
Catalytic oxidation of benzene over ruthenium–cobalt bimetallic catalysts and study of its mechanism†
Xiaolong
Liu
 *a,
Junlin
Zeng
a,
Wenbo
Shi
ab,
Jian
Wang
a,
Tingyu
Zhu
*ac and
Yunfa
Chen
a
*a,
Junlin
Zeng
a,
Wenbo
Shi
ab,
Jian
Wang
a,
Tingyu
Zhu
*ac and
Yunfa
Chen
a
aBeijing Engineering Research Center of Process Pollution Control, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuxl@ipe.ac.cn; tyzhu@ipe.ac.cn; Fax: +86 010 82544821; Tel: +86 010 82544821
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum (Beijing), Beijing 102249, China
cCenter for Excellence in Regional Atmospheric Environment, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China
First published on 28th November 2016
Abstract
In this work, ruthenium-based bimetallic catalysts of the formula Ru–5M/TiO2 (M = Mn, Co, Ce, Cu, Fe) were prepared and evaluated in benzene oxidation, and Ru–5Co/TiO2 exhibited the lowest complete oxidation temperature (220 °C), demonstrating Co3O4 as the best dopant. The influence of the Ru and Co contents on the catalytic activity was also studied, and 1 wt% Ru and 5 wt% Co3O4 were confirmed. Water vapor showed an inhibition effect on benzene oxidation at 210 °C, whereas the catalytic efficiency was barely influenced at a higher temperature (230 °C). STEM analyses revealed that Ru–Co bimetallic species showed similar distributions on the catalytic surface. Based on the correlation of the catalytic results with various characterization techniques, the synergistic effect between Ru and Co was well demonstrated, and a four-step transformation in the oxidation process was concluded. Besides, systematic in situ FTIR studies for benzene oxidation over Ru/TiO2, Co/TiO2, and Ru–5Co/TiO2 were conducted and compared, and many organic intermediates were observed in the in situ FTIR spectra. Accordingly, the reaction mechanism for benzene oxidation over the Ru–Co bimetallic catalyst was proposed.
1. Introduction
Volatile organic compounds (VOCs) emitted from industries and manufacturing plants are recognized as great contributors to air pollution.1,2 Therefore, various strategies based on physical and chemical methods have been developed for VOC removal, such as thermal incineration, catalytic oxidation, and adsorption.3–5 Among these techniques, catalytic oxidation is regarded as the most promising method to reduce VOC emissions due to its advantages of high efficiency, low temperature, and low cost.5–8 Accordingly, a number of catalysts have been explored for the complete oxidation of VOCs.9–13 Platinum and palladium catalysts have been well demonstrated,14–17 whereas the drawback of high cost is a great limit for their wide applications. Meanwhile, transition metal oxides have been also increasingly studied,18–21 usually showing lower catalytic activity than noble metal catalysts. Designing applicable catalysts for VOC removal is still a meaningful field to explore.Bimetallic catalysts commonly show higher catalytic activity, selectivity, and anti-poisoning ability than monometallic materials,22–26 due to the ensemble effect, electronic effect, and the generation of new active sites with the doping of a second metal species. The promotion of performance is generally defined as a bimetallic synergistic effect.27,28 O'Shea et al. prepared Pd–Mn materials and employed these catalysts in methane oxidation, and a Pd–Mn synergistic effect was observed.29 Jodaei et al. reported Ag–M (M = Fe, Co, Mn)-ZSM-5 catalyzed ethyl acetate oxidation, and Ag–Fe-ZSM-5 showed the highest catalytic activity.30 These results revealed that selecting appropriate bimetallic species might be a vital factor for accomplishing the bimetallic synergistic effect.
Ruthenium catalysts have attracted increasing importance in CO oxidation,31 the Deacon process,32 and catalytic oxidation of VOCs.33,34 However, ruthenium-catalyzed VOC oxidations are surprisingly far less investigated than other noble metals. Recently, complete oxidation of ethyl acetate,35 propane,36,37 toluene,38 chlorobenzene,39,40 and trichloroethylene41 has been well established. Nonetheless, it has to be also pointed out that Ru materials still showed slightly lower activity than Pt and Pd catalysts for some common VOCs, such as toluene and benzene, which belong to an important class of VOCs.17,38,42 Doping a transition metal oxide with high oxygen mobility into a Ru catalyst may be an effective strategy to increase its catalytic activity.
Cobalt oxide has been regarded as an attractive material in VOC purification due to its oxygen mobility in Co3+/Co2+ redox, which has been proven to be an important factor in the oxidation process.43 For VOC oxidation, it is generally concluded that its great oxygen transfer ability is attributed to the weak Co–O within the spinel Co3O4 structure.44 Co3O4 has been tested as an active species,19 dopant,22,45 or support.46–48 However, efforts should be continuously devoted to further improve its applicability.
Hence, using Co3O4 as the dopant for noble metals, such as Ru, is very conducive to optimizing the catalytic performance of the present Ru-based catalysts. To the best of our knowledge, complete oxidation of VOCs over a Ru–Co bimetallic catalyst has not been reported. Herein, a series of Ru–Co/TiO2 samples were prepared and evaluated in the catalytic oxidation of benzene. Their catalytic activities were well correlated with the physicochemical properties. Besides, in situ FTIR studies were also conducted, and the reaction mechanism was accordingly proposed.
2. Experimental section
2.1. Catalyst preparation
The Ru–5M/TiO2 (M = Mn, Co, Ce, Cu, Fe) catalysts were prepared through a step-impregnation method. The percentage of the Ru element was 1 wt%, and that of MOx was 5 wt%. In a typical preparation of Ru–5Co/TiO2, commercial P25 TiO2 (Degussa, SBET = 57.5 m2 g−1) was mixed with a solution of Co(NO3)2 (Aladdin), and the mixture was stirred for 5 h at room temperature. The solvent was removed using a rotary evaporator, and the resulting solid was dried at 110 °C for 5 h. Then, the solid was calcined at a ramp of 5 °C min−1 from 30 °C to 300 °C and kept at this temperature for 5 h, giving a dark grey powder of 5Co/TiO2. Subsequently, an aqueous solution of Ru(NO)(NO3)3 (1.5 mg mL−1, Aladdin) was mixed with 5Co/TiO2, and the mixture was stirred for 5 h at room temperature. Then, the solvent was removed using a rotary evaporator, and the resulting solid was dried at 110 °C for 5 h. Finally, the solid was calcined at a ramp of 5 °C min−1 from 30 °C to 300 °C and kept at this temperature for 5 h, giving the final product denoted as Ru–5Co/TiO2.Ru–(y)Co/TiO2 (y = 1, 3, 5, 10) and (y)Ru–5Co/TiO2 (y = 0.05, 0.1, 0.2, 0.5, 1.0, 2.0) were prepared using the same method by adjusting the contents of Ru and Co3O4. 5Co–Ru/TiO2 was synthesized with the sequence of Ru first and Co3O4 second under the same conditions. (Ru–5Co)/TiO2 was obtained using a co-impregnation method under the same conditions with just one impregnation step.
2.2. Catalyst characterization
The Ru content was measured with an inductively coupled plasma-optical emission spectrometer (ICP-OES, Optima 5300DV, Perkin Elmer). X-ray diffraction (XRD) patterns of the samples were recorded on a powder diffractometer (Rigaku D/Max-RA) using Cu Kα radiation (40 kV and 120 mA). The porous texture was characterized by N2 adsorption at 77 K in an automatic surface area and porosity analyzer (Autosorb iQ, Quantachrome). Transmission electron microscopy (TEM), high resolution transmission electron microscopy (HR-TEM), and high-angle annular dark-field scanning TEM (HAADF-STEM) images were collected on a FEI Tecnai G2 F20 field emission electron microscope operating at 200 kV. Hydrogen temperature-programmed reduction (H2-TPR) measurements were conducted on a chemical adsorption analyzer (Autochem II 2920, Micromeritics). The samples (100 mg) were pretreated in Ar at 300 °C for 1 h before TPR analysis. After being cooled under the same atmosphere to room temperature, TPR experiments were carried out in a flow of 10% H2/Ar heated from RT to 600 °C at a ramp of 10 °C min−1. The alteration in H2 concentration of the effluent was monitored on-line by the chemical adsorption analyzer. The hydrogen consumption was calibrated using TPR of copper oxide under the same conditions. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Thermo escalab 250Xi X-ray using an Al Kα source.2.3. Catalytic evaluation
The catalytic activities of the catalysts were evaluated in a continuous flow fixed-bed quartz microreactor (i.d. = 4 mm) from 100 to 300 °C with 100 mg of catalyst (60–80 mesh). Benzene was introduced into the gas cylinder, and its concentration in the total flow of the reactant mixture (500 ppm benzene + 20% O2 + Ar (balance)) was calibrated by GC through a by-pass. The total flow ratio of the reactant mixture was 100 mL min−1 with a weight hourly space velocity (WHSV) of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1. The reactants and products were analyzed on-line with a gas chromatograph (GC 2010 Plus, Shimadzu) equipped with a flame ionization detector (FID). CO2 and CO at the outlet were detected using another FID with a methanizer (MTH, Shimadzu) furnace. The conversion of benzene was calculated using eqn (1).
000 mL g−1 h−1. The reactants and products were analyzed on-line with a gas chromatograph (GC 2010 Plus, Shimadzu) equipped with a flame ionization detector (FID). CO2 and CO at the outlet were detected using another FID with a methanizer (MTH, Shimadzu) furnace. The conversion of benzene was calculated using eqn (1). | (1) |
2.4. In situ FTIR experiments
The FTIR spectra were collected using an FTIR spectrometer (Nicolet 6700, Thermo) equipped with an MCT detector (cooled by liquid nitrogen) and a stainless steel IR cell (CaF2 windows). Firstly, the IR cell was flushed with flowing Ar (100 mL min−1 for 2 h). Then the sample (about 45 mg) was pressed into a supported disk and oxidized at 300 °C for 1 h in a 20% O2/Ar (100 mL min−1) stream. Subsequently, the sample was cooled to 240 °C and a background spectrum was collected in a flowing 20% O2/Ar stream. Eventually, the IR spectra were collected at different times in a flowing stream of 500 ppm benzene/20% O2/Ar (100 mL min−1).3. Results and discussion
3.1. Catalytic oxidation of benzene
The catalytic oxidation of benzene over all catalysts was established under the conditions of 500 ppm benzene at a WHSV of 60000 mL g−1 h−1 within the range 150–300 °C. As shown in Fig. 1A, the catalytic activity increased in the sequence of Ru–5Cu/TiO2, Ru–5Mn/TiO2, Ru–5Fe/TiO2, Ru/TiO2, Ru–5Ce/TiO2, and Ru–5Co/TiO2 with a 90% conversion temperature (T90) of 266 °C, 233 °C, 232 °C, 229 °C, 226 °C, and 215 °C, respectively. It can be seen that Ru–5Co/TiO2 and Ru–5Ce/TiO2 showed higher catalytic efficiencies than Ru/TiO2, whereas other dopants showed a negative effect. It is noteworthy that Ru–5Co/TiO2 gave the lowest complete oxidation temperature of 220 °C, which is comparable to a reported Pd catalyst,17 revealing that Co3O4 was the most appropriate dopant in benzene oxidation. Its higher catalytic activity was tentatively ascribed to the oxygen mobility in the Co3+/Co2+ redox, which might play an important role in the bimetallic catalyzed oxidation. Hence, the Ru–Co bimetallic catalytic system was selected and employed in the following research.
 | ||
| Fig. 1 Benzene conversion as a function of reaction temperature over (A) Ru/TiO2 and Ru–5M/TiO2 (M = Mn, Co, Ce, Cu, Fe), (B) Ru–5Co/TiO2, 5Co–Ru/TiO2, and (Ru–5Co)TiO2, (C) Ru–(y)Co/TiO2 (y = 1, 3, 5, 10), (D) (y)Ru–5Co/TiO2 (y = 0.05, 0.1, 0.2, 0.5, 1.0, 2.0) under the conditions of benzene concentration = 500 ppm, benzene/O2 molar ratio = 1/400, and WHSV = 60000 mL g−1 h−1. | ||
In order to determine the best preparation method, Ru–5Co/TiO2, 5Co–Ru/TiO2, and (Ru–5Co)/TiO2 were prepared and compared in benzene oxidation. As shown in Fig. 1B, Ru–5Co/TiO2 exhibited the highest catalytic activity, and 5Co–Ru/TiO2 and (Ru–5Co)/TiO2 were almost comparable with each other within the full temperature range, giving the same T90 of 227 °C (Table S1†). It can be seen that the impregnation sequence plays an important role in the synthesis of bimetallic catalysts with a synergistic effect.
A series of Ru–Co/TiO2 catalysts with different Ru or Co3O4 contents were then synthesized and employed in benzene oxidation (Fig. 1C and D). For Ru–(y)Co/TiO2 (y = 1, 3, 5, 10), Ru–5Co/TiO2 gave the highest catalytic efficiency. The T90 of Ru–1Co/TiO2, Ru–10Co/TiO2, and Ru–3Co/TiO2 were 220 °C, 225 °C, and 227 °C, respectively (Table S1†). For (y)Ru–5Co/TiO2, the catalytic activity apparently increased with the improvement of the Ru content from 0.05% to 2%, and their T90 were 295 °C, 278 °C, 249 °C, 229 °C, 215 °C, and 210 °C, respectively. However, considering the applicability of Ru catalysts, 1 wt% Ru and 5 wt% Co3O4 were confirmed and employed in the following studies.
The anti-moisture performance of the catalyst is crucial to its applicability in VOC purification for industrial off-gas. Hence, the catalytic oxidation of benzene was carried out in the presence of 1 vol% H2O, and the results are summarized in Fig. 2. When the temperature was 230 °C, introduction of H2O led to a very slight decrease of the conversion, and the catalytic efficiency was recovered with the cut off of H2O. When the temperature was set at 210 °C, benzene conversion was stabilized at around 80%. Addition of H2O led to a sharp decrease of benzene conversion by ca. 40%, revealing that H2O exhibited an inhibition effect on the catalytic oxidation process by occupying the reactive sites on the catalyst surface. When H2O was cut off, at first, the catalyst gave an apparently higher benzene conversion than before possibly due to the unbalanced heat release. Gradually, the catalytic efficiency was balanced.
 | ||
| Fig. 2 Effect of water vapor on benzene conversion at different temperatures over Ru–5Co/TiO2 under the conditions of benzene concentration = 500 ppm, benzene/O2 molar ratio = 1/400, and WHSV = 60000 mL g−1 h−1. | ||
3.2. Catalyst characterization
The XRD patterns of Ru/TiO2 and Ru–5M/TiO2 (M = Mn, Co, Ce, Cu, Fe) are shown in Fig. 3A. The diffraction patterns of the Ru-based catalysts are consistent with those of the unloaded P25 support, with the peaks at 25.4°, 48.1°, 53.6°, and 55.2° (2θ) due to anatase TiO2 (PDF 21-1272), and the 27.5°, 36.2°, and 54.4° (2θ) peaks are attributable to rutile TiO2 (PDF 21-1276). Any peaks attributable to ruthenium species were not observed possibly due to their good dispersion. In the XRD patterns of Ru–5Co/TiO2 and Ru–5Fe/TiO2, the peaks at 31.3°, 44.8°, 59.4°, and 65.2° (2θ) are ascribed to Co3O4 (PDF 43-1003), whereas the peak at 36.2° (2θ) is ascribed to Fe2O3 (PDF 33-0664). Besides, a series of Ru–(y)Co/TiO2 (y = 1, 3, 5, 10) were then prepared and characterized. As shown in Fig. 3B, characteristic peaks of Co3O4 were observed when the Co3O4 content was above 5 wt%. The peak intensities increased with the improvement of the Co3O4 content. | ||
| Fig. 3 XRD patterns of (A) Ru/TiO2 and Ru–5M/TiO2 (M = Mn, Co, Ce, Cu, Fe), and (B) Ru–(y)Co/TiO2 (y = 1, 3, 5, 10). | ||
TEM and HR-TEM were conducted to observe the morphology of Ru–(y)Co/TiO2 catalysts. As shown in Fig. 4, ruthenium species were not observed. HRTEM images were collected and presented in Fig. 4b, e, h, and k. Interplanar lattice spacings for rutile (110) and anatase (101) were observed for TiO2, and cobalt oxide with clear lattice spacings was not observed. To analyze the Ru and Co species, their corresponding EDX spectra were also collected (Fig. 4c, f, i, and l). The EDX spectra confirmed the presence of Ti, O, Ru, and Co elements. With the increase of Co3O4 loading, the Co intensity strengthened obviously. Additionally, TEM and EDX characterization were also recorded for Ru–5Co/TiO2, 5Co–Ru/TiO2, and (Ru–5Co)/TiO2, respectively, and no obvious difference was observed (Fig. S2†).
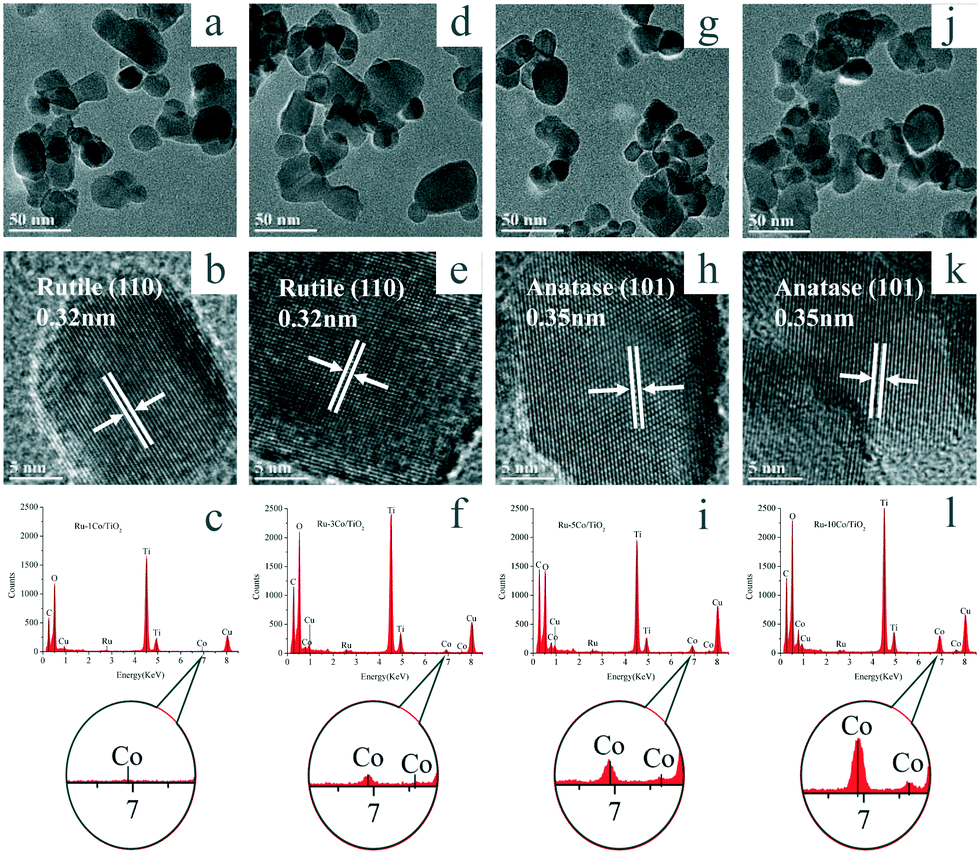 | ||
| Fig. 4 TEM images (a, d, g, j represent y = 1, 3, 5, 10, respectively), HRTEM images (b, e, h, k represent y = 1, 3, 5, 10, respectively), and EDX spectra (c, f, i, l, represent y = 1, 3, 5, 10) of Ru–(y)Co/TiO2. | ||
The dispersion of the active species is commonly correlated with the activity of the catalyst. For many ruthenium oxide-containing materials, the ruthenium oxide exists as thin layers,32 which were hard to observe in the TEM images due to their low contrast. However, STEM methods are commonly used to explore the element distributions, which might afford the interpretation of the catalytic results. Hence, HAADF-STEM and STEM-EDS mapping images were collected for Ru–5Co/TiO2, which exhibited the highest catalytic activity in benzene oxidation. As shown in Fig. 5, a dark field-scanning mode was employed. The support was clearly observed, whereas the bright spots were not confirmed to be the ruthenium species or caused by overlapping TiO2 particles. Accordingly, STEM-EDS mapping was collected. Fig. 5c–e present the distributions of Ru (green), Co (red), and Ti (yellow), respectively. The superimposition of Ru and Co revealed that Ru and Co exhibited similar distribution behavior (Fig. 5f), which might induce the bimetallic synergistic effect to a great extent, leading to an outstanding catalytic performance. Besides, Fig. 5g shows the superimposition of Ru, Co, and Ti, revealing an inhomogeneous distribution of Ru–Co species on TiO2 (P25), which is consistent with previous reports.41,42
 | ||
| Fig. 5 Representative HAADF-STEM images (a and b) and STEM-EDS mapping images of (c) Ru, (d) Co, (e) Ti, (f) Ru–Co, and (g) Ru–Co/Ti for the Ru–5Co/TiO2 catalyst. | ||
H2-TPR experiments were conducted to investigate the reducibility of Ru/TiO2, 5Co/TiO2, and Ru–5Co/TiO2. As shown in Fig. 6, the Ru/TiO2 sample gave a single reduction peak at 95 °C, which can be assigned to the reduction of RuO2 to Ru.36 For 5Co/TiO2, two main reduction peaks appeared at 294 °C and 419 °C, corresponding to the reduction of Co3O4 to CoO and CoO to metallic Co, respectively.22 As for the bimetallic catalyst Ru–5Co/TiO2, the reduction peak of RuO2 still remained at 95 °C, whereas the reduction peaks of cobalt oxides appeared at 142 °C and 274 °C, respectively. Obviously, the reducibility of cobalt oxides was enhanced, suggesting a synergistic effect between Ru and Co in Ru–5Co/TiO2. This phenomenon is generally defined as the hydrogen spillover effect, and it is commonly observed in the low-temperature reduction of a transition metal oxide in the presence of a noble metal.28
 | ||
| Fig. 6 H2-TPR profiles of Ru/TiO2, 5Co/TiO2, and Ru–5Co/TiO2. | ||
The XPS spectra for Ru–(y)Co/TiO2 (y = 1, 3, 5, 10), 5Co–Ru/TiO2, and (Ru–5Co)/TiO2 were collected, and the Ru 3d and O 1s spectra are summarized in Fig. 7. The assignments and definitions of Ru peaks were controversial and inconsistent with previous studies.49,50 In this research, the Ru 3d spectra were tentatively deconvoluted into three peaks at 280.2, 281.4, and 282.6 eV, assignable to Rucus (coordinatively unsaturated site), Rucus + Oot (Rucus bonded with on-top O), and Ru4+satellite (satellite peak of Ru4+), respectively.42,51 Rucus has been demonstrated to be a catalytically active site, since the adsorbed O species and organic reactants could be easily transferred onto Rucus due to its unsaturated property.42,51 As illustrated in Fig. 7A and Table S2,† Ru–5Co/TiO2 had the most abundant Rucus, which would greatly facilitate the oxidation process.
 | ||
| Fig. 7 XPS spectra of (A) Ru 3d and (B) O 1s for Ru–Co bimetallic catalysts (a, b, c, d, e, and f represent Ru–1Co/TiO2, Ru–3Co/TiO2, Ru–5Co/TiO2, Ru–10Co/TiO2, 5Co–Ru/TiO2, and (Ru–5Co)/TiO2, respectively). | ||
As shown in Fig. 7B, O 1s spectra were deconvoluted into two peaks at 529.8 eV and 531.8 eV, assignable to Olatt (lattice oxygen) and Oads (adsorbed oxygen, e.g., O2−, O22−, or O−), respectively.52 It has been generally regarded that the electrophilic Oads species play an important role in the catalytic oxidation of VOCs.53 As summarized in Table S2,† Ru–5Co/TiO2 exhibited the highest Oads/Olatt molar ratio of 0.116. For 5Co–Ru/TiO2 and (Ru–5Co)/TiO2, their Oads/Olatt values were almost equal (0.071), and their catalytic curves were basically overlapped (Fig. 1B). Hence, a four-step transformation in the oxidation process was proposed. Firstly, a Co(III)/Co(II) transformation was initiated, and Oads was released (eqn (2)). Then, Co(II) was oxidized into Co(III) (eqn (3)). Subsequently, Oads and organic reactants (Rorg) were easily transferred onto Rucus due to its unsaturated property, generating Ru–O–Rorg species (eqn (4)). Finally, Ru–O–Rorg species decomposed into CO2 and H2O through a series of complicated steps (eqn (5)).
| Co(III) − O → Co(II) + Oads | (2) |
| Co(II) + [O] → Co(III) − O | (3) |
| Rucus + Oads + Rorg → Ru − O − Rorg | (4) |
| Ru − O − Rorg + [O] → Rucus + CO2 + H2O | (5) |
3.3. In situ FTIR studies and reaction mechanism
In order to further explore the detailed reaction mechanism of benzene oxidation over Ru–Co catalysts, in situ FTIR spectra of Ru–5Co/TiO2, Ru/TiO2, and Co/TiO2 were collected (Fig. 8). It can be seen that Ru–5Co/TiO2 and Ru/TiO2 showed very similar spectra, which are apparently different from those of Co/TiO2, revealing that the Ru–Co catalytic oxidation mainly proceeded through a Ru-centered process. For Ru–5Co/TiO2, the band at 1617 cm−1 was attributed to the vibration of the aromatic ring adsorbed parallel to the surface,54 showing little change over time. The band at 1582 cm−1 was assigned to the phenolate species.55 The band at 1720 cm−1 was tentatively ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from quinone or other ketone species according to previous research.56 The bands at 1532–1519, 1429, 1313–1303 cm−1 were attributed to maleate species.54,57 The band at 1558 cm−1 was assigned to the COO– stretching vibration of surface acetates, and the band at 1375 cm−1 was attributed to the CH3 stretching vibration of surface acetates.54,57 For Co/TiO2, the broad band at 1537 cm−1 should be attributed to the superposition of the maleate and acetate species. Notably, it was confusing to assign the bands at 1863 and 1451 cm−1 in the spectra of Ru–5Co/TiO2 and Ru/TiO2, since these bands were totally missing for Co/TiO2.
O from quinone or other ketone species according to previous research.56 The bands at 1532–1519, 1429, 1313–1303 cm−1 were attributed to maleate species.54,57 The band at 1558 cm−1 was assigned to the COO– stretching vibration of surface acetates, and the band at 1375 cm−1 was attributed to the CH3 stretching vibration of surface acetates.54,57 For Co/TiO2, the broad band at 1537 cm−1 should be attributed to the superposition of the maleate and acetate species. Notably, it was confusing to assign the bands at 1863 and 1451 cm−1 in the spectra of Ru–5Co/TiO2 and Ru/TiO2, since these bands were totally missing for Co/TiO2.
 | ||
| Fig. 8 In situ FTIR spectra collected at 240 °C after (a) 1, (b) 3, (c) 10, (d) 20, (e) 30, and (f) 60 min in a 500 ppm benzene/20% O2/Ar stream for (A) Ru–5Co/TiO2, (B) Ru/TiO2, and (C) Co/TiO2, and (D) spectra collected after 60 min for these three catalysts. | ||
To demonstrate the unidentified bands, in situ FTIR experiments for (y)Ru–5Co/TiO2 (y = 0.05, 0.1, 0.2, 0.5, 1.0) were also conducted (Fig. 9). Most bands could be identified as discussed above, and the two main sets of the bands between 1600 and 1400 cm−1 showed a representative variation tendency with the improvement of Ru content. Surprisingly, the band intensity at 1863 cm−1 decreased with the improvement of Ru content. In previous studies, the bands between 1900 and 1750 cm−1 were commonly regarded as stretching vibrations of CO for anhydride (OC–O–CO, 1870 cm−1),58 acyl chloride (OCCl, 1842 cm−1),59 or aldehyde species (R–CHO, 1755 cm−1).54 In this research, no chlorine was introduced, and they were not quite consistent with the wavenumbers for aldehyde species. Hence, they should be due to the anhydride species, such as maleic anhydride. As for the decreasing tendency, it was tentatively proposed that most Ru species dispersed as small RuOx clusters at lower loadings (≤0.2 wt%), and the π donor of maleic anhydride could be easily coordinated to Ruδ+ at the corner sites of the small RuOx clusters, whereas the coordination behavior could not be established in the Co/TiO2 system. With more Ru addition, Ru nanoparticles were formed, and most Ru species were present as RuO2, which were inclined to adsorb other organic salt species, leading to the weakening of the band intensity for maleic anhydride. Besides, it was reasonable to assign the band at 1451 cm−1 to the COO– stretching vibration of surface acetates adsorbed on Ru centers.55
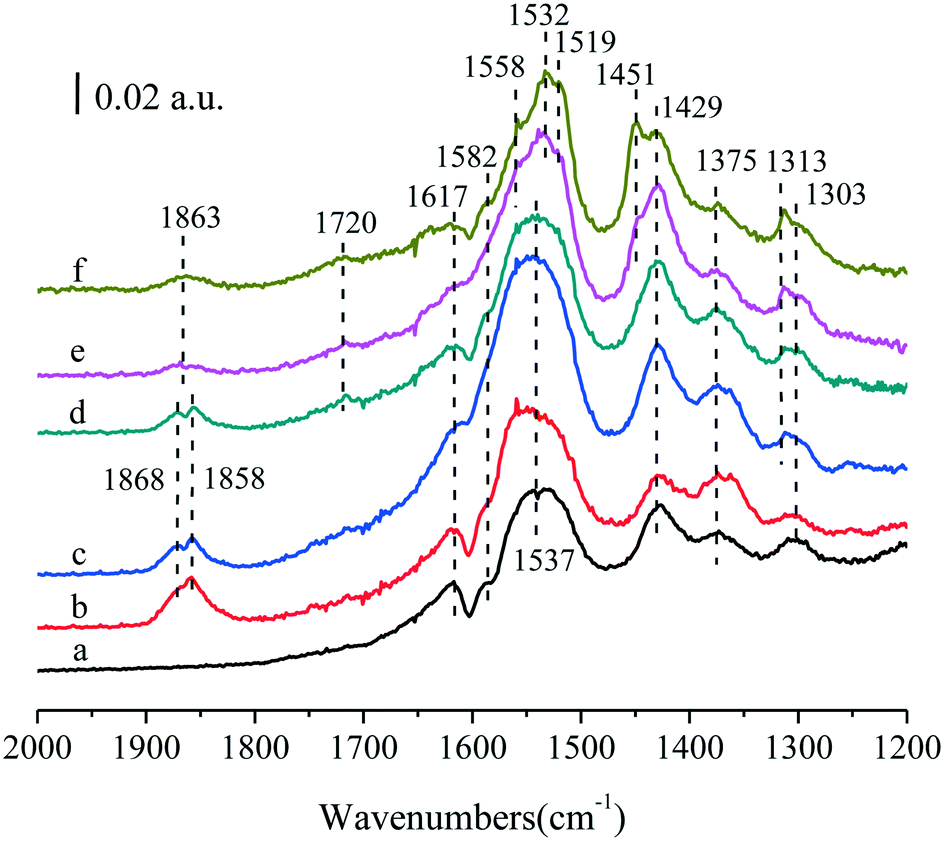 | ||
| Fig. 9 In situ FTIR spectra collected at 240 °C after 60 min in a 500 ppm benzene/20% O2/Ar stream for (a) 5Co/TiO2, (b) 0.05Ru–5Co/TiO2, (c) 0.1Ru–5Co/TiO2, (d) 0.2Ru–5Co/TiO2, (e) 0.5Ru–5Co/TiO2, and (f) Ru–5Co/TiO2. | ||
In order to confirm the oxidation-initiation step, an in situ FTIR study during the adsorption of benzene/Ar at 240 °C over Ru–5Co/TiO2 was conducted, and the results are summarized in the ESI.† As shown in Fig. S3,† three main broad groups of bands at 1600–1500 cm−1, 1450–1400 cm−1, and 1375–1350 cm−1, were observed. It is noteworthy that the spectra displayed similar band regions to those in Fig. 8A, and the band intensities slowly increased over time. The band intensities were relatively weaker than those of the spectra collected with O2 possibly due to the oxygen deficiency along with the oxygen-consuming process. Hence, it was believed that the oxidation was initiated by the activation of lattice oxygen (eqn (2)) but not from the activation of molecular oxygen.
As shown in Fig. 10, the reaction mechanism was proposed. On the Ru–Co catalyst (A) surface, benzene (B) first reacts with the Ru–Co centers, generating the phenolate species (C). The oxygen-containing group is generally regarded as an electron-donating group and an ortho–para position director. Hence, the phenolate species are easily transformed into o-benzoquinone (D) and p-benzoquinone (E). With the promoting effect of Ru–Co, a ring-opening proceeds under the attack of active oxygen species, and some small organic intermediates are generated, such as maleates (F), acetates (G), and ketones (H). Eventually, the intermediates are oxidized into inorganic products CO2 and H2O, and the Ru–Co catalyst is recovered.
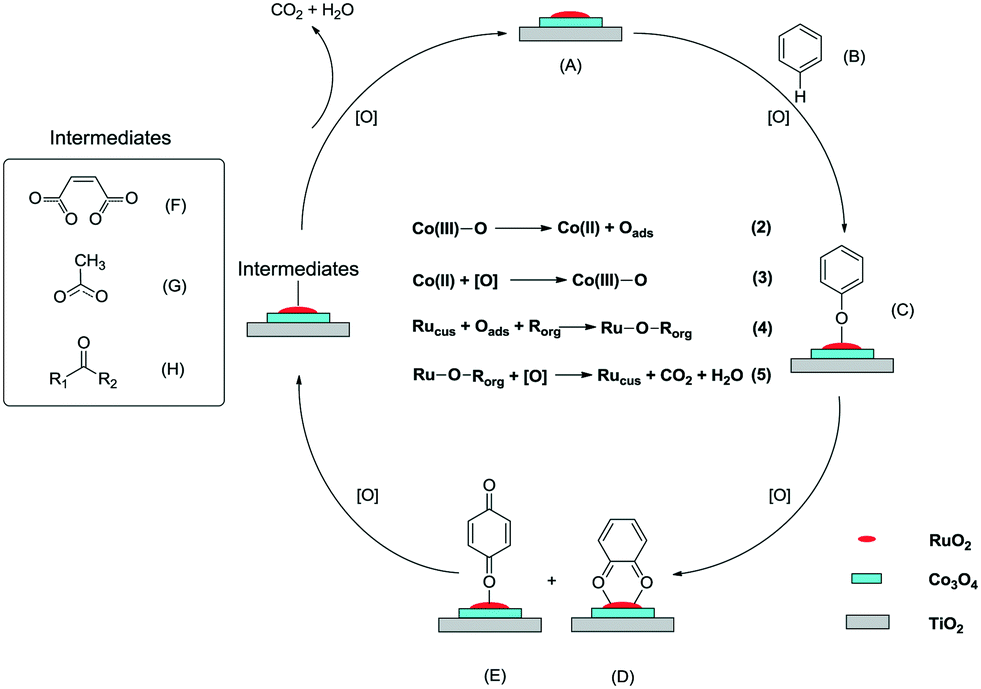 | ||
| Fig. 10 Proposed mechanism for the catalytic oxidation of benzene over a Ru–Co bimetallic catalyst. | ||
4. Conclusions
In summary, among the bimetallic catalysts of the formula Ru–5M/TiO2 (M = Mn, Co, Ce, Cu, Fe) for benzene oxidation, Ru–5Co/TiO2 exhibited the highest catalytic efficiency. The best contents of Ru and Co3O4 were confirmed to be 1 wt% and 5 wt%, respectively. Water vapor showed an inhibition effect on benzene oxidation at 210 °C, and this inhibition effect was weakened at 230 °C. The characterization was well-correlated with the catalytic results, and the Ru–Co bimetallic synergistic effect was proposed. Ru–Co species showed similar distributions on the TiO2 support. The reducibility of cobalt oxides was enhanced with Ru addition, suggesting a hydrogen spillover effect. Ru–5Co/TiO2 showed the highest Oads/Olatt molar ratio. Based on the results, a four-step transformation promoted by Ru–Co was concluded. Many organic intermediates such as phenolate species, quinone, maleates, acetates, and ketones were observed in the in situ FTIR spectra. Accordingly, a reaction mechanism for the catalytic oxidation of benzene over a Ru–Co bimetallic catalyst was proposed.Acknowledgements
This work was supported by the Natural Science Foundation of China (21607154) and the Beijing Natural Science Foundation (8164063).Notes and references
- J. Deng, S. He, S. Xie, H. Yang, Y. Liu, G. Guo and H. Dai, Environ. Sci. Technol., 2015, 49, 11089–11095 CrossRef CAS PubMed
.
- M. Ousmane, L. F. Liotta, G. Di Carlo, G. Pantaleo, A. M. Venezia, G. Deganello, L. Retailleau, A. Boreave and A. Giroir-Fendler, Appl. Catal., B, 2011, 101, 629–637 CrossRef CAS
- R. Fiorenza, M. Bellardita, L. Palmisano and S. Scirè, J. Mol. Catal. A: Chem., 2016, 415, 56–64 CrossRef CAS
- J. Li, R. Lu, B. Dou, C. Ma, Q. Hu, Y. Liang, F. Wu, S. Qiao and Z. Hao, Environ. Sci. Technol., 2012, 46, 12648–12654 CrossRef CAS PubMed
- K. Everaert and J. Baeyens, J. Hazard. Mater., 2004, 109, 113–139 CrossRef CAS PubMed
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 CAS
- S. Ojala, S. Pitkäaho, T. Laitinen, N. N. Koivikko, R. Brahmi, J. Gaalova, L. Matejova, A. Kucherov, S. Paivarinta, C. Hirschmann, T. Nevanpera, M. Riihimaki, M. Pirila and R. L. Keiski, Top. Catal., 2011, 54, 1224–1256 CrossRef CAS
- C. E. Hetrick, F. Patcas and M. D. Amiridis, Appl. Catal., B, 2011, 101, 622–628 CrossRef CAS
- T. K. Nevanperä, S. Ojala, N. Bion, F. Epron and R. L. Keiski, Appl. Catal., B, 2016, 182, 611–625 CrossRef
- S. Scirè and L. F. Liotta, Appl. Catal., B, 2012, 125, 222–246 CrossRef
- M. Hosseini, T. Barakat, R. Cousin, A. Aboukais, B. L. Su, G. De Weireld and S. Siffert, Appl. Catal., B, 2012, 111, 218–224 CrossRef
- R. Wang and J. Li, Environ. Sci. Technol., 2010, 44, 4282–4287 CrossRef CAS PubMed
- S. Scirè, P. M. Riccobene and C. Crisafulli, Appl. Catal., B, 2010, 101, 109–117 CrossRef
- T. Barakat, J. C. Rooke, R. Cousin, J. F. Lamonier, J. M. Giraudon, B. L. Su and S. Siffert, New J. Chem., 2014, 38, 2066–2074 RSC
- T. Barakat, J. C. Rooke, H. L. Tidahy, M. Hosseini, R. Cousin, J. F. Lamonier, J. M. Giraudon, G. De Weireld, B. L. Su and S. Siffert, ChemSusChem, 2011, 4, 1420–1430 CrossRef CAS PubMed
- L. F. Liotta, Appl. Catal., B, 2010, 100, 403–412 CrossRef CAS
- C. He, J. Li, P. Li, J. Cheng, Z. Hao and Z. Xu, Appl. Catal., B, 2010, 96, 466–475 CrossRef CAS
- B. Solsona, T. García, R. Sanchis, M. D. Soriano, M. Moreno, E. Rodríguez-Castellón, S. Agouram, A. Dejoz and J. M. L. Nieto, Chem. Eng. J., 2016, 290, 273–281 CrossRef CAS
- L. F. Liotta, H. J. Wu, G. Pantaleo and A. M. Venezia, Catal. Sci. Technol., 2013, 3, 3085–3102 CAS
- T. Garcia, D. Sellick, F. Varela, I. Vazquez, A. Dejoz, S. Agouram, S. H. Taylor and B. Solsona, Appl. Catal., A, 2013, 450, 169–177 CrossRef CAS
- F. G. Durán, B. P. Barbero, L. E. Cadús, C. Rojas, M. A. Centeno and J. A. Odriozola, Appl. Catal., B, 2009, 92, 194–201 CrossRef
- S. Zhao, K. Li, S. Jiang and J. Li, Appl. Catal., B, 2016, 181, 236–248 CrossRef CAS
- B. Darif, S. Ojala, L. Pirault-Roy, M. Bensitel, R. Brahmi and R. L. Keiski, Appl. Catal., B, 2016, 181, 24–33 CrossRef CAS
- S. Xie, J. Deng, S. Zang, H. Yang, G. Guo, H. Arandiyan and H. Dai, J. Catal., 2015, 322, 38–48 CrossRef CAS
- T. Barakat, V. Idakiev, R. Cousin, G. S. Shao, Z. Y. Yuan, T. Tabakova and S. Siffert, Appl. Catal., B, 2014, 146, 138–146 CrossRef CAS
- X. Wang, J. Stöver, V. Zielasek, L. Altmann, K. Thiel, K. Al-Shamery, M. Baumer, H. Borchert, J. Parisi and J. Kolny-Olesiak, Langmuir, 2011, 27, 11052–11061 CrossRef CAS PubMed
- R. Fiorenza, C. Crisafulli, G. G. Condorelli, F. Lupo and S. Scirè, Catal. Lett., 2015, 145, 1691–1702 CrossRef CAS
- J. Feng, Y. Zhang, W. Xiong, H. Ding and B. He, Catalysts, 2016, 6, 51 CrossRef
- V. A. D. O'Shea, M. C. Alvarez-Galvan, J. Requies, V. L. Barrio, P. L. Arias, J. F. Cambra, M. B. Guemez and J. L. G. Fierro, Catal. Commun., 2007, 8, 1287–1292 CrossRef
- A. Jodaei, D. Salari, A. Niaei, M. Khatamian and N. Caylak, Environ. Technol., 2011, 32, 395–406 CrossRef CAS PubMed
- K. Qadir, S. H. Joo, B. S. Mun, D. R. Butcher, J. R. Renzas, F. Aksoy, Z. Liu, G. A. Somorjai and J. Y. Park, Nano Lett., 2012, 12, 5761–5768 CrossRef CAS PubMed
- W. Shi, X. Liu, J. Zeng, J. Wang, Y. Wei and T. Zhu, Chin. J. Catal., 2016, 37, 1181–1192 CrossRef CAS
- J. Okal and M. Zawadzki, Appl. Catal., B, 2009, 89, 22–32 CrossRef CAS
- T. Mitsui, K. Tsutsui, T. Matsui, R. Kikuchi and K. Eguchi, Appl. Catal., B, 2008, 81, 56–63 CrossRef CAS
- N. Kamiuchi, T. Mitsui, H. Muroyama, T. Matsui, R. Kikuchi and K. Eguchi, Appl. Catal., B, 2010, 97, 120–126 CrossRef CAS
- D. P. Debecker, B. Farin, E. M. Gaigneaux, C. Sanchez and C. Sassoye, Appl. Catal., A, 2014, 481, 11–18 CrossRef CAS
- J. Okal and M. Zawadzki, Appl. Catal., B, 2011, 105, 182–190 CrossRef CAS
- X. Liu, J. Wang, J. Zeng, X. Wang and T. Zhu, RSC Adv., 2015, 5, 52066–52071 RSC
- H. Huang, Q. Dai and X. Wang, Appl. Catal., B, 2014, 158, 96–105 CrossRef
- Q. Dai, S. Bai, X. Wang and G. Lu, Appl. Catal., B, 2013, 129, 580–588 CrossRef CAS
- J. Wang, X. Liu, J. Zeng and T. Zhu, Catal. Commun., 2016, 76, 13–18 CrossRef CAS
- X. Liu, J. Zeng, J. Wang, W. Shi and T. Zhu, Catal. Sci. Technol., 2016, 6, 4337–4344 CAS
- L. F. Liotta, M. Ousmane, G. Di Carlo, G. Pantaleo, G. Deganello, G. Marcì, L. Retailleau and A. Giroir-Fendler, Appl. Catal., A, 2008, 347, 81–88 CrossRef CAS
- T. Garcia, S. Agouram, J. F. Sánchez-Royo, R. Murillo, A. M. Mastral, A. Aranda, I. Vázquez, A. Dejoz and B. Solsona, Appl. Catal., A, 2010, 386, 16–27 CrossRef CAS
- P. Petrova, T. Tabakova, G. Munteanu, R. Zanella, M. Tsvetkov and L. Ilieva, Catal. Commun., 2013, 36, 84–88 CrossRef CAS
- Y. Liu, H. Dai, J. Deng, S. Xie, H. Yang, W. Tan, W. Han, Y. Jiang and G. Guo, J. Catal., 2014, 309, 408–418 CrossRef CAS
- B. Solsona, E. Aylón, R. Murillo, A. M. Mastral, A. Monzonis, S. Agouram, T. E. Davies, S. H. Taylor and T. Garcia, J. Hazard. Mater., 2011, 187, 544–552 CrossRef CAS PubMed
- J. Łojewska, A. Kołodziej, J. Zak and J. Stoch, Catal. Today, 2005, 105, 655–661 CrossRef
- P. A. Cox, J. B. Goodenough, P. J. Tavener, D. Telles and R. G. Egdell, J. Solid State Chem., 1986, 62, 360–370 CrossRef CAS
- H. J. Lewerenz, S. Stucki and R. Kötz, Surf. Sci., 1983, 126, 463–468 CrossRef CAS
- H. Over, A. P. Seitsonen, E. Lundgren, M. Smedh and J. N. Andersen, Surf. Sci., 2002, 504, L196–L200 CrossRef CAS
- Q. Ye, J. Zhao, F. Huo, D. Wang, S. Cheng, T. Kang and H. Dai, Microporous Mesoporous Mater., 2013, 172, 20–29 CrossRef CAS
- D. Y. C. Leung, X. Fu, D. Ye and H. Huang, Kinet. Catal., 2012, 53, 239–246 CrossRef CAS
- J. Lichtenberger and M. D. Amiridis, J. Catal., 2004, 223, 296–308 CrossRef CAS
- C. E. Hetrick, J. Lichtenberger and M. D. Amiridis, Appl. Catal., B, 2008, 77, 255–263 CrossRef CAS
- S. Lomnicki, J. Lichtenberger, Z. T. Xu, M. Waters, J. Kosman and M. D. Amiridis, Appl. Catal., B, 2003, 46, 105–119 CrossRef CAS
- J. Zeng, X. Liu, J. Wan, H. Lv and T. Zhu, J. Mol. Catal. A: Chem., 2015, 408, 221–227 CrossRef CAS
- G. Busca, G. Ramis and V. Lorenzelli, J. Mol. Catal., 1989, 50, 231–240 CrossRef CAS
- P. S. Chintawar and H. L. Greene, J. Catal., 1997, 165, 12–21 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Various characterization analyses and evaluation data for the catalysts, and CO2 selectivity of benzene oxidation over Ru–5Co/TiO2. See DOI: 10.1039/c6cy02141g |
| This journal is © The Royal Society of Chemistry 2017 |