
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pillar[4]pyridinium: a square-shaped molecular box†
Sandra
Kosiorek
a,
Bartłomiej
Rosa
a,
Tomasz
Boinski
b,
Helena
Butkiewicz
a,
Marek P.
Szymański
b,
Oksana
Danylyuk
 a,
Agnieszka
Szumna
*b and
Volodymyr
Sashuk
*a
a,
Agnieszka
Szumna
*b and
Volodymyr
Sashuk
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-248 Warsaw, Poland. E-mail: vsashuk@ichf.edu.pl; Web: http://groups.ichf.edu.pl/sashuk
bInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-248 Warsaw, Poland. E-mail: aszumna@icho.edu.pl
First published on 27th November 2017
Abstract
Numerous applications of Stoddart's ‘blue-box’, a pyridinium containing macrocycle of rectangular shape, encouraged us to seek successors of this amazing molecule. Using a one-step cyclization reaction we synthesized a square-shaped cyclic tetramer consisting of 4-methylenepyridinium units – pillar[4]pyridinium (P[4]P). Pillar[4]pyridinium is a quadruply positively charged water-soluble macrocycle with a highly symmetric, strained structure and an electron-deficient cavity. These features impel the macrocycle to assemble into channel networks in the solid state and render it an effective fluoride receptor in water.
Macrocycles which are inherently ionic are quite rare, especially those bearing a permanent electric charge. Most of them belong to cyclophanes composed of alternating neutral and charged units.1–12 Macrocycles made exclusively of charged units, yet of the same type, are represented only by calix[n]pyridiniums (CPs)13,14 and calix[n]imidazoliums (CIs).15,16 Being the structural analogs of calix[n]arenes (CAs), both are cone-shaped.17 We report here pillar[4]pyridinium (P[4]P) (Fig. 1), which represents a new class of cationic macrocycles, pillar[n]pyridiniums (PPs). This is the first charged macrocycle that structurally resembles pillar[n]arenes (PAs).18 The compound is a cyclic tetramer consisting of pyridyl units linked through nitrogen and para carbon with methylene bridges, being an electron-deficient analog of a missing electron-rich pillar[4]arene. It is the most compact macrocycle among the reported multiply charged macrocycles with a highly strained ring geometry. In this study, we show its synthesis, structure and physicochemical characterization.
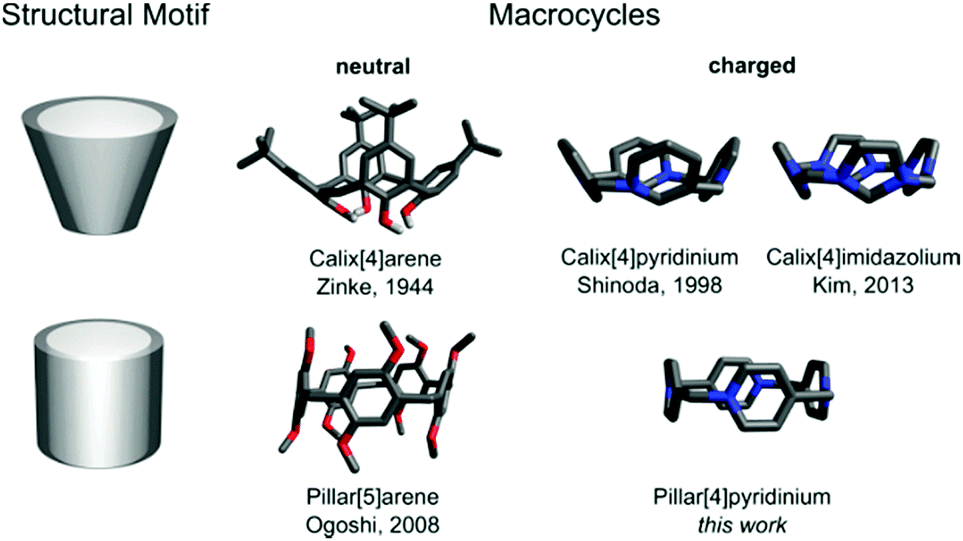 | ||
| Fig. 1 Macrocycles and their structural motifs. | ||
P[4]P is formed in the condensation reaction of 4-(bromomethyl)pyridine hydrobromide carried out under basic conditions in the presence of ammonium hexafluorophosphate. Previous studies on the analogous reaction without the hexafluorophosphate additive revealed the formation of linear oligomeric products only.19 We found that the reaction outcome is crucially dependent on the type and amount of a base. We tested DBU, Et3N, pyridine and sodium bicarbonate. We found that severalfold excess of base leads to the formation of mainly oligomeric products. The cyclomerization reaction starts dominating when the base is employed in a nearly stoichiometric amount. The reaction in DMF gave mainly polymeric products, while the reaction in water caused substrate hydrolysis. The highest yield of P[4]P (50% according to NMR) was found in the reaction carried out in acetonitrile with stoichiometric amounts of NaHCO3 and NH4PF6 (Fig. 2a). The crude product obtained under these optimized conditions was purified by recrystallization from aqueous potassium bromide solutions. Despite a surfeit of bromide ions in the solution, the macrocycle crystallizes as P[4]P·(PF6)4 salt, indicating a tight ion pairing between P[4]P4+ and hexafluorophosphate ions and the role of the latter as a template in the macrocycle formation. The second batch of P[4]P was obtained as iodide salt, P[4]P·(I)4, through the addition of potassium iodide to the remaining mother liquor.
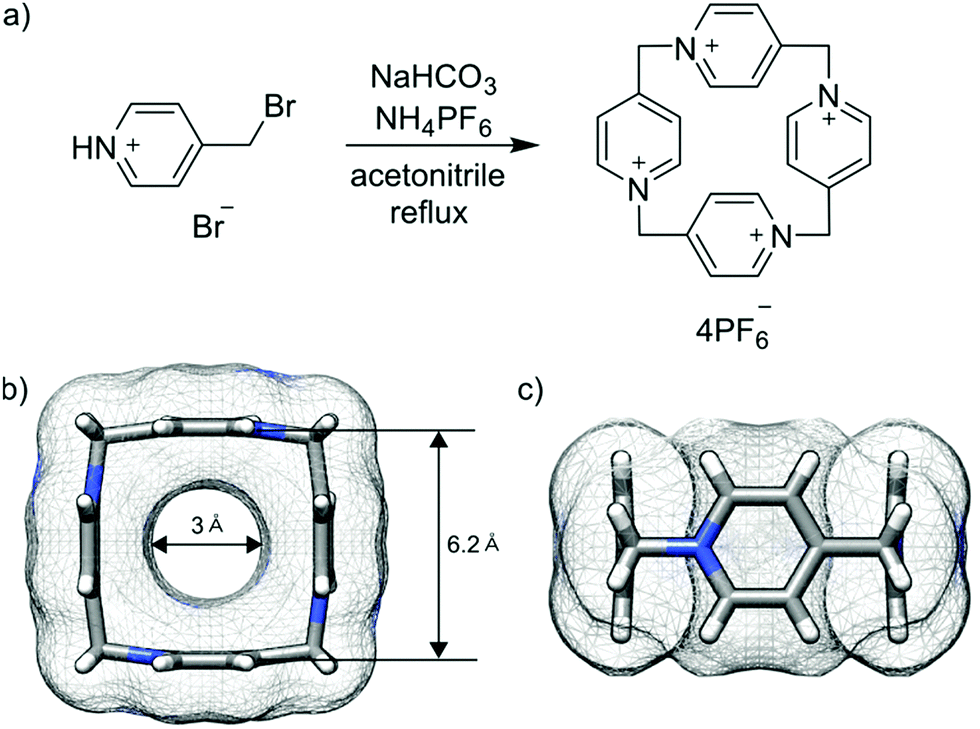 | ||
| Fig. 2 Reaction scheme (a). Top (b) and side (c) views of P[4]P (carbon atoms in grey, nitrogen atoms in blue and hydrogen atoms in white). | ||
The isolated P[4]P salts are sparingly soluble in common solvents, e.g. P[4]P·(PF6)4 dissolves slightly in DMSO, wet acetonitrile and aqueous solutions of sodium sulfate. The solubility of P[4]P significantly increases after anion exchange: NTf2 ions make the macrocycle soluble in acetonitrile, whereas its chloride salt readily dissolves in pure water. Crystals suitable for X-ray analyses were grown for hexafluorophosphate and iodide salts.
P[4]P·(PF6)4 crystallizes in the orthorhombic unit cell, Immm space group. The asymmetric unit consists of one eighth of the tetracationic macrocycle and hexafluorophosphate anions in three crystallographically non-equivalent positions (quarter and two of one-eight occupancies). The P[4]P adopts a highly symmetric square box-like conformation (Fig. 2b and c). The size of the quadruply positively charged macrocyclic ring is 6.2 Å (measured between the centroids of the pyridinium rings). The strain within the macrocycle is manifested in the reduction of the valence angles N+–CH2–C to 106.6 and 107.2° from the tetrahedral angle of 109.5°. The inner cavity of the P[4]P is empty, while the interactions with PF6− are realized via both rims of the macrocycle (Fig. 3a). There are close contacts between F atoms and P[4]P rims, witnessing (C–H)⋯F hydrogen bonds (C⋯F distances of 3.12 and 3.14 Å). The tubular assembly is built by perfect stacking of P[4]P rings with sandwiched PF6− anions (Fig. 3c). The remaining hexafluorophosphate anions separate adjacent tubes in the crystal lattice (Fig. 3b), interacting with the outer surface of the macrocycles via ionic (C–H)⋯F hydrogen bonds, CH2⋯F hydrogen bonds, and π+⋯anion interactions between pyridinium moieties of P[4]P and PF6− anions.
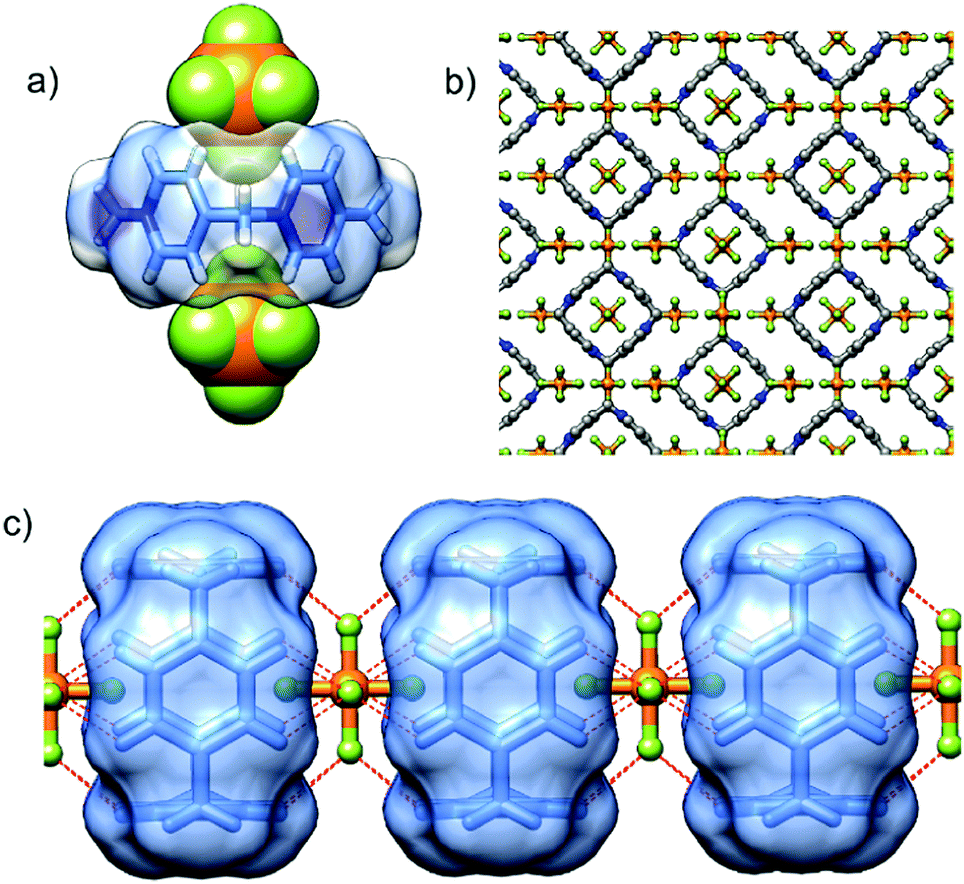 | ||
| Fig. 3 (a) The ion pairing between P[4]P and two PF6− anions; (b) crystal packing of P[4]P·(PF6)4 salt viewed from the a direction; and (c) tubular assembly realized by perfect stacking of P[4]P macrocycles sandwiched by PF6− anions (ionic (C–H)+⋯F− hydrogen bonds are represented as dashed red lines). | ||
P[4]P·(I)4 crystallizes in the tetragonal space group I4/m. The unit cell consists of one eighth of the macrocycle residing on the 4-fold axes and perpendicular mirror plane, half of the iodide anion and water molecules. The valence angle N+–CH2–C is 105.2°, witnessing even more considerable strain than in the hexafluorophosphate complex. The size of the inner cavity is just enough to fit a water molecule (Fig. 4a). Another two water molecules sit at the rims of the macrocycle interacting with the inner cavity water via hydrogen bonds (O–H⋯O distance of 2.67 Å). Again, the tubular assembly is formed where rigid macrocyclic tetracations are perfectly stacked with alternating iodide anions sandwiched at all vertexes of the macrocyclic boxes (Fig. 4c). Besides electrostatic non-directional interactions between P[4]P and iodide anions, the (C–H)⋯I hydrogen bonds between the pyridinium rings of adjacent macrocycles and iodide ions stabilize the tubular assembly. Such a tubular channel structure built from the alternatively charged entities is spanned by water wires running through the central pore of the assembly (Fig. 4b).
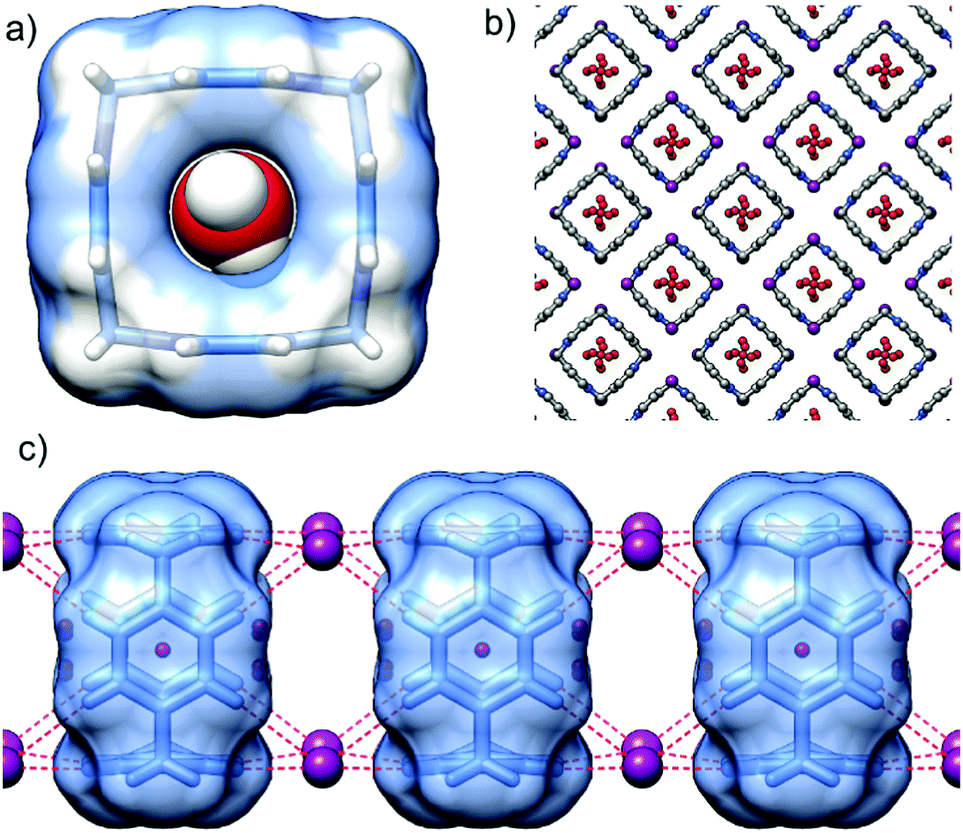 | ||
| Fig. 4 (a) The inclusion of a water molecule into a P[4]P macrocyclic cavity in the P[4]P·(I)4 salt; (b) crystal packing of P[4]P·(I)4 salt viewed from the c direction, note the water wires inside supramolecular tubes; and (c) tubular assembly of P[4]P macrocycles sandwiched by I− anions (ionic (C–H)+⋯I− hydrogen bonds are represented as dashed red lines). | ||
The D4v symmetry of the P[4]P macrocyclic ring in solution is corroborated by NMR experiments (Fig. 5). The 1H NMR spectrum consists of two doublets and one singlet. The doublets come from aromatic protons, whereas the singlet is ascribed to the CH2 bridge. The 13C NMR spectrum is also simple. It consists of four singlets, three of which are aromatic and one is aliphatic. In contrast, mass spectra are far more complex because of a highly charged nature of the macrocycle (for an ESI spectrum see the ESI,† Fig. S1).15
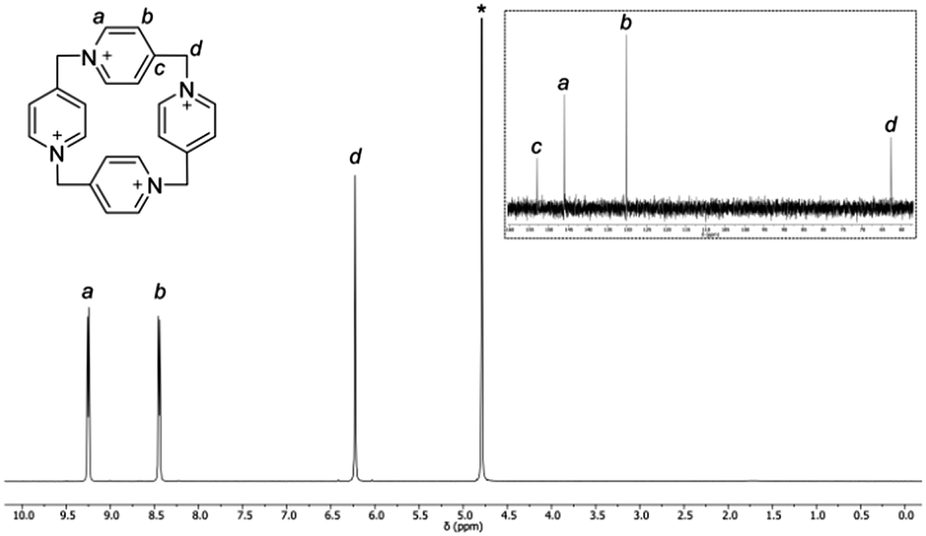 | ||
| Fig. 5 1H and 13C NMR (inset) spectra of P[4]P·(PF6)4 in D2O (1 M Na2SO4 solution) at 293 K. Residual water is marked with an asterisk. | ||
The geometric fit indicates that the size of the macrocyclic ring may be suitable for complexation of fluoride anions (Fig. 6b). However, for P[4]P·(PF6)4 the cavity is blocked by PF6− ions and complexation of fluorides is not observed. When PF6− ions are exchanged with chlorides the macrocycle efficiently recognizes fluoride in water. The complexation process was monitored using 19F NMR (Fig. 6a). The titrations were repeated three times in different concentration ranges. Non-linear curve fitting to the titration points reveals a mixed H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G stoichiometry of complexation and a chemically reasonable fit was obtained using KHG = 1.45 × 104 M−1, KHG2 = 0.91 × 103 M−1, KHG3 = 1.05 × 103 M−1 and KHG4 = 1.15 × 102 M−1 (D2O, 298 K). Simpler models (HG only and mixed HG with HG2) show substantial systematic deviations in non-linear curve fitting, especially in the region of high guest concentrations, which is the most indicative for higher stoichiometries (Fig. S8 and S9, ESI†). It should be noted that the association constant for 1:1 complexation of fluoride by P[4]P·Cl4 is of the same order of magnitude as was reported for calix[n]imidazolium macrocycles.15
:G stoichiometry of complexation and a chemically reasonable fit was obtained using KHG = 1.45 × 104 M−1, KHG2 = 0.91 × 103 M−1, KHG3 = 1.05 × 103 M−1 and KHG4 = 1.15 × 102 M−1 (D2O, 298 K). Simpler models (HG only and mixed HG with HG2) show substantial systematic deviations in non-linear curve fitting, especially in the region of high guest concentrations, which is the most indicative for higher stoichiometries (Fig. S8 and S9, ESI†). It should be noted that the association constant for 1:1 complexation of fluoride by P[4]P·Cl4 is of the same order of magnitude as was reported for calix[n]imidazolium macrocycles.15
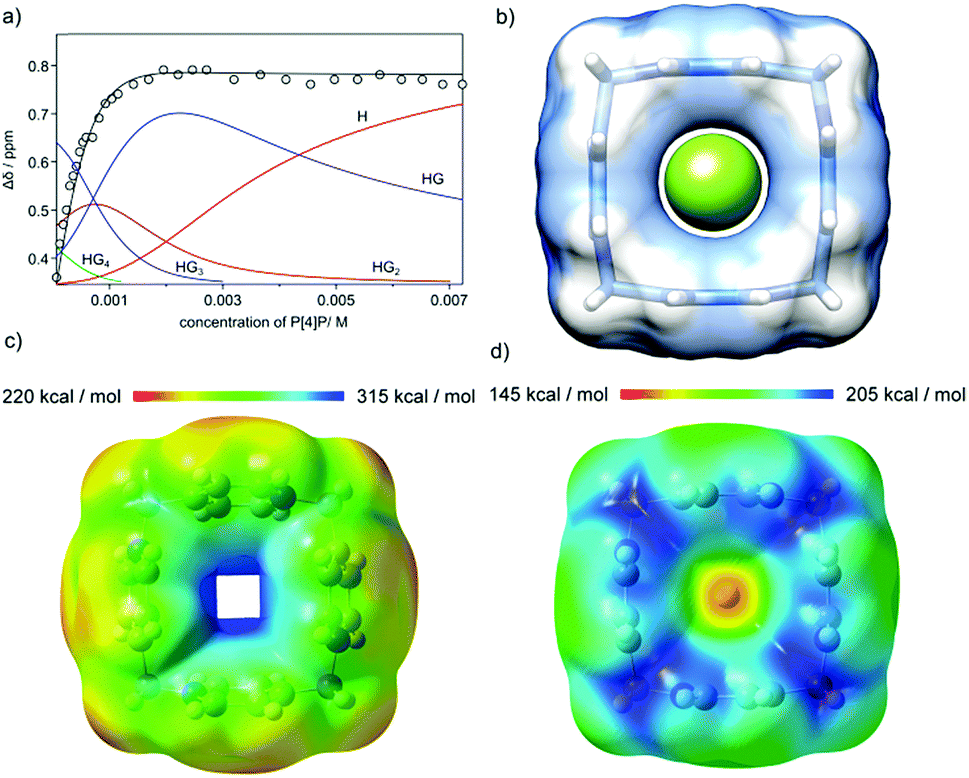 | ||
| Fig. 6 (a) 19F chemical shift changes during the titration of TBAF with P[4]P·Cl4, the titration curve fitted using a model with mixed HG–HG4 species and complexes’ distribution (D2O, 298 K); (b) optimized geometry of the F− ⊃ P[4]P4+C4v-symmetric complex; (c) optimized geometry and an electrostatic potential mapped onto the total electron density isosurface for P[4]P4+; and (d) an electrostatic potential mapped onto the total electron density isosurface for the F− ⊃ P[4]P4+ complex (DFT/B3LYP 6-31+G(d)). | ||
Geometry optimizations and calculations of an electrostatic potential were performed for various charged and neutral species: P[4]P4+ (H), P[4]P4+F− (HG), P[4]P4+F2− (HG2), P[4]P4+F3− (HG3) and P[4]P4+F4− (HG4) at the DFT/B3LYP 6-31+G(d) level in water (PCM/CPCM).20 The results indicate that for the P[4]P4+ cation the aromatic rings are tilted by 10° (S4 symmetry, Fig. 6c), while the pillar-shaped structure (C4v-symmetric, tilt angle 0°) is unstable (negative frequencies). The positive electrostatic potential for P[4]P4+ has the highest values in the cavity. For the P[4]P4+F− species (HG complex) both C4v-symmetric (Fig. 6d) and tilted S4-symmetric structures are stable. For species with more anions the calculations indicate that the anions locate next to the methylene hydrogen atoms. For example, the starting HG2 sandwich-type structure (with anions located above and below the cavity) converges to a structure with anions located next to methylene bridges (Fig. S12, ESI†). The same was observed for the HG4 complex (Fig. S14, ESI†). These results support the hypothesis that the first complexation event (HG formation) proceeds in the cavity, while the remaining anions are complexed externally.
The stability of P[4]P under various conditions was monitored by NMR. In analogy to Stoddart's ‘blue-box’, it is susceptible to the action of nucleophiles, bases and reducing agents, leading to a ring rupture.21,22 An illustrative example is the decomposition of P[4]P·(PF6)4 in DMSO. When the sample of P[4]P·(PF6)4 was kept at 120 °C in DMSO for 2 h, the NMR spectrum (Fig. S2, ESI†) indicated that the ring of the macrocycle is opened and the generated sulfonium ion is cleaved to an aldehyde. The reaction resembles Kornblum and Kröhnke oxidation reactions.23,24 It is important to note that the reaction does not stop at this stage, and the linear product undergoes further fragmentation until the formation of isonicotinic aldehyde. The macrocycle is also sensitive to the presence of iodide even only as a counterion, e.g. P[4]P·(I)4 decomposes in the boiling water, while P[4]P·(PF6)4 remains nearly intact. It is stable in neutral, acidic and slightly basic solutions but rapidly decomposes at pH > 10. Furthermore, the irreversible structural changes occur during attempts to reduce P[4]P·(PF6)4 electrochemically or with SnCl2. Remarkably, the degradation of the macrocycle can be easily identified by the appearance of an intense color. The solutions turn red with strong alkali, blue in hot DMSO and violet in the presence of SnCl2 (Fig. S4–S6, ESI†). Another property of P[4]P is its unusual acidity. The macrocycle, as revealed by hydrogen–deuterium exchange studies (Fig. S3, ESI†), readily loses protons from the carbons next to nitrogen. At near neutral pH the deprotonation occurs primarily on the methylene bridges (Hd, Fig. 5). At higher pH the reaction also takes place on the methine positions (Ha, Fig. 5). The increased acidity of these protons can be accounted for by the ring strain, and also by resonance and inductive effects.
In summary, we have, for the first time, presented the synthesis and characterization of a polycationic macrocycle of pillar shape. The macrocycle has a high concentration of a positive charge and it is able to complex fluoride in its cavity in aqueous solutions. Current efforts are underway in our lab to prepare higher homologs of P[4]P, which might be useful as receptors for electron-rich molecules, as catalysts for reactions occurring via negatively-charged transition states, and as stators for molecular machines. According to NMR, one of such congeners is formed along with P[4]P in the presence of NH4PF6. Our present studies are focused on its isolation and characterization.
This work was financed by the National Science Centre of Poland (OPUS grant no. UMO-2016/23/B/ST5/02937) and the Wroclaw Centre for Networking and Supercomputing (grant no. 299).
Conflicts of interest
There are no conflicts to declare.References
- P. J. Altmann, C. Jandl and A. Pothig, Dalton Trans., 2015, 44, 11278–11281 RSC.
- B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1988, 27, 1547–1550 CrossRef.
- M. Bühner, W. Geuder, W.-K. Gries, S. Hünig, M. Koch and T. Poll, Angew. Chem., Int. Ed., 1988, 27, 1553–1556 CrossRef.
- J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183–192 CrossRef CAS PubMed.
- M. Juríček, J. C. Barnes, E. J. Dale, W.-G. Liu, N. L. Strutt, C. J. Bruns, N. A. Vermeulen, K. C. Ghooray, A. A. Sarjeant, C. L. Stern, Y. Y. Botros, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 12736–12746 CrossRef PubMed.
- C. J. Serpell, J. Cookson, A. L. Thompson and P. D. Beer, Chem. Sci., 2011, 2, 494–500 RSC.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed.
- B. Shirinfar, N. Ahmed, Y. S. Park, G.-S. Cho, I. S. Youn, J.-K. Han, H. G. Nam and K. S. Kim, J. Am. Chem. Soc., 2013, 135, 90–93 CrossRef CAS PubMed.
- N. Ahmed, B. Shirinfar, I. S. Youn, A. Bist, V. Suresh and K. S. Kim, Chem. Commun., 2012, 48, 2662–2664 RSC.
- K. Chellappan, N. J. Singh, I.-C. Hwang, J. W. Lee and K. S. Kim, Angew. Chem., Int. Ed., 2005, 44, 2899–2903 CrossRef CAS.
- S. Kumar, P. Singh, A. Mahajan and S. Kumar, Org. Lett., 2013, 15, 3400–3403 CrossRef CAS PubMed.
- S. T. J. Ryan, J. del Barrio, R. Suardíaz, D. F. Ryan, E. Rosta and O. A. Scherman, Angew. Chem., Int. Ed., 2016, 55, 16096–16100 CrossRef CAS PubMed.
- S. Shinoda, M. Tadokoro, H. Tsukube, S. Shinoda and R. Arakawa, Chem. Commun., 1998, 181–182 RSC.
- K. Wang, J.-H. Cui, S.-Y. Xing and H.-X. Dou, Org. Biomol. Chem., 2016, 14, 2684–2690 CAS.
- Y. Chun, N. Jiten Singh, I.-C. Hwang, J. Woo Lee, S. U. Yu and K. S. Kim, Nat. Commun., 2013, 4, 1797 CrossRef PubMed.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed.
- A. Zinke and E. Ziegler, Ber. Dtsch. Chem. Ges., 1944, 77, 264–272 CrossRef.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- S. Monmoton, H. Lefebvre, F. Costa-Torro and A. Fradet, Macromol. Chem. Phys., 2008, 209, 2374–2381 CrossRef CAS.
- M. Frisch, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- O. Š. Miljanić and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12966–12970 CrossRef PubMed.
- I. Aprahamian, W. R. Dichtel, T. Ikeda, J. R. Heath and J. F. Stoddart, Org. Lett., 2007, 9, 1287–1290 CrossRef CAS PubMed.
- N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand and W. M. Weaver, J. Am. Chem. Soc., 1957, 79, 6562 CrossRef CAS.
- F. Kröhnke, Angew. Chem., Int. Ed., 1963, 2, 380–393 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1571841 and 1571842. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc08562a |
| This journal is © The Royal Society of Chemistry 2017 |