwell time of 2 s per step, using 0.1 mm quartz cells (SUPRASIL, Hellma GmbH, Germany). The spectra were recorded in the wavelength range of 170–330 nm. Camphor-sulfonic acid served as a calibration material for the instruments. The substances were dissolved in distilled water and the pH was adjusted by HClO4 and NaOH solutions, for final metal ion and peptide concentration of ∼5.0 × 10−3 M (I = 0.1 M NaClO4). The temperature was controlled by a built in Peltier unit. From raw spectra the water baseline was subtracted.Microcalorimetric measurements
The calorimetric data were recorded on a Mettler-Toledo 822e differential scanning calorimeter. Known quantities of the samples (∼10 μl) were placed in the sample holder (∼120 μl medium pressure stein steel crucibles sealed with the O-ring). The same amount of the reference material was prepared in the same way. The difference of the amount of heat was recorded (i.e. DSC scanning) as the function of temperature in the range of 303.2–373.2 K with the heating rate of 1.0 K min−1. In the samples the concentrations of Cu(ClO4)2 and NaClO4 were 0.01958 M and 0.1990 M, respectively, while the concentration of His-Ala was 0.02283 M. The reference samples contained only NaClO4 with the concentrations of 0.2186 M or 0.1990 M.Data evaluation
The series of temperature dependent EPR spectra were simulated by the “two-dimensional” simulation program EPRTEMP.90 The program fits the isotropic EPR parameters of the components derived for 273.2 K (g0, A0 copper hyperfine coupling, aN nitrogen superhyperfine couplings) and the relaxation parameters α, β, γ, which define the linewidths in the equation σMI = α + βMI + γMI2, where MI denotes the magnetic quantum number of copper nucleus. Since natural copper(II) chloride was used for the measurements, the calculated spectra were composed from the spectra of 63Cu and 65Cu by taking into account the ratio of hyperfine couplings according to the magnetic moments and the respective components were weighed by their natural abundances. The copper (referring to 63Cu) and nitrogen hyperfine couplings and relaxation parameters are given in field dimensions (gauss [G] = 10−4 T). The EPR parameters at a given temperature Q(T) can be calculated by a power expansion containing no more than four terms:where T0 = 273.2 K, Q0 is the value at 273.2 K and Qn (n = 1–3), the coefficients that can be fitted to describe the temperature dependence of Q(T). The thermodynamic parameters ΔS and ΔH were also fitted. The details of the fitting used for the investigated systems are described in the EPR Measurements section.

For each spectrum, the noise-corrected regression parameter (Rj for the jth spectrum) is derived from the average square deviation (SQD) between the experimental and the calculated intensities. For the series of spectra, the fit is characterized by the overall regression coefficient R, calculated from the overall average SQD. The R value was 0.9965 for the series of His-Gly and 0.9970 for the Gly-Gly temperature dependent EPR spectra. The details of the statistical analysis were published previously.64,67,90
The isomer ratios were calculated by the van't Hoff relationship:  . In the equation, K is the isomerization stability coefficient at the temperature given, i.e., K equals to the ratio of the concentrations of histamine and the peptide type complex species. ΔH and ΔS are the reaction enthalpy and entropy for the formation of the histamine-type complex, respectively:
. In the equation, K is the isomerization stability coefficient at the temperature given, i.e., K equals to the ratio of the concentrations of histamine and the peptide type complex species. ΔH and ΔS are the reaction enthalpy and entropy for the formation of the histamine-type complex, respectively:
| isomer p ⇌ isomer h. |
The same van't Hoff relationship combined with the mass-balance equations was applied for the evaluation of the CD, SRCD and calorimetric data. The Zita program package was applied for the latter calculations.91
Matrix rank analysis was performed by the MRA program.92
Results and discussion
pH-Metric titrations
In principle, the shift in microscopic equilibria between the coordination isomers can be monitored by temperature dependent spectroscopic measurements. The calculation of the individual spectra of the expected isomers from these data is only straightforward, if the distribution of the macrospecies is essentially unaffected during these experiments. This was verified by pH-potentiometric titrations of the equimolar copper(II)–His-Gly system at 298.2 and 328.2 K. The evaluation of the titration curves demonstrated that the [CuL]+ complex was dominant at pH ∼ 5.0 at both temperatures (Fig. S1†).EPR measurements
The chemical exchange between the isomeric species is slow on the EPR time-scale and thus the EPR spectra of microspecies can be fitted by the superimposition of isotropic spectra of the respective species. The averaged EPR spectroscopic parameters (g0, A0, copper hyperfine coupling and α, β, γ relaxation parameters) can be used to describe the individual spectrum at a certain temperature. The temperature dependence of these parameters can be taken into account by a power expansion (see the Data evaluation section). A reference spectral series were first measured to determine the minimum required number of the coefficients necessary for the description of the temperature-dependent EPR spectra. The [CuLH−1] complex of Gly-Gly dipeptide was selected with {NH2,N−,COO−} peptide type coordination being the predominant species at pH ∼ 7.67 This type of coordination is expected for the isomer p of the His-Gly [CuL]+ complex, too. Simulations and parameter fitting procedures revealed that the linear dependence of g0, A0 and α, β parameters with fixed values for the nitrogen superhyperfine couplings and γ relaxation parameter was sufficient for the description of the temperature dependent spectra (Fig. S2†). The calculated parameters are shown in Table 1.| L = GG | L = HG | |||||
|---|---|---|---|---|---|---|
| Isomer p | Isomer h | |||||
| Q 0 | Q 1/° | Q 0 | Q 1/° | Q 0 | Q 1/° | |
| g 0 | 2.1202(1) | −5.1(5) × 10−4 | 2.115(1) | 1.3(5) × 10−4 | 2.1437(5) | 6.8(1) × 10−5 |
| A 0/G | 67.2(2) | −0.007(6) | 63(1) | 0.04(3) | 60.4(3) | −0.053(3) |
| a N1/G | 14.1(2) | — | 14(1) | — | 12.5(6) | — |
| a N2/G | 11.7(2) | — | 14(1) | — | 11.5(6) | — |
| α/G | 14.7(2) | 0.153(1) | 21(2) | 0.24(8) | 34.1(9) | −0.08(2) |
| β/G | −6.7(1) | 0.072(2) | −10(1) | 0.06(4) | −16.6(4) | 0.15(1) |
| γ/G | 0.8(2) | — | 2(1) | — | 1.9(2) | — |
The linear temperature dependence of the above parameters was taken into account in the simulation of the superimposed EPR spectra for both components of the His-Gly [CuL]+ complex. Our temperature-dependent pH-potentiometric titrations showed that the [CuL]+ complex was dominant at pH ∼ 5.0, with only a small percent of the [CuLH]2+ and [Cu2L2H−2] complexes present. The EPR spectrum of [CuLH]2+ is very similar to the spectrum of the histamine-type [CuL]+ isomer, and the spectrum of the dimer [Cu2L2H−2] complex is a broad singlet, therefore, the new well resolved component with EPR parameters close to those of Gly-Gly [CuLH−1] could only be considered as a peptide-type linkage isomer of [CuL]+. The temperature dependent spectral series were then fitted by taking into account the two microspecies of the [CuL]+ complex (Table 1 and Fig. 2). The coordination modes in Gly-Gly [CuLH−1] and His-Gly [CuL]+ peptide-type complexes are the same, but slight differences in their EPR parameters were observed owing to the presence of the His side-chain in the latter complex, which modifies the electron density at the donor atoms of the ligand and the rotation correlation time, as well. By this approach we could obtain more reliable parameters for the minor species as compared to our earlier results (g0 = 2.1323, A0 = 54.5 G, aN1 = 14.0 G and aN2 = 13.0 G).84 The EPR parameters of the predominant histamine-type isomer are in good accordance with previously determined data,84 as well as, with those obtained for the histamine [CuL]2+ complex (g0 = 2.1436, A0 = 63.4 G, aN1 = 12.7 G and aN2 = 11.4 G)93 supporting the hypothesized type of coordination in this isomer. This model resulted in a good agreement between the measured and simulated EPR spectra (Fig. 2c).
 | ||
| Fig. 2 Temperature dependent EPR spectra calculated for (a) peptide- and (b) histamine-type isomer of the His-Gly [CuL]+ complex at pH = 5.05. Spectra were calculated at 283.2 K (blue), at 328.2 K (red) and in between (black). (c) Comparison of measured (black) and simulated (red) EPR spectra. A vertical line helps to follow the appearance of the sharpest line of the peptide-type isomer. | ||
At first sight, the temperature dependence of the parameters provided surprising results. The higher the temperature the lower the bond strength and thus, the increase of the g0 and decrease of the A0 values are expected. Indeed, for g0 this was the case for the two His-Gly complex isomers but not for the [CuLH−1] complex of Gly-Gly. We may resolve this apparent contradiction by the analysis of the equation  (where α is the molecular orbital coefficient, n is the quantum mechanical coefficient, λ is the spin–orbit coupling and ΔE is the energy difference between the ground state and the excited states), g0 depends on the ratio of the spin–orbit coupling and the separation of the energy levels. We may assume that at elevated temperature the decrease of the spin–orbit coupling, as the consequence of more diffuse molecular orbitals, can exceed the effect of the decrease in the separation of energy levels. Accordingly, the g0 value may decrease with an increase in temperature. The temperature dependence of the A0 value is in addition, connected with the Fermi contact term related to the 3d–4s orbital mixing.94 For the “histamine-type” isomer of the Hys-Gly complex A0 decreases but for the “peptide-type” species A0 increases upon increase of the temperature. We assume that a slightly different symmetry distortion of the molecular orbitals may influence the 3d–4s orbital mixing in a different manner.
(where α is the molecular orbital coefficient, n is the quantum mechanical coefficient, λ is the spin–orbit coupling and ΔE is the energy difference between the ground state and the excited states), g0 depends on the ratio of the spin–orbit coupling and the separation of the energy levels. We may assume that at elevated temperature the decrease of the spin–orbit coupling, as the consequence of more diffuse molecular orbitals, can exceed the effect of the decrease in the separation of energy levels. Accordingly, the g0 value may decrease with an increase in temperature. The temperature dependence of the A0 value is in addition, connected with the Fermi contact term related to the 3d–4s orbital mixing.94 For the “histamine-type” isomer of the Hys-Gly complex A0 decreases but for the “peptide-type” species A0 increases upon increase of the temperature. We assume that a slightly different symmetry distortion of the molecular orbitals may influence the 3d–4s orbital mixing in a different manner.
From the temperature dependence of the isomer ratio (see Table S1† and Fig. 3) the standard enthalpy and entropy changes of the isomer equilibrium could be determined as ΔH = −21.4 kJ mol−1 and ΔS = −62 J mol−1 K−1 with the standard errors of 0.4 kJ mol−1 and 1 J mol−1 K−1, respectively. Deviations could not be correctly calculated due to the unknown correlation values. The thermodynamic data show that the higher temperature is favourable for peptide–amide deprotonation, while the side-chain coordination becomes predominant at low temperature.
 | ||
| Fig. 3 The temperature dependence of the ratio of the histamine and peptide type isomers of the [CuL]+ complex of His-Gly, according to the calculations based on the change of the EPR spectra. | ||
The EPR spectrum of the His-Gly [CuL]+ complex recorded in frozen solution at 77 K showed characteristic features of a dimer copper(II) complex formation (Fig. S3†), but distinct from that of the [Cu2L2H−2] complex.84 This indicates that the macroscopic equilibrium shifts under such conditions, and the anisotropic EPR spectrum does not provide further insight into the equilibrium of the histamine- and peptide-type linkage isomers of the [CuL]+ complex.
CD measurements in the wavelength range of the visible light
Our initial CD measurements in the equimolar copper(II) His-Gly system yielded too small a deviation between the spectra recorded at 293.2 and 333.2 K under conditions of the isomer formation (Fig. S4†). The reason for this is that the C-terminal Gly residue of the ligand is optically inactive thus, the transmission of the ligand optical activity to the copper(II) chromophore is ineffective.95,96 As the quantitative evaluation of these spectra would be ambiguous, we applied the His-Ala dipeptide with a chiral amino acid at its C-terminus in further CD investigations. The methyl side chain of alanine does not influence the coordination properties towards the copper(II) ion. Thus, the equilibrium constants, i.e. the species distribution, are essentially the same as in the copper(II)–His-Gly system.97–99 The CD pattern of this system changed substantially within the 283.2–333.2 K temperature range (Fig. 4a). It can also be seen that the spectrum recorded at a low temperature is similar to those of the copper(II) complexes with histamine-type, i.e. {NH2,Nim} coordination96 in agreement with the EPR measurements. At the same time, on increasing the temperature the negative band around 650 nm becomes more pronounced. This band is characteristic of the copper(II) complexes with peptide-type {NH2,N−,COO−} coordination.95 Thus, the ratio of the two isomers clearly changes: the complexation of the chelating backbone atoms including the deprotonated amide nitrogen is favoured at elevated temperature against the side-chain type coordination. | ||
| Fig. 4 Temperature dependent CD spectra recorded in the 300–800 nm wavelength range for equimolar systems of copper(II) and (a) His-Ala with the [CuL]+ complex dominating at pH = 5.00, (b) Gly-Leu with the peptide-type [CuLH−1] complex dominating at pH = 7.00 in the equilibrium system. The copper(II) ion concentration was ∼5.0 × 10−3 M in a cuvette with 1 cm optical path length. | ||
The Gly-Leu dipeptide containing a chiral amino acid with non-coordinating side-chain at the C-terminus was selected for comparison. The CD spectrum of its copper(II) complex was recorded at pH 7.0, where the {NH2,N−,COO−} coordinated peptide-type [CuLH−1] species dominates in the solution.97,99 The temperature change in the same range as for the His-Ala complex did not significantly affect the CD pattern (Fig. 4b). This suggests that the temperature change, and in parallel the eventual shift of the conformational population of the bound ligand molecule had no or minor influence on the Cotton effect obtained in the wavelength range that is characteristic of d–d transitions. This is also reflected by the success of the matrix rank analysis of the temperature dependent CD spectral series of the copper(II)–His-Ala system. The MRA program92 was able to fit the data by hypothesizing the presence of only two species in the solution (Fig. S5†).
Together with the pH metric results on the validity of macrospeciation, these observations support that the change of the CD pattern in the copper(II)–His-Ala system is indeed due to the shift of the microequilibria upon the change of the temperature. Thus, the CD data obtained in the copper(II)–His-Ala system as a function of the temperature were used to fit the thermodynamic data by non-linear parameter estimation. For the fitting procedure, the previously mentioned van't Hoff equation, the Beer–Lambert law and the mass-balance equations were applied. Fitting the CD data for the isomerization of the [CuL]+ complex resulted in a good agreement between the measured and simulated spectra (Fig. S6†). Together with ΔH and ΔS, the individual molar ellipticities of the two isomers were also obtained (Fig. 5).
 | ||
| Fig. 5 The calculated molar CD spectra for the [CuL]+ isomers of His-Ala. The spectrum of the histamine-type isomer is in blue, while that of the peptide-type isomer is in red. | ||
The molar spectrum of one of the isomers corresponds well to those published previously for monohistamine-type copper(II) complexes (λmax = 746–752 nm, Δε = 0.09–0.11 M−1 cm−1).96 The spectrum of the other isomer resembles the [CuLH−1] complexes of Cu(II) Gly-Ala dipeptide (λmin = 650 nm, Δε = −0.35 M−1 cm−1)100 and the Ala-rich tetrapeptides (λmin = 685 nm, −Δε = 0.24–0.32 M−1 cm−1)101 with {NH2,N−,CO} type of coordination, as well as that of the Gly-Leu complex, although with smaller intensity. Thus, the calculated spectra can be assigned to the “histamine-type” and “peptide-type” isomers of the [CuL]+ complex of His-Ala, respectively. The calculated ΔH and ΔS values for the formation of isomer h were (−22.9 ± 3.5) kJ mol−1 and (−65 ± 11) J mol−1 K−1, respectively in accordance with the EPR result for the copper(II)–His-Gly system. The relatively high deviation values arose from the fact that thermodynamic parameters cannot inherently be determined more accurately. The obvious explanation of this statement is shown in Fig. 6.
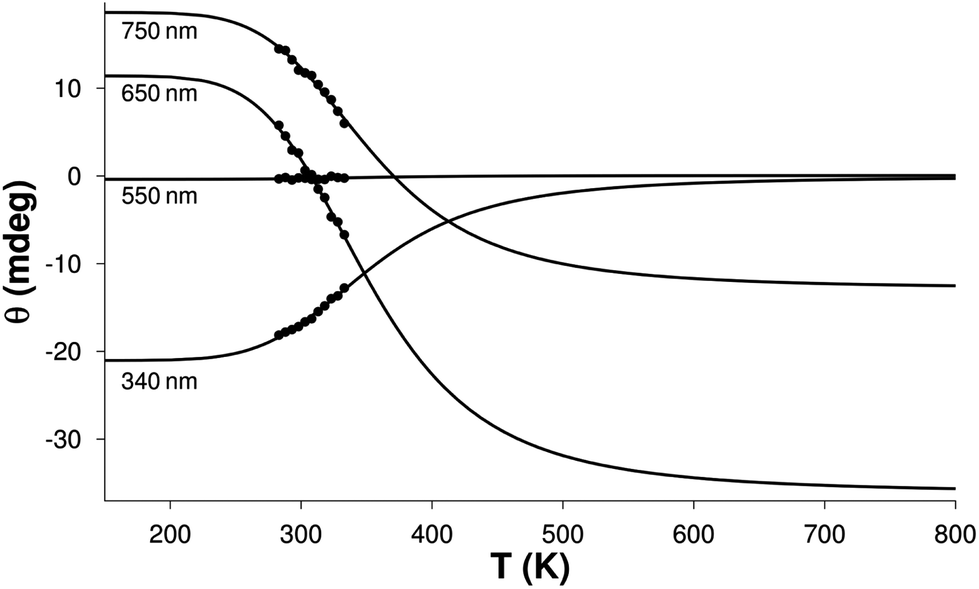 | ||
| Fig. 6 The measured (filled circles) and the calculated (solid lines) ellipticity values of the [CuL]+ complex of His-Ala as a function of temperature at four different wavelengths. The copper(II) ion concentration was ∼5.0 × 10−3 M in a cuvette with 1 cm optical path length. | ||
The temperature range of the detectable ellipticity change is about 300 K. Accurate determination of the influencing parameters would require the investigation of this whole temperature range to get sufficient information about the shape of the θ(T) curves. However, only a small part of this range is experimentally achievable in aqueous solutions, leading to large correlations between the parameters. Nevertheless, the above values are in agreement with those calculated from the EPR data.
SRCD measurements
Previous SRCD measurements showed that solutions of small peptides show a characteristic and pH sensitive CD pattern in the far UV wavelength region.102 This observation can be explained in terms of the unique peptide conformation population in each of the different protonation states. The metal ion coordination also greatly affects the spectra through emerging charge transfer transitions and by changing the ligand conformation population. Therefore, we expected that the change in the coordination type in the isomeric equilibrium will also have a significant effect on the spectra, and carried out temperature dependent SRCD measurements with His-Gly, His-Ala and Gly-Leu as well as with their copper(II)-complexes in the 283.2–333.2 K range, at pH 5.07 for the first two dipeptides and at 7.00 for the latter, respectively. The series of the SRCD spectra for His-Gly and its [CuL]+ complex are depicted in Fig. 7, and those measured for His-Ala and Gly-Leu are included in the ESI (Fig. S7†). | ||
| Fig. 7 Temperature dependent SRCD spectra recorded (a) for the His-Gly dipeptide and (b) for its [CuL]+ complex at pH = 5.00 in the equilibrium system. The optical path length was 0.1 mm. | ||
While the spectra were recorded in the 170–330 nm wavelength range, the plots were reduced to the 175–330 nm range, as below 175 nm the absorption of the distilled water already resulted in maximum high tension voltage at the photomultiplier unit, causing the data to be unreliable (Fig. S8†). The minimum wavelength limit of the maximum high tension was typically between 176–178 nm (Fig. S9†) and only the CD data obtained above the limiting values were taken into account in the evaluation process.
Generally, the spectra of the free ligands differ from those of the metal containing systems at all temperatures examined. While only slight variation of the SRCD spectra can be observed within the series recorded for the His-Gly or His-Ala free ligands, the spectra of their [CuL]+ complexes exhibit more pronounced temperature dependent changes. On the other hand, the extent of the spectral variation upon temperature change is similar in the case of the free form and the copper(II)-complex of Gly-Leu peptide. As no linkage isomers of this peptide can form in its [CuLH−1] complex, the modest temperature-dependent spectral variations reflect only the increased flexibility of the molecule at elevated temperatures. Taken together, this indicates that the shift of the coordination isomer microequilibrium has a major contribution to the conformational changes of the ligands in copper(II)–His-Gly or His-Ala complexes.
The results of the MRA calculations on the temperature dependent SRCD spectral series obtained for the metal complexes are shown in Table S2.† It can clearly be seen that the first two diagonal elements are significant in comparison with their standard deviation values for the His-Gly and His-Ala complexes, while there is only one diagonal element of this kind for the copper(II)–Gly-Leu system. This again supports the presence of the isomeric species in the former complexes. The detailed analysis of the residual curves (Fig. S10†) revealed that none of the above systems can be perfectly interpreted with the MRA program, indicating that there is a continuous change in the shape of the spectra of individual species as a function of the temperature. The chiral effect leading to the observed SRCD spectra stems from the sum of the inherent configurational chirality and the contribution of the linkage and ligand conformational isomers. All of these may simultaneously change with temperature, increasing the complexity of the data analysis. No common algorithm is available yet to handle the SRCD data by an appropriate quantum and physico-chemical approach, similar to that applied above for the EPR spectra. The development of such a program would, however, be of great importance, as it could provide additional information on the ligand conformation in its complexed form in aqueous solutions.
Microcalorimetric experiments
The isomerization enthalpy was calculated from the differential scanning calorimetric data by the combination of the van't Hoff equation and the basic energy conservation equations. The repeated experiments led to the −ΔH = (19–27) kJ mol−1 range for the formation of the h isomer from the p isomer. This is in agreement with the values determined by spectroscopic methods, although, this range seems wide as it was the case for the CD experiments, too. Limited data are available in the literature for isomeric equilibria. An example is the cis to trans isomer conversion of N-phosphoryl thioureas, where similar −ΔH = (13–15) kJ mol−1 values were obtained.103The negative value in our system reveals that the conversion of the {NH2,N−,COO−} peptide-type copper(II) complex to its {NH2,Nim} histamine-type linkage isomer is an exothermic process. This can be explained by the fact that the protonation of the backbone amide group is favoured at pH ∼ 5, taken that its basicity is much higher compared to the imidazole side-chain nitrogen. Accordingly, the formation of peptide-type coordination mode in the reaction of Gly-Gly with copper(II) was shown to be slightly endothermic (1.6–2.8 kJ mol−1).103 In addition, the positive mesomeric effect of the imidazole ring increases the electron density on the nitrogen donor group, strengthening the copper(II)–Nim bond, as demonstrated by the negative standard reaction enthalpy related to the formation of the copper(II)–histamine [CuL]2+ complex (−54.3 kJ mol−1).104
Conclusions
Our analysis of temperature dependent spectroscopic data showed direct evidence for the existence of the peptide- and histamine-type linkage-isomers of His-Gly and His-Ala [CuL]+ complexes, followed by their characterization – without applying close derivatives of the ligands. According to the results of the EPR and CD measurement, the histamine-type complex (∼80%) is predominant at room temperature but its amount decreased with the increase of the temperature, and the percentage of the peptide-type complex could reach 40% at 328.2 K. The thermodynamic parameters (i.e. the standard enthalpy and entropy changes) of the isomerisation process – determined by both calorimetric and spectroscopic methods – revealed essentially the same numerical values with similar uncertainty. Based on these results, we can discuss the behaviour of the isomers. The strong binding of the imidazole nitrogen donor group to the copper(II) ion lowers the enthalpy of the histamine-type complex. On the other hand, the tridentate coordination in the case of the peptide-type isomer results in the higher number of freely moving atoms in the uncoordinated histidine side-chain, and the release of an additional water molecule from the coordination sphere of the copper(II) ion, leading to the entropy increase. Thus, the conversion of a peptide-type isomer of [CuL]+ into a histamine-type linkage isomer seems to be promoted by the negative standard reaction enthalpy at low temperatures, but the opposite reaction is favourable at high temperatures due to the entropy increase.The experimental methods for the detection, structural and thermodynamic characterization of microspecies in labile metal complex systems has to be carefully selected. EPR for paramagnetic complexes, NMR for diamagnetic ones, UV-Vis for those with a sensitive chromophore and CD for those possessing chirality is an obvious choice. In specific systems, the monocomplexes formed by each of the multiple binding sites offering the same donor atom set (e.g. {3 × N−,NIm} for copper(II) in certain proteins) can be considered as linkage isomers.71–73,75 In such cases CD spectroscopy can perform better than EPR because it is sensitive to the differences in the chiral environment provided by the amino acid side chains. Another advantage of the CD spectroscopic measurements carried out in the wavelength range of the d–d transitions is that the spectra of single species (isomers) are not sensitive to the temperature change, allowing for a simple evaluation of the data. The CD measurements in the UV range could be more generally performed for all complexes containing chiral ligands (most of the bioligands). By developing a common algorithm to parametrize and fit these spectra, not only the presence of the isomers, but also the conformation of the complexed ligands could be monitored using this method, requiring minimal stoichiometric amounts of substances. Such experiments would complement NMR e.g. for the paramagnetic metal complexes.
With the forewarning in mind that the widest accessible temperature range in aqueous solutions is an inherent limitation of the experiments, our study shed light on the potential of temperature-dependent measurements by properly chosen spectroscopic methods to directly characterize linkage-isomers of kinetically labile metal complexes.
Acknowledgements
This work has received support through the Hungarian National Research Development and Innovation Office (Grants No. K120130, GINOP 2.3.2-15-2016-00038 and K119442) and the Hungarian Scientific Research Fund (OTKA No. K115762). The support of the European Union Research Infrastructure Action FP7 from the CALIPSO Programme (FP7/2007-2013, grant No. 312284) is also acknowledged. The authors thank László Janovák's help during the calorimetric measurements as well as Tamás Jakusch and Emrah Özel for helpful discussions.References
- P. Comba, G. N. DeIuliis, G. A. Lawrance, S. M. Luther, M. Maeder, A. L. Nolan, M. J. Robertson and P. Turner, Dalton Trans., 2003, 2188–2193 RSC.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406–4422 RSC.
- D. Furushou, T. Hashibe, T. Fujinami, K. Nishi, H. Hagiwara, N. Matsumoto, Y. Sunatsuki, M. Kojima and S. Iijima, Polyhedron, 2013, 52, 1489–1498 CrossRef.
- M. B. Rossi, P. Alborés and L. M. Baraldo, Inorg. Chim. Acta, 2011, 374, 334–340 CrossRef CAS.
- S. K. Chawla, M. Arora, K. Nättinen and K. Rissanen, Mendeleev Commun., 2006, 16, 88–90 CrossRef.
- A. J. Blake, N. R. Brooks, N. R. Champness, M. Crew, A. Deveson, D. Fenske, D. H. Gregory, L. R. Hanton, P. Hubberstey and M. Schröder, Chem. Commun., 2001, 1432–1433 RSC.
- M. Du, X.-J. Zhao, J.-H. Guo and S. R. Batten, Chem. Commun., 2005, 4836–4838 RSC.
- A. V. Ivanov, V. I. Mitrofanova, M. Kritikos and O. N. Antzutkin, Polyhedron, 1999, 18, 2069–2078 CrossRef CAS.
- B. Noszál, W. Guo and D. L. Rabenstein, J. Phys. Chem., 1991, 95, 9609–9614 CrossRef.
- M. Kraszni, Z. Szakács and B. Noszál, Anal. Bioanal. Chem., 2004, 378, 1449–1463 CrossRef CAS PubMed.
- G. Parkin, Chem. Rev., 1993, 93, 887–911 CrossRef CAS.
- M. Melnik and C. E. Holloway, Coord. Chem. Rev., 2006, 250, 2261–2270 CrossRef CAS.
- Y. Jean, A. Lledos, J. K. Burdett and R. Hoffmann, J. Am. Chem. Soc., 1988, 110, 4506–4516 CrossRef CAS.
- P. Gütlich, H. A. Goodwin and D. N. Hendrickson, Angew. Chem., Int. Ed. Engl., 1994, 33, 425–427 CrossRef.
- E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 1998, 173, 133–165 CrossRef CAS.
- T. Zhang, M. Ozbil, A. Barman, T. J. Paul, R. P. Bora and R. Prabhakar, Acc. Chem. Res., 2015, 48, 192–200 CrossRef CAS PubMed.
- B. Noszál, D. Visky and M. Kraszni, J. Phys. Chem. B, 2006, 110, 14507–14514 CrossRef PubMed.
- T. N. Parac and N. M. Kostić, J. Am. Chem. Soc., 1996, 118, 5946–5951 CrossRef CAS.
- R. P. Bora, A. Barman, X. Zhu, M. Ozbil and R. Prabhakar, J. Phys. Chem. B, 2010, 114, 10860–10875 CrossRef CAS PubMed.
- J. K. Bashkin, Curr. Opin. Chem. Biol., 1999, 3, 752–758 CrossRef CAS PubMed.
- J. Lee, A. Y. Kovalevsky, I. V. Novozhilova, K. A. Bagley, P. Coppens and G. B. Richter-Addo, J. Am. Chem. Soc., 2004, 126, 7180–7181 CrossRef CAS PubMed.
- B. S. Lim and R. H. Holm, Inorg. Chem., 1998, 37, 4898–4908 CrossRef CAS PubMed.
- E. Reisner, V. B. Arion, A. Rufińska, I. Chiorescu, W. F. Schmid and B. K. Keppler, Dalton Trans., 2005, 2355–2364 RSC.
- R. Cini, S. Defazio, G. Tamasi, M. Casolaro, L. Messori, A. Casini, M. Morpurgo and M. Hursthouse, Inorg. Chem., 2007, 46, 79–92 CrossRef CAS PubMed.
- G. Grasso and S. Bonnet, Metallomics, 2014, 6, 1346–1357 RSC.
- A. H. Krotz, L. Y. Kuo, T. P. Shields and J. K. Barton, J. Am. Chem. Soc., 1993, 115, 3877–3882 CrossRef CAS.
- M. T. Ma, H. N. Hoang, C. C. G. Scully, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2009, 131, 4505–4512 CrossRef CAS PubMed.
- B. Ding, Y. Y. Liu, Y. Q. Huang, W. Shi, P. Cheng, D. Z. Liao and S. P. Yan, Cryst. Growth Des., 2009, 9, 593–601 CAS.
- A. M. Rich, E. Bombarda, A. D. Schenk, P. E. Lee, E. H. Cox, A. M. Spuches, L. D. Hudson, B. Kieffer and D. E. Wilcox, J. Am. Chem. Soc., 2012, 134, 10405–10418 CrossRef CAS PubMed.
- Y. Berezovskaya, C. T. Armstrong, A. L. Boyle, M. Porrini, D. N. Woolfson and P. E. Barran, Chem. Commun., 2011, 47, 412–414 RSC.
- J. Hoang and R. S. Prosser, Biochemistry, 2014, 53, 5727–5736 CrossRef CAS PubMed.
- M. A. Wilson and A. T. Brunger, J. Mol. Biol., 2000, 301, 1237–1256 CrossRef CAS PubMed.
- M. A. Schumacher, A. F. Rivard, H. P. Bächinger and J. P. Adelman, Nature, 2001, 410, 1120–1124 CrossRef CAS PubMed.
- S. J. Philips, M. Canalizo-Hernandez, I. Yildirim, G. C. Schatz, A. Mondragón and T. V. O'Halloran, Science, 2015, 349, 877–881 CrossRef CAS PubMed.
- C. C. Chang, L. Y. Lin, X. W. Zou, C. C. Huang and N. L. Chan, Nucleic Acids Res., 2015, 43, 7612–7623 CrossRef CAS PubMed.
- M. A. Wilson and A. T. Brunger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, D59, 1782–1792 CrossRef CAS.
- U. Heinz, L. Hemmingsen, M. Kiefer and H. W. Adolph, Chem. – Eur. J., 2009, 15, 7350–7358 CrossRef CAS PubMed.
- E. Kopera, T. Schwerdtle, A. Hartwig and W. Bal, Chem. Res. Toxicol., 2004, 17, 1452–1458 CrossRef CAS PubMed.
- M. J. Tamás, S. K. Sharma, S. Ibstedt, T. Jacobson and P. Christen, Biomolecules, 2014, 4, 252–267 CrossRef PubMed.
- T. Jacobson, C. Navarrete, S. K. Sharma, T. C. Sideri, S. Ibstedt, S. Priya, C. M. Grant, P. Christen, P. Goloubinoff and M. J. Tamás, J. Cell Sci., 2012, 125, 5073–5083 CrossRef CAS PubMed.
- M. Nagaoka, J. Kuwahara and Y. Sugiura, Biochem. Biophys. Res. Commun., 1993, 194, 1515–1520 CrossRef CAS PubMed.
- M. a. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed.
- P. Faller and C. Hureau, Coord. Chem. Rev., 2012, 256, 2127–2128 CrossRef CAS.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198–1211 CrossRef CAS PubMed.
- K. Mazák and B. Noszál, J. Med. Chem., 2012, 55, 6942–6947 CrossRef PubMed.
- S. D. Tsotsoros, Y. Qu and N. P. Farrell, J. Inorg. Biochem., 2015, 143, 117–122 CrossRef CAS PubMed.
- X. Wang, H. Li, X. Du, J. Harris, Z. Guo and H. Sun, Chem. Sci., 2012, 3, 3206–3215 RSC.
- J. Kasparkova, S. Pospisilova and V. Brabec, J. Biol. Chem., 2001, 276, 16064–16069 CrossRef CAS PubMed.
- V. B. Arion, E. Reisner, M. Fremuth, M. A. Jakupec, B. K. Keppler, V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chem., 2003, 42, 6024–6031 CrossRef CAS PubMed.
- A. C. G. Hotze, S. E. Caspers, D. De Vos, H. Kooijman, A. L. Spek, A. Flamigni, M. Bacac, G. Sava, J. G. Haasnoot and J. Reedijk, J. Biol. Inorg. Chem., 2004, 9, 354–364 CrossRef CAS PubMed.
- J. C. Chen, J. Li, L. Qian and K. C. Zheng, J. Mol. Struct. (THEOCHEM), 2005, 728, 93–101 CrossRef CAS.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- F. S. Mackay, J. A. Woods, H. Moseley, J. Ferguson, A. Dawson, S. Parsons and P. J. Sadler, Chem. – Eur. J., 2006, 12, 3155–3161 CrossRef CAS PubMed.
- X. Ma, J. Jia, R. Cao, X. Wang and H. Fei, J. Am. Chem. Soc., 2014, 136, 17734–17737 CrossRef CAS PubMed.
- V. Brabec and O. Nováková, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS PubMed.
- U. Kalinowska-Lis, J. Ochocki and K. Matlawska-Wasowska, Coord. Chem. Rev., 2008, 252, 1328–1345 CrossRef CAS.
- H. Song, W. Li, R. Qi, L. Yan, X. Jing, M. Zheng and H. Xiao, Chem. Commun., 2015, 51, 11493–11495 RSC.
- J. L. Burmeister, Coord. Chem. Rev., 1968, 3, 225–245 CrossRef CAS.
- T. G. Appleton, J. F. J. Pesch, M. Wienken, S. Menzer and B. Lippert, Inorg. Chem., 1992, 31, 4410–4419 CrossRef CAS.
- T. Kiss and A. Gergely, Inorg. Chim. Acta, 1983, 78, 247–254 CrossRef CAS.
- M. Kato, K. Unoura, T. Takayanagi, Y. Ikeda, T. Fujihara and A. Nagasawa, Inorg. Chem., 2013, 52, 13375–13383 CrossRef CAS PubMed.
- A. Rockenbauer, T. Szabó-Plánka, Z. Árkosi and L. Korecz, J. Am. Chem. Soc., 2001, 123, 7646–7654 CrossRef CAS PubMed.
- M. Fainerman-Melnikova, T. Szabó-Plánka, A. Rockenbauer and R. Codd, Inorg. Chem., 2005, 44, 2531–2543 CrossRef CAS PubMed.
- R. Šípoš, T. Szabó-Plánka, A. Rockenbauer, N. V. Nagy, J. Šima, M. Melník and I. Nagypál, J. Phys. Chem. A, 2008, 112, 10280–10286 CrossRef PubMed.
- N. V. Nagy, T. Szabó-Plánka, A. Rockenbauer, G. Peintler, I. Nagypál and L. Korecz, J. Am. Chem. Soc., 2003, 125, 5227–5235 CrossRef CAS PubMed.
- N. V. Nagy, T. Szabó-Plánka, G. Tircsó, R. Király, Z. Árkosi, A. Rockenbauer and E. Brücher, J. Inorg. Biochem., 2004, 98, 1655–1666 CrossRef CAS PubMed.
- B. S. Creaven, E. Czeglédi, M. Devereux, É. A. Enyedy, A. Foltyn-Arfa Kia, D. Karcz, A. Kellett, S. McClean, N. V. Nagy, A. Noble, A. Rockenbauer, T. Szabó-Plánka and M. Walsh, Dalton Trans., 2010, 39, 10854–10865 RSC.
- T. Kiss and A. Gergely, J. Chem. Soc., Dalton Trans., 1984, 1951–1957 RSC.
- M. Klewpatinond and J. H. Viles, FEBS Lett., 2007, 581, 1430–1434 CrossRef CAS PubMed.
- S. Timári, I. Turi, K. Várnagy and I. Sóvágó, Polyhedron, 2014, 79, 72–79 CrossRef.
- K. Ősz, J. Inorg. Biochem., 2008, 102, 2184–2195 CrossRef PubMed.
- T. Gajda, B. Henry and J.-J. Delpuech, J. Chem. Soc., Perkin Trans. 2, 1994, 157–164 RSC.
- L. Hong, T. M. Carducci, W. D. Bush, C. G. Dudzik, G. L. Millhauser and J. D. Simon, J. Phys. Chem. B, 2010, 114, 11261–11271 CrossRef CAS PubMed.
- R. R. Garipov, V. G. Shtyrlin, D. A. Safin, Y. I. Zyavkina, F. D. Sokolov, A. L. Konkin, A. V. Aganov and A. V. Zakharov, Chem. Phys., 2006, 320, 59–74 CrossRef CAS.
- T. Gajda, B. Gyurcsik, T. Jakusch, K. Burger, B. Henry and J.-J. Delpuech, Inorg. Chim. Acta, 1998, 275–276, 130–140 CrossRef CAS.
- N. Hasani and A. Eslami, Polyhedron, 2015, 85, 412–428 CrossRef CAS.
- P. Coppens, I. I. Vorontsov, T. Graber, A. Y. Kovalevsky, Y. S. Chen, G. Wu, M. Gembicky and I. V. Novozhilova, J. Am. Chem. Soc., 2004, 126, 5980–5981 CrossRef CAS PubMed.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–883 CrossRef CAS PubMed.
- D. Schaniel, T. Woike, N. R. Behrnd, J. Hauser, K. W. Krämer, T. Todorova and B. Delley, Inorg. Chem., 2009, 48, 11399–11406 CrossRef CAS PubMed.
- S. Lee, T. Carlson, N. Christian, K. Lea, J. Kedzie, J. P. Reilly and J. J. Bonner, Mol. Biol. Cell, 2000, 11, 1753–1764 CrossRef CAS PubMed.
- S. D. Westerheide, R. Raynes, C. Powell, B. Xue and V. N. Uversky, Curr. Protein Pept. Sci., 2012, 13, 86–103 CrossRef CAS PubMed.
- T. Szabó-Plánka, N. V. Nagy, A. Rockenbauer and L. Korecz, Inorg. Chem., 2002, 41, 3483–3490 CrossRef.
- F. J. C. Rosotti and H. Rosotti, The determination of stability constants, McGraw-Hill Book Co., New York, 1962 Search PubMed.
- E. Högfeldt, in Stability Constants of Metal-Ion Complexes, Pergamon, New York, 1982, p. 32 Search PubMed.
- M. Whitfield, in Chemical Oceanography, ed. J. P. Riley and G. Skirrow, Academic, New York, 2nd edn, 1975, pp. 44–171 Search PubMed.
- L. Zékány, I. Nagypál and G. Peintler, Technical Software Distributors, Baltimore MD, 1991.
- P. Limão-Vieira, A. Giuliani, J. Delwiche, R. Parafita, R. Mota, D. Duflot, J. P. Flament, E. Drage, P. Cahillane, N. J. Mason, S. V. Hoffmann and M. J. Hubin-Franskin, Chem. Phys., 2006, 324, 339–349 CrossRef.
- A. Rockenbauer, N. V. Nagy, F. Le Moigne, D. Gigmes and P. Tordo, J. Phys. Chem. A, 2006, 110, 9542–9548 CrossRef CAS PubMed.
- G. Peintler, Zita, A Comprehensive Program Package for Fitting Parameters of Chemical Reaction Mechanisms, Versions 2.1–5.0, Department of Physical Chemistry, University of Szeged, Szeged, Hungary, 2012.
- G. Peintler, I. Nagypal, A. Jancso, I. R. Epstein and K. Kustin, J. Phys. Chem. A, 1997, 101, 8013–8020 CrossRef CAS.
- T. Szabó-Plánka, A. Rockenbauer, L. Korecz and D. Nagy, Polyhedron, 2000, 19, 1123–1131 CrossRef.
- A. Rockenbauer, J. Magn. Reson., 1979, 35, 429–438 CAS.
- B. Gyurcsik, I. Vosekalna and E. Larsen, J. Inorg. Biochem., 2001, 85, 89–98 CrossRef CAS PubMed.
- N. I. Jakab, B. Gyurcsik, T. Körtvélyesi, I. Vosekalna, J. Jensen and E. Larsen, J. Inorg. Biochem., 2007, 101, 1376–1385 CrossRef CAS PubMed.
- H. Sigel, Inorg. Chem., 1975, 14, 1535–1540 CrossRef CAS.
- H. Kozłowski, W. Bal, M. Dyba and T. Kowalik-Jankowska, Coord. Chem. Rev., 1999, 184, 319–346 CrossRef.
- I. Sóvágó, in Biocoordination Chemistry, ed. K. Burger, Ellis Horwood, New York, 1990, pp. 135–184 Search PubMed.
- J. M. Tsangaris and R. B. Martin, J. Am. Chem. Soc., 1970, 92, 4255–4260 CrossRef CAS PubMed.
- G. Formicka-Kozlowska, H. Kozlowska, I. Z. Siemion, K. Sobczyk and E. Nawrocka, J. Inorg. Biochem., 1981, 15, 201–212 CrossRef CAS.
- J. Šebek, B. Gyurcsik, J. Šebestík, Z. Kejík, L. Bednárová and P. Bouř, J. Phys. Chem. A, 2007, 111, 2750–2760 CrossRef PubMed.
- L. A. Kochergina and A. V. Emel'yanov, Russ. J. Phys. Chem. A, 2015, 89, 580–585 CrossRef CAS.
- A. Gergely and I. Sóvágó, Inorg. Chim. Acta, 1976, 20, 19–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables and figures. See DOI: 10.1039/c7dt00884h |
| This journal is © The Royal Society of Chemistry 2017 |