
In vitro toxicity evaluation of silica-coated iron oxide nanoparticles in human SHSY5Y neuronal cells
Gözde
Kiliç
ab,
Carla
Costa
cd,
Natalia
Fernández-Bertólez
ab,
Eduardo
Pásaro
a,
João Paulo
Teixeira
cd,
Blanca
Laffon
*a and
Vanessa
Valdiglesias
a
aDICOMOSA Group, Department of Psychology, Area of Psychobiology, Universidade da Coruña, Research Services Building, Campus Elviña s/n, 15071-A Coruña, Spain. E-mail: blaffon@udc.es; Fax: +34 981167172; Tel: +34 981167000
bDepartment of Cell and Molecular Biology, University of A Coruña, Faculty of Sciences, Campus A Zapateira s/n, 15071-A Coruña, Spain
cDepartment of Environmental Health, Portuguese National Institute of Health, Rua Alexandre Herculano 321, Porto 4000-055, Portugal
dEPIUnit - Institute of Public Health, University of Porto, Rua das Taipas no. 135, Porto 4050-600, Portugal
First published on 23rd October 2015
Abstract
Iron oxide nanoparticles (ION) have been widely used in biomedical applications, for both diagnosis and therapy, due to their unique magnetic properties. They are intensively explored in neuromedicine mostly because of their ability to cross the blood brain barrier. Hence, their potential harmful effects on neuronal cells need to be carefully assessed. The objective of this study was to evaluate the toxicity of silica-coated ION (S-ION) (10–200 μg ml−1) on human neuronal SHSY5Y cells. Alterations in the cell cycle, cell death by apoptosis or necrosis, and membrane integrity were assessed as cytotoxicity parameters. Genotoxicity was determined by a γH2AX assay, a micronucleus (MN) test, and a comet assay. Complementarily, possible effects on DNA damage repair were also analysed by means of a DNA repair competence assay. All analyses were performed in complete and serum-free cell culture media. Iron ion release from the nanoparticles was notable only in complete medium. Despite being effectively internalized by the neuronal cells, S-ION presented in general low cytotoxicity; positive results were only obtained in some assays at the highest concentrations and/or the longest exposure time tested (24 h). Genotoxicity evaluations in serum-free medium were negative for all conditions assayed; in complete medium, dose and time-dependent increase in DNA damage not related to the production of double strand breaks or chromosome loss (according to the results of the γH2AX assay and MN test), was obtained. The presence of serum slightly influenced the behaviour of S-ION; further studies to investigate the formation of a protein corona and its role in nanoparticle toxicity are necessary.
Introduction
Since the birth of nanotechnology, iron oxide nanoparticles (ION) have gained interest for a wide variety of applications. Due to their unique magnetic properties ION have been widely utilized in various biomedical applications for both diagnosis and therapy, such as contrast agents in magnetic resonance imaging,1–3 heating mediators for cancer therapy,4 carriers for delivery of drugs5,6 and genes.7,8 Apart from these main applications, ION are intensively explored in neuromedicine mostly because they have the ability to cross the blood brain barrier.9,10 This ability, together with their limited toxic potential, makes them very suitable for use as promising diagnostic and therapeutic tools for nervous system malignancies.Considering that iron oxides occur naturally as nanosized crystals in the earth's crust,11 and that ION have already been used in clinical applications,12 it could seem that there is no underlying risk associated with these nanoparticles. Although some studies in the literature have shown that ION are less toxic than other types of metal nanoparticles,11,13,14 systematic studies on their effects on the human nervous system are rare, and their results have been inconsistent.15 Hence, considering the relevant uses and promising applications of ION in the neuromedicine field, their potential harmful effects on neuronal cells need to be carefully assessed.
Naked ION tend to form agglomerates becoming unstable over certain periods of time; they can be easily trapped by the immune system as foreign materials, which means that they cannot reach the desired target; and are chemically highly active and easily oxidized in air, resulting frequently in loss of magnetism and dispensability.16,17 To solve these problems, the surface of nanoparticles may be modified by coating with a number of materials for different purposes, and surface functionalisation may play a key role not only in regulating the cell-membrane penetration but also in affecting the cell activity.18 ION functionalized with different surface chemicals have been tested in different neuronal cell lines, showing conflicting results. While plain ION show a low health hazard,19 surface functionalisation can trigger very different cellular responses.20 Thus, exposure to dimercaptosuccinic acid-coated ION (maghemite) caused a dose-dependent reduction of viability and capacity of PC12 rat cells to extend neurites in response to the nerve growth factor.21 Similar results were obtained for ION coated with dextran, carboxydextran, lipid and citrate in the same cell type.22 Besides, in the same study different cytotoxic potentials were observed on c17.2 mouse neural progenitor cells; the citrate-coated ION were the most toxic and the lipid-coated ones were the least toxic, under the experimental conditions used. In contrast, polyethylene glycol-coated ION increased the efficiency of neurite outgrowth in a dose-dependent manner in nerve growth factor-stimulated PC12 cells.14 Thus, further and more detailed studies, particularly those employing human neural cells, are required to identify any potential toxicity associated with the use of ION with particular surface coatings.
Among all the possible surface modifications for ION, silica (SiO2) coating has several advantages that make it especially suitable to be employed for medical purposes. For instance, silica-coated ION (S-ION) are negatively charged at blood pH helping to avoid aggregate formation in body fluids,5 and its transparent matrix allows the efficient passage of excitation and emission light, which is a relevant property for imaging diagnosis.23 However, their potential toxicity on the nervous system has not been deeply addressed.
Therefore, the objective of this study was to evaluate the toxicity of S-ION on human neuronal SHSY5Y cells. To that aim, alterations in the cell cycle, cell death by apoptosis or necrosis, and membrane integrity were assessed as cytotoxicity parameters. Besides, genotoxicity was determined by evaluating the levels of phosphorylation of H2AX histone (γH2AX assay), a micronucleus (MN) test, and a comet assay. Complementarily, possible effects of S-ION on DNA damage repair were also analysed by means of a DNA repair competence assay.
Materials and methods
Chemicals
Bleomycin (BLM) (CAS no. 9041-93-4), camptothecin (Campt) (CAS no. 7689-03-4), mytomycin C (MMC) (CAS no. 50-07-7), hydrogen peroxide (H2O2), and propidium iodide (PI) were purchased from Sigma-Aldrich Co. BLM, Campt and MMC were dissolved in sterile distilled water prior use.Nanoparticle preparation
Silica-coated magnetite nanoparticles were synthesized and prepared as stable water suspensions (5 mg ml−1) as described by Yi et al.24 Prior to each treatment, a stock suspension (1 mg ml−1) was prepared in incomplete (serum-free) or complete SHSY5Y culture medium (see composition below), and was sonicated in a water bath for 5 min. Serial dilutions were carried out to obtain the different test concentrations, and sonicated in the water bath for an additional 5 min period. Physicochemical characteristics of these nanoparticles (particle size and morphology, surface chemistry, and hydrodynamic size and zeta potential in water, complete and incomplete SHSY5Y medium) were previously determined.25Dissolved iron concentrations in the cell culture media and artificial lysosomal fluid
To analyse the release of iron ions from the S-ION, suspensions (5, 10, 25, 50, 75, 100, 150, 175, 200, 250 and 300 μg ml−1) were incubated in incomplete or complete cell culture medium and in artificial lysosomal fluid (ALF) for 3, 6 and 24 h at 37 °C in a humidified 5% CO2 environment. The ALF solution (pH 4.5) was prepared as previously described by Midander et al.26 After centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min, the S-ION solid phase was removed from the liquid medium. Flame atomic absorption spectroscopy (FAAS) (Thermoelemental Solaar S4 v.10.02) was used to quantify the iron content in the supernatant. Cell culture media without nanoparticles being subjected to the same experimental conditions were used as negative controls.
000 rpm for 30 min, the S-ION solid phase was removed from the liquid medium. Flame atomic absorption spectroscopy (FAAS) (Thermoelemental Solaar S4 v.10.02) was used to quantify the iron content in the supernatant. Cell culture media without nanoparticles being subjected to the same experimental conditions were used as negative controls.
Cell culture and S-ION treatment
Human neuroblastoma SHSY5Y cells (European Collection of Cell Cultures) were grown in the nutrient mixture, EMEM/F12 (1:1) with 1% non-essential amino acids, 1% antibiotic and an antimycotic solution, supplemented with 10% heat inactivated foetal bovine serum (FBS) (all from Invitrogen) in a humidified incubator at 37 °C with 5% CO2. To carry out the experiments, cells were seeded in 96-well plates (5–6 × 104 cells per well), and allowed to adhere for 24 h at 37 °C prior to the experiments. For each experiment, these cells were incubated with four different S-ION concentrations (10, 50, 100 and 200 μg ml−1) at 37 °C for 3 or 24 h, in incomplete or complete cell culture medium.
The treatment doses and exposure times were selected from the previous results of cell viability assays, as producing a maximum decrease in viability of 30%.25 Incomplete or complete cell culture media were used as negative controls in all experiments. The following chemicals were used as positive controls: Campt (10 μM) for apoptosis; Triton X-100 (1%) for membrane integrity; MMC (1.5 μM) for the cell cycle and MN test; and BLM (1 μg ml−1) for the comet assay and γH2AX analysis.
Cellular uptake
The potential of the S-ION to be taken up by the cells was evaluated by means of flow cytometry using a FACSCalibur flow cytometer (Becton Dickinson). After the treatments, the analysis was carried out on the basis of size and complexity of the cells by measuring the forward scatter (FSC) and the side scatter (SSC), according to the protocol described.27Cell cycle analysis
The cell distribution along the different phases of the cell cycle was examined in cells treated with S-ION or the controls by measuring the relative cellular DNA content with a flow cytometry technique (FACSCalibur flow cytometer, Becton Dickinson), following the previously published protocol.28 An estimated DNA content of approximately 104 events was measured from the PI signal. Cell cycle histograms were analysed by Cell Quest Pro software (Becton Dickinson). Results are reported as the average percentage of cells in each phase of the cell cycle (G0/G1, S and G2/M). Complementarily, the subG1 region of the cell cycle distribution was also evaluated, indicative of the late stages of apoptosis.29Apoptosis and necrosis detection
A commercially available kit (BD Pharmingen™ Annexin-V FITC apoptosis kit) was used to quantify the various modes of cell death that may be potentially induced by S-ION treatment, following the manufacturer's recommendations. Approximately 104 events were acquired with a FASCalibur flow cytometer (Becton Dickinson). Data from Annexin V-Fluorescein isothiocyanate (FITC) and PI were analysed using Cell Quest Pro software (Becton Dickinson). Early apoptosis and late apoptosis/necrosis were expressed as the percentages of Annexin V+/PI− and Annexin V+/PI+ cells, respectively.Membrane integrity
A commercial kit (Roche Diagnostics) was used to measure the lactate dehydrogenase (LDH) activity in cell culture media, according to the manufacturer's instructions. After exposure, half the amount of the cell culture medium was collected for LDH measurement. Absorption was measured at 490 nm with a reference wavelength of 655 nm using a Cambrex ELx808 microplate reader (Biotek, KC4). Triton X-100 (1%) was taken as positive control and set as 100% cytotoxicity. LDH release was calculated as follows:where [A]sample, [A]medium and [A]positive control denote the absorbance of the sample, the corresponding medium negative control and Triton X-100 positive control, respectively.

γH2AX assay
At the end of the exposure period, the level of phosphorylation of H2AX histone at the serine 139 residue (γH2AX) was evaluated using an anti-human γH2AX-Alexa Fluor 488-conjugated antibody (Becton Dickinson), following previously published protocols,30,31 with some modifications.32 A minimum of 104 events were acquired with a flow cytometer (FACSCalibur, Becton Dickinson) to obtain data from Alexa Fluor 488 and PI fluorescence intensities. Data were analysed using Cell Quest Pro software (Becton Dickinson). Results are reported in each case as the percentage of events positive for both dyes, Alexa Fluor 488 and PI.Micronuclei evaluation
After treatments with S-ION and controls, the cells were cultured for an additional period of 24 h in fresh complete medium. Next, a suspension of nuclei and MN was prepared according to a previously reported procedure.33 The final suspension was analysed with a FACSCalibur flow cytometer (Becton Dickinson) as previously described.34Comet assay
To evaluate DNA damage induction the alkaline comet assay was performed after treating the cells with S-ION or the controls, following the previously published protocol35 with minor modifications.36 Comet IV Software (Perceptive Instruments) was used for image capture and analysis. In all cases 100 cells were scored (50 from each replicate slide), and the percentage of DNA in the comet tail (tDNA%) was used as the DNA damage parameter. Nevertheless, in order to be sure that the nanoparticles tested do not interfere with the comet assay protocol, a modified version of the comet assay was performed initially following the previously described protocol.37 Briefly, untreated cells were centrifuged at 200g for 5 min at 4 °C. After removing the supernatant, 40 μl S-ION were added directly to the cells just before mixing them with 40 μl of 2% low-melting-point (LMP) agarose, so that the final concentration of S-ION was 200 μg ml−1, the highest dose to be tested for genotoxicity. Then the alkaline comet assay was carried out following the general protocol described above.DNA repair competence assay
The experimental design previously described was followed to evaluate the effects of S-ION on DNA damage repair.38 It consisted of three consecutive phases: (i) in phase A (pre-treatment) cells were incubated for 3 or 24 h in the presence or absence of S-ION (50 μg ml−1) at 37 °C; (ii) in phase B (DNA damage induction) cells were challenged with H2O2 (100 μM) for 5 min at 37 °C in the presence or absence of S-ION (50 μg ml−1); and (iii) in phase C (repair) cells were washed in fresh medium to remove treatment, and incubated with or without S-ION (50 μg ml−1) for 30 min at 37 °C to allow DNA repair. The alkaline comet assay was then performed after phase B (data labelled as “before repair”) and phase C (data labelled as “after repair”) as described previously.Additionally, the cells were treated with S-ION (50 μg ml−1) for 30 min and the comet assay was performed immediately after. This was done to test whether 30 min incubation with S-ION (as occurs in phase C) might induce significant damage to DNA.
Statistical analysis
Statistical analyses were performed using SPSS for Windows statistical package (version 20.0). A minimum of three independent experiments were performed for each experimental condition tested, and each condition was always run in duplicate and under blind conditions. Experimental data were expressed as mean ± standard error. Differences between the groups were tested by using the Kruskal–Wallis test and Mann–Whitney U-test. The associations between the two variables were analysed by Spearman's correlation. A P-value of less than 0.05 was considered significant.Results
The S-ION used in this study were previously characterized regarding their physical–chemical features.25 In brief, transmission electron microscopy measurements showed that they are spherical particles with a mean diameter of 20.2 nm, and the dynamic light scattering results revealed that they aggregate to a low extent in both types of culture media (141.6 ± 1.4 nm in incomplete medium and 111.1 ± 1.1 nm in complete medium). They have a negative zeta potential in water (−31.8 mV) which decreases in cell culture media (−21.7 and −10.3 mV in serum-free and complete media, respectively).The release of iron ions from the S-ION was studied in incomplete and complete cell culture media, and also in ALF with respect to the influence of pH on nanoparticle degradation. It was found to be very low in incomplete medium and ALF solution when tested three times (3, 6 and 24 h) (Fig. 1). Nevertheless, important concentrations of dissolved iron were observed when S-ION were suspended in complete media, generally increasing with exposure time and nanoparticle dose.
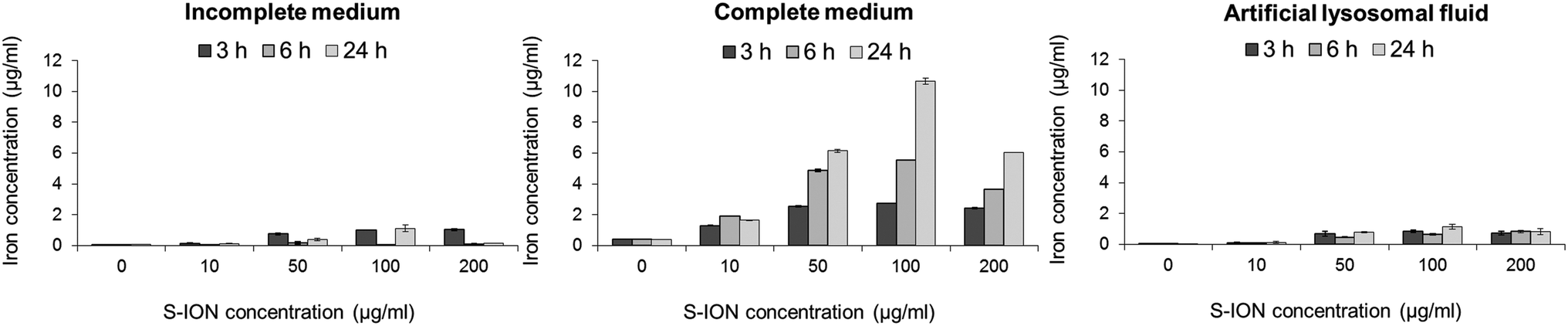 | ||
| Fig. 1 Analysis of iron ions released from S-ION in (A) incomplete cell culture medium, (B) complete cell culture medium, and (C) artificial lysosomal fluid. Bars represent the mean standard error. | ||
Results obtained by testing the ability of S-ION to enter the human neuroblastoma cells are shown in Fig. 2. The nanoparticles were effectively internalized by the cells under all conditions tested in a dose-dependent manner (incomplete medium: r = 0.824, P < 0.01 for 3 h treatment and r = 0.877, P < 0.01 for 24 h treatment; complete medium: r = 0.737, P < 0.01 for 3 h treatment and r = 0.692, P < 0.01 for 24 h treatment). However, uptake was slightly higher in serum-free medium than in complete medium, and for the highest dose tested it was more prominent at 3 h than at 24 h treatment.
 | ||
| Fig. 2 (A) Neuronal cell uptake of S-ION prepared in incomplete and complete medium. Bars represent the mean standard error. **P < 0.01, significant difference with regard to the corresponding negative control. (B) Flow cytometry plot of control cells (left), and cells treated with 200 μg ml−1 S-ION for 3 h in incomplete medium (right). R2: Region of cells containing nanoparticles. | ||
Fig. 3 shows the cell distribution during the various phases of the cell cycle after exposing the neuronal cells to S-ION. The 3 h treatments, regardless of the medium employed, did not modify the cell cycle, and significant alterations at 24 h treatments were only observed for the 200 μg ml−1 concentration (decrease in the G2/M phase and notable although not significant increase in the S phase for treatment in incomplete medium, and increase in the S phase for treatment in complete medium). Besides, the subG1 region of the cell cycle distribution was also evaluated, since DNA fragmentation, indicative of the late stages of apoptosis, results in the appearance of PI-stained events containing subG1 levels;29 results are shown in Fig. 4. No significant increase in the subG1 fraction was observed except for the cells exposed in serum-free medium to the highest S-ION dose for 24 h.
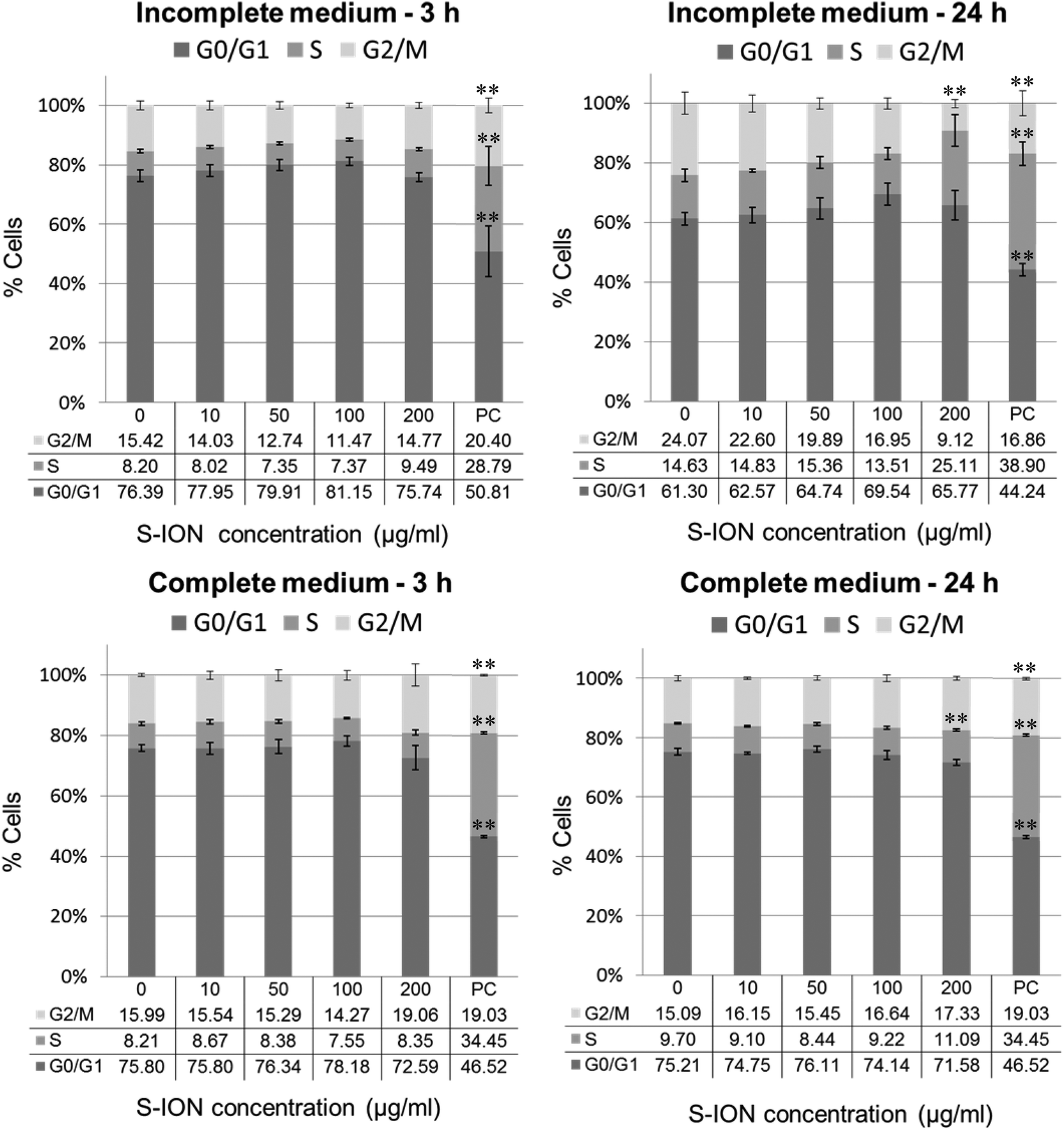 | ||
| Fig. 3 Analysis of SHSY5Y cell cycle after 3 and 24 h of treatment with S-ION prepared in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (MMC 1.5 μM). **P < 0.01, significant difference with regard to the corresponding negative control. | ||
 | ||
| Fig. 4 Apoptosis (% of cells in the subG1 region of cell cycle distribution) in neuronal cells treated with S-ION prepared in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (MMC 1.5 μM). *P < 0.05, significant difference with regard to the corresponding negative control. | ||
To further investigate whether treatments with S-ION were able to induce cell death by apoptosis or necrosis, a double stain with Annexin V and PI was carried out. Results obtained from the analyses showed that S-ION did not induce early apoptosis (events positive for Annexin V but negative for PI) at any concentration after 3 h of exposure regardless of the medium used (Fig. 5). After 24 h of treatment significant increases in the apoptosis rate could only be observed for the highest doses assayed (200 μg ml−1 in incomplete medium and 100 and 200 μg ml−1 in complete medium). No significant induction of necrosis/late apoptosis (events positive for both Annexin V and PI) was obtained under any experimental conditions tested.
 | ||
| Fig. 5 Apoptosis and necrosis cell rates (%) after exposure of neuronal cells to S-ION for 3 and 24 h prepared in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (Campt 10 μM). *P < 0.05, **P < 0.01, significant difference with regard to the corresponding negative control. | ||
The potential alterations in the neuronal cell membrane integrity caused by S-ION exposure were assessed by measuring LDH activity in extracellular medium, since LDH is released when the cell membrane is damaged. Results obtained in this test are shown in Fig. 6. No significant alteration in the percentage of LDH activity was observed at any medium, concentration or treatment time tested.
 | ||
| Fig. 6 Results of membrane integrity assessment (LDH assay) in SHSY5Y cells exposed to S-ION in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (Triton X-100 1%). **P < 0.01, significant difference with regard to the corresponding negative control. | ||
The genotoxic potential of the S-ION was examined using different approaches. As a rapid screening method for genotoxicity, we first analysed H2AX phosphorylation, an early cellular response to the induction of DNA double-strand breaks (DSB). As can be clearly seen in Fig. 7, S-ION did not induce γH2AX at either of the conditions analysed.
 | ||
| Fig. 7 Phosphorylation of H2AX histone after treatment of neuronal cells with S-ION prepared in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (BLM 1 μg ml−1). **P < 0.01, significant difference with regard to the corresponding negative control. | ||
Next, we applied a relatively less specific approach, the MN test scored by flow cytometry, in order to quantify chromosomal alterations. The results of MN evaluation showed that no significant changes were produced in the MN ratio after treatment of the neuronal cells with the S-ION (Fig. 8).
 | ||
| Fig. 8 MN induction in neuronal cells after treatment with S-ION prepared in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (MMC 1.5 μM). *P < 0.05, significant difference with regard to the corresponding negative control. | ||
The comet assay (single cell gel electrophoresis) was used for measuring primary DNA damage in SHSY5Y cells caused by exposure to S-ION. Due to the distinct physicochemical characteristics of nanomaterials, several possible interferences may occur with the comet assay methodology.39 Thus, a comprehensive test for detecting these interferences was carried out before starting DNA damage evaluation, following Magdolenova et al.37 As can be observed from Fig. 9, addition of S-ION to the cells just before the lysis step was found not to interfere with the subsequent steps of the experimental protocol, since no differences were found between the DNA damage measured in the absence and in the presence of the nanoparticles. When the comet assay was applied to neuronal cells treated with S-ION in serum-free medium, no significant alteration in %tDNA was detected (Fig. 10). Nevertheless, dose-dependent induction of DNA damage was observed in complete medium (r = 0.948, P < 0.05 for 3 h treatment, and r = 0.842, P < 0.05 for 24 h treatment).
 | ||
| Fig. 9 Results of interference testing between S-ION (200 μg ml−1) and comet assay methodology in incomplete and complete medium. Bars represent the mean standard error. | ||
 | ||
| Fig. 10 DNA damage induction in neuronal cells after treatment with S-ION prepared in incomplete and complete medium. Bars represent the mean standard error. PC: Positive control (BLM 1 μg ml−1). *P < 0.05, **P < 0.01, significant difference with regard to the corresponding negative control. | ||
Results obtained in the DNA repair competence assay are shown in Fig. 11. When cells were challenged with H2O2 and no exposure to S-ION was carried out, there was a significant decrease in the level of DNA damage after the 30 min repair period in both media tested. When incubation with S-ION was carried out before damage induction by H2O2 (phase A, either 3 or 24 h), no repair was observed in serum-free medium but %tDNA was significantly reduced in complete medium, more pronounced in incubation for 24 h than for 3 h. Similar results were obtained when S-ION were applied only during the 30 min repair period (phase C), although treatment of cells for 30 min with only S-ION increased significantly the DNA damage over the control level in both cell culture media. However, the opposite occurred for experiments where treatment with H2O2 and S-ION were performed simultaneously (phase B), i.e., significant repair was observed in incomplete medium and no repair in complete medium.
 | ||
| Fig. 11 Effects of S-ION on repair of H2O2-induced DNA damage in neuronal cells. Incubation with S-ION was carried out either before H2O2 treatment (phase A), simultaneously (phase B), or during the repair period (phase C). Bars represent the mean standard error. *P < 0.05, **P < 0.01, significant difference with regard to the same treatment before repair; #P < 0.05, significant difference with regard to the control. | ||
Discussion
Magnetic nanoparticles are one of the first nanomaterials to be approved for clinical use.40 Besides, because of their ability to overcome the restraints of the blood–brain barrier,41 they have been used as carriers for the transport of drugs, siRNA, or DNA into the brain. To date, little is known on the effects of ION on the human nervous system. Few studies have been published regarding the in vitro neurotoxicity of ION on human neural cell lines, and results obtained are not consistent.42–44Due to its chemical stability, coating with silica can transform ION by increasing their biocompatibility and offering the capacity to functionalize their surface without affecting their magnetic properties.45–47 Moreover, silica-coating helps to convert hydrophobic nanoparticles into hydrophilic water-soluble particles.48 Nevertheless, despite the numerous advantages and potential applications in neuromedicine, the possible neurotoxicity of S-ION has not been completely discarded yet.
There is a consensus that in vitro methods can provide useful information concerning basic biological processes underlying neurotoxicity, and specific information about the mechanisms of action of neurotoxicants.49 Therefore human SHSY5Y neuroblastoma cells were chosen as an appropriate cell model widely used for studying neurotoxicity, since they maintain many biochemical and functional properties of neurons.50 The toxicity of S-ION on SHSY5Y cells was evaluated using a range of concentrations (10–200 μg ml−1) and two treatment times (3 and 24 h). As physicochemical properties of nanomaterials may be quite different depending on how they are suspended, we also considered the possible differences in toxicity of S-ION when they were prepared under biologically relevant conditions (absence or presence of FBS).
Analysis of iron ion release from the S-ION suspensions showed a different behaviour of the nanoparticles depending on the media composition. Low concentrations of iron were detected in incomplete medium and ALF solution, whereas the release of ions was notable in the presence of serum (complete medium), in general increasing with time and S-ION concentration. Release of iron ions from ION was previously described in a number of studies.51 However this release can vary depending on the suspension conditions (e.g. pH) and the nanoparticle surface coating.52 Results obtained here suggest that degradation of the studied S-ION is not pH-dependent as observed for other S-ION.52 Furthermore this degradation is also not dependent on the particle size, since similar hydrodynamic diameters obtained in different media showed very different dissolution rates.
The chemical synthesis, as well as the presence of a coating, which surrounds and isolates the magnetic material from the environment, and its physicochemical properties, may influence the degradation rate of the particles and so the release of iron ions.53,54 This would explain the differences found in our study, since ION suspended in complete medium may externally interact with serum proteins, thus favouring the silica coating degradation and causing a higher iron release from the nanoparticle core. In fact, proteins may increase the dissolution rates of iron oxides through both aqueous complexation and ligand-enhanced dissolution.55
The evaluation of cellular uptake of nanoparticles by flow cytometry using the side scatter parameter (indicative of cell granularity/complexity) is suitable for initial screening of nanotoxicity.56 Experimental data confirmed that the S-ION were effectively taken up by neuroblastoma cells, and the uptake was higher when exposure was performed in the absence of serum. These findings agree with previous studies in other cell types showing that S-ION are quickly internalized by macrophages,57 and by A549 and HeLa cells.52 Differences in nanoparticle uptake were previously reported by Krais et al., who studied the role of serum proteins on ION uptake, and observed that the presence of a protein ‘corona’ may indeed influence the cellular uptake of folic acid-functionalized ION.58 Agreeing with our results, Salvati et al. speculated that excessive binding of serum proteins may prevent selective, ligand-mediated uptake of nanoparticles.59 Besides, our in vitro studies revealed a remarkable lower degree of internalization for the highest S-ION concentration at 24 h when compared to the one obtained at 3 h. This is likely due to the progressive nanoparticle agglomeration at this high concentration, which causes a more noticeable interference with the uptake process at the longest exposure period.
Cell cycle machinery corresponds to a series of events which lead the cell to its division and duplication.60 Results obtained from the analysis of the cell cycle showed that exposure for 3 h to S-ION did not alter it at any concentration, which agrees with previous findings from some other studies using bare or differently coated ION.61 However, significant mitotic arrest (increase in the rate of cells in the S phase and/or decrease in the rate of cells in the G2/M phase) was observed for 24 h treatments in both culture media at the highest dose tested. Similar alterations in the cell cycle were observed by Namvar et al. after exposing Jurkat cells to bare magnetite nanoparticles prepared by green biosynthesis (using a brown seaweed), but the dose used was much lower (6.4 μg ml−1, corresponding to the inhibitory concentration 50 [IC50], calculated by the MTT assay).62 In the previous cell viability assays with our S-ION,25 viabilities obtained for treatments up to 200 μg ml−1 were always higher than or equal to 70% as calculated by MTT, neutral red uptake and alamar blue assays; therefore cytotoxicity of the current nanoparticles, at least to SHSY5Y cells, was much lower than cytotoxicity of the ION used by Namvar et al. in Jurkat cells. These observations agree with the general assumption that ION coated with silica are indeed less toxic that bare ION. Besides, in a very recently published study, Couto et al. were unable to find any alteration in the cell cycle when testing polyacrylic acid (PAA)-coated and non-coated ION on human T lymphocytes (48 h treatments).63
In order to evaluate apoptosis, which is critical in many physiological and pathological processes, we used two alternative strategies: analysis of the subG1 region of the cell cycle distribution, indicative of DNA fragmentation at the late stages of apoptosis, and Annexin V/PI staining for sensitive detection of early stage apoptosis. Results obtained with the two strategies were quite similar. The only significant increase in apoptosis rate observed was for the highest S-ION concentration after 24 h treatments in both media tested. The exception was the subG1 region in complete medium, which did not show any significant alteration. This difference may be explained on the basis of the methodological differences between the two techniques used. Annexin V/PI staining and measurement are carried out just after the treatments, and reflect early stage apoptosis, meanwhile subG1 region analysis is performed after an additional 24 h incubation period following the treatments, and is indicative of late stage apoptosis. Hence, probably the cells undergoing early apoptosis detected by Annexin V/PI staining have already been mostly removed when the subG1 region was analysed. Similar apoptosis results were reported by Jeng and Swanson, who found that ION only induce apoptosis in mouse Neuro-2A neuroblastoma cells after exposure to concentrations higher than 100 μg ml−1.64 Contrarily, Namvar et al. described time-dependent (from 12 to 48 h) increases in the apoptosis rates in Jurkat cells treated with bare magnetite nanoparticles (6.4 μg ml−1), evaluated by the two same methodologies used in the current work.62 Likewise, significant apoptosis induction (evaluated by means of mitochondrial membrane potential, JC-1 assay) in cervical and lung cells exposed to 2.5 nM S-ION (magnetite) for 48 h was reported.52 This concentration is equivalent to approximately 30 μg ml−1 of our S-ION, dose which produced negative results under all conditions tested in this study.
Cell death by necrosis (and/or late apoptosis) was also determined by Annexin V/PI staining, and no alterations in this rate were found in SHSY5Y cells exposed to S-ION in any of the conditions assayed. Namvar et al. obtained contrary results again: time-dependent increase in the necrosis/late apoptosis rate in Jurkat cells treated with ION but, as mentioned above, toxicity of these nanoparticles (in terms of cell viability decrease) was much higher.62
Possible effects of S-ION exposure on cell membrane integrity of neurons were evaluated by the LDH leakage assay. Negative results were obtained for all experimental conditions evaluated, which is essentially in agreement with most results obtained for cell cycle and apoptosis or necrosis induction, and also with previous results from the same nanoparticles, doses and cell line using MTT and alamar blue viability assays.25 Nevertheless, positive LDH leakage results were obtained by Malvindi et al. after treatment of A549 and HeLa cells for 48 and 96 h with 2.5 nM S-ION (as already mentioned, equivalent to 30 μg ml−1 of the current S-ION), according to their apoptosis assessment results and confirming the lower cytotoxicity of our silica-coated nanoparticles.52
Taking all cytotoxicity results together, S-ION tested showed low cytotoxic potential; data from serum-free medium indicate a slightly larger harmful potential [viability reduction,25 cell cycle alterations and apoptosis induction (present study)], agreeing with the faintly higher entrance of the nanoparticles into the cells.
For testing the potential of S-ION to induce damage on the genetic material, we used a battery of genotoxicity tests, i.e., the γH2AX assay, the MN test, and the comet assay. As a response to the formation of DNA DSB, H2AX flanking the DSB sites are rapidly phosphorylated at the serine 139 residue to become γH2AX. Under normal conditions, γH2AX foci appear within few minutes after the lesion, reach maximum levels after about 30 min, and their half-life has been estimated to be 2–7 h (reviewed in Valdiglesias et al.).65 Reliability and specificity of the γH2AX assay as a biomarker of DNA damage have already been proved.66,67 We used the γH2AX assay evaluated by flow cytometry since it provides an automated, fast, practical, and reproducible high-throughput platform that increases considerably the number of cells evaluated, diminishing the variability and enhancing the statistical power of the results.68 No significant increase in the γH2AX levels was observed in SHSY5Y cells after exposure to S-ION. No other study employing the γH2AX assay for testing genotoxicity caused by any type of ION could be found in the literature; however this cell line showed significant H2AX phosphorylation activity when treated with ZnO nanoparticles69 but not when exposed to different types of TiO2 nanoparticles.70
The purpose of the MN test is to identify chromosome aberrations, since MN may contain lagging chromosome fragments or whole chromosomes; therefore it detects both clastogenic and aneugenic events. After S-ION treatment no effects were observed in the neuronal cells in terms of MN formation. To our knowledge no studies have been reported on MN induction by S-ION in any type of cells so far, but passiveness of other types of ION on MN formation have been documented in cells from a different origin in in vitro71–73 and in vivo studies.71,74,75
The comet assay is one of the most frequently used methods for genotoxicity testing. Since it is simple, versatile, and able to detect different DNA lesions, it has been claimed to be the most promising assay to measure potential genotoxicity of nanomaterials.76 Thus, we applied the alkaline comet assay to examine primary DNA damage induced by S-ION. But before that, we confirmed that these nanoparticles do not interfere with the assay methodology, since interference of different nanomaterials had been previously reported.37,77,78 When treatments were carried out in serum-free medium no significant induction of DNA damage was observed. However, in complete medium S-ION induced dose- and time-dependent increase in the comet parameter, in agreement with the iron ion dissolution determination, which showed significant amounts of ions released from the nanoparticles in complete medium. Although the human body contains relatively high concentrations of iron, the presence of this metal at concentrations higher than physiological can lead to deleterious effects. Iron ions are able to interact with DNA in-between the bases, thereby unwinding the double-helix79 and causing single strand breaks (SSB) and oxidative base modification.80 This kind of damage, especially SSB, is detected by the standard alkaline comet assay but is not related to phosphorylation of H2AX or MN production. Therefore, this may help to explain the positive results obtained in the comet assay and the negative ones from the H2AX assay and the MN test. According to the current results, the type of DNA damage induced by S-ION on neuronal cells is likely not related to DSB but mostly to repairable DNA lesions (alkali labile sites and SSB), indicating recent damage.81 Similar increases in comet assay parameters (tail length and tail DNA intensity) were reported by Malvindi et al. in A549 and HeLa cells treated with S-ION,52 by Hong et al. in murine L-929 fibroblast cells exposed to ION coated with (3-aminopropyl)trimethoxysilane (APTMS), tetraethyl-orthosilicate (TEOS)–APTMS, or citrate,82 and by Bhattacharya et al. in human lung IMR-90 fibroblasts and bronchial epithelial BEAS-2B cells treated with bare hematite.83 Moreover, no induction of chromosomal aberrations (which require DSB production) was observed in human T lymphocytes treated with PAA-coated and non-coated ION,63 which further supports our results.
Possible effects of S-ION on DNA repair processes were tested by the DNA repair competence assay using H2O2 as the challenging agent. H2O2 causes damage to DNA by generating hydroxyl-free radicals (OH˙).84 These radicals attack DNA at the sugar residue of the DNA backbone, leading to SSB.85
Rejoining of SSB induced by H2O2 is a simple cellular process; thousands of breaks per cell can be repaired in a matter of half an hour in typical cultured mammalian cells.81 In the current study, repair of approximately one-third of the DNA damage observed after H2O2 treatment was obtained during a 30 min period, both in incomplete and in complete media. Incubation with the nanoparticles was carried out at three different stages of the assay: before inducing DNA damage (pre-treatment or phase A, for 3 or 24 h), during DNA damage induction (phase B), or during the repair period (phase C). Results obtained were different depending on the presence of serum in the medium. In incomplete medium no significant decrease in the DNA damage during the repair phase was observed when S-ION incubation was carried out before DNA damage induction, or during the repair phase. Since incubation only with S-ION for 30 min caused a significant increase in the comet parameter, maybe the negative repair result obtained for phase C is related to DNA damage induced directly by the S-ION instead of (or in addition) the actual disturbance on the repair machinery. When treatment with H2O2 and the nanoparticles was performed simultaneously the repair process occurred normally; a possible explaining reason is the short time for this incubation (only 5 min), insufficient to cause any alteration in the repair systems.
The results observed in complete medium suggested scarce effects on DNA repair, since significant decreases in the DNA damage were observed after the repair period under all conditions tested, except for phase B. As indicated before, a notable release of iron ions from the S-ION took place in complete medium. The deleterious effects of transition metal ions, such as iron, to DNA are greatly enhanced by the presence of oxygen and related species; thus, iron ions readily associate with DNA and, in the presence of hydrogen peroxide, a high ratio of DSB to SSB is generated.86 It is well known that the repair of DSB can take hours.87 Therefore, the result obtained for phase B in complete medium is probably more related to the type of DNA damage induced (more DSB than SSB), for which the 30 min repair period tested was not long enough, than to alterations in the repair process. To the best of our knowledge, this is the first study addressing the potential effects of ION on cellular repair systems. Therefore, further investigations are required to go into detail about all these findings.
Conclusion
Despite being effectively internalized by the neuronal cells, S-ION presented general low cytotoxicity; positive results were only obtained in some assays at the highest concentrations and/or the longest exposure time tested. Genotoxicity evaluations in incomplete medium were negative for all conditions assayed; in complete medium dose and time-dependent increase in DNA damage, not related to the production of DSB or chromosome loss (according to the results of the γH2AX assay and the MN test), was obtained. Differences in the three genotoxicity assays applied, regarding their sensitivity to detect different types of genetic damage, confirm the need for using them in combination, since they complement one another.The medium composition (presence or absence of serum) influenced the behaviour of S-ION, although not to a great extent. Uptake of the nanoparticles by the cells, cytotoxicity, and effects on DNA repair were more pronounced in the absence of serum. On the contrary, iron ion release and primary DNA damage were only observed in complete medium. Formation of a protein corona in the presence of serum has probably an important role in these differences. Further studies are needed to determine the protein corona formation and to elucidate the possible role of redox imbalance in the generation of harmful effects, particularly those related to DNA damage.
Our study demonstrates how the preparation conditions, beyond the intrinsic properties of the nanoparticles, may determine the cytotoxic and genotoxic outcomes. Results obtained in this work also highlight the importance of screening of possible interactions of nanoparticles with the living systems, especially with the nervous system components, in order to increase the knowledge on the effects of nanoparticles at different levels to be able to guarantee manufacturers’ and consumers’ safety.
Acknowledgements
This work was funded by Xunta de Galicia (EM 2012/079) and by TD1204 MODENA COST Action. G. Kiliç was supported by a fellowship from the University of A Coruña.References
- Y.-K. Peng, C. N. P. Lui, T.-H. Lin, C. Chang, P.-T. Chou, K. K. L. Yung and S. C. E. Tsang, Faraday Discuss., 2014, 175, 13–26 CAS.
- S. Xue, Y. Wang, M. Wang, L. Zhang, X. Du, H. Gu and C. Zhang, Int. J. Nanomed., 2014, 9, 2527–2538 Search PubMed.
- L. Zhang, Y. Wang, Y. Tang, Z. Jiao, C. Xie, H. Zhang, P. Gu, X. Wei, G.-Y. Yang, H. Gu and C. Zhang, Nanoscale, 2013, 5, 4506–4516 RSC.
- M. Dan, Y. Bae, T. A. Pittman and R. A. Yokel, Pharm. Res., 2014, 32, 1615–1625 CrossRef PubMed.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- H. Wang, J. Yi, S. Mukherjee, P. Banerjee and S. Zhou, Nanoscale, 2014, 6, 13001–13011 RSC.
- D. Li, X. Tang, B. Pulli, C. Lin, P. Zhao, J. Cheng, Z. Lv, X. Yuan, Q. Luo, H. Cai and M. Ye, Int. J. Nanomed., 2014, 9, 3347–3361 Search PubMed.
- S. C. McBain, U. Griesenbach, S. Xenariou, A. Keramane, C. D. Batich, E. W. F. W. Alton and J. Dobson, Nanotechnology, 2008, 19, 405102 CrossRef CAS PubMed.
- S. D. Kong, J. Lee, S. Ramachandran, B. P. Eliceiri, V. I. Shubayev, R. Lal and S. Jin, J. Controlled Release, 2012, 164, 49–57 CrossRef CAS PubMed.
- J. Wang, Y. Chen, B. Chen, J. Ding, G. Xia, C. Gao, J. Cheng, N. Jin, Y. Zhou, X. Li, M. Tang and X. M. Wang, Int. J. Nanomed., 2010, 5, 861–866 CAS.
- N. Singh, G. J. S. Jenkins, R. Asadi and S. H. Doak, Nano Rev., 2010, 1 Search PubMed.
- P. Reimer and T. Balzer, Eur. J. Radiol., 2003, 13, 1266–1276 Search PubMed.
- H. L. Karlsson, P. Cronholm, J. Gustafsson and L. Möller, Chem. Res. Toxicol., 2008, 21, 1726–1732 CrossRef CAS PubMed.
- J.-E. Kim, J.-Y. Shin and M.-H. Cho, Arch. Toxicol., 2012, 86, 685–700 CrossRef CAS PubMed.
- V. Valdiglesias, G. Kiliç, C. Costa, N. Fernández-Bertólez, E. Pásaro, J. P. Teixeira and B. Laffon, Environ. Mol. Mutagen., 2015, 56, 125–148 CrossRef CAS PubMed.
- C.-W. Lu, Y. Hung, J.-K. Hsiao, M. Yao, T.-H. Chung, Y.-S. Lin, S.-H. Wu, S.-C. Hsu, H.-M. Liu, C.-Y. Mou, C.-S. Yang, D.-M. Huang and Y.-C. Chen, Nano Lett., 2007, 7, 149–154 CrossRef CAS PubMed.
- P. B. Santhosh and N. P. Ulrih, Cancer Lett., 2013, 336, 8–17 CrossRef CAS PubMed.
- A. B. Davila-Ibanez, V. Salgueirino, V. Martinez-Zorzano, R. Mariño-Fernández, A. García-Lorenzo, M. Maceira-Campos, M. Muñoz-Ubeda, E. Junquera, E. Aicart, J. Rivas, F. J. Rodriguez-Berrocal and J. L. Legido, ACS Nano, 2012, 6, 747–759 CrossRef CAS PubMed.
- S. Y. Liu, Y. Han, L. P. Yin, L. Long and R. Liu, Adv. Mater. Res., 2008, 47–50, 1097–1100 CrossRef CAS.
- E. Ying and H.-M. Hwang, Sci. Total Environ., 2010, 408, 4475–4481 CrossRef CAS PubMed.
- T. R. Pisanic 2nd, J. D. Blackwell, V. I. Shubayev, R. R. Fiñones and S. Jin, Biomaterials, 2007, 28, 2572–2581 CrossRef PubMed.
- S. J. H. Soenen, U. Himmelreich, N. Nuytten and M. De Cuyper, Biomaterials, 2011, 32, 195–205 CrossRef CAS PubMed.
- R. Alwi, S. Telenkov, A. Mandelis, T. Leshuk, F. Gu, S. Oladepo and K. Michaelian, Biomed. Opt. Express, 2012, 3, 2500–2509 CrossRef CAS PubMed.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- C. Costa, F. Brandão, M. J. Bessa, S. Costa, V. Valdiglesias, G. Kiliç, N. Fernández-Bertólez, P. Quaresma, E. Pereira, E. Pásaro, B. Laffon and J. P. Teixeira, J. Appl. Toxicol., 2015 DOI:10.1002/jat.3213.
- K. Midander, J. Pan, I. O. Wallinder and C. Leygraf, J. Environ. Monit., 2007, 9, 74–81 RSC.
- H. Suzuki, T. Toyooka and Y. Ibuki, Environ. Sci. Technol., 2007, 41, 3018–3024 CrossRef CAS PubMed.
- V. Valdiglesias, B. Laffon, E. Pásaro and J. Méndez, J. Environ. Monit., 2011, 13, 1831–1840 RSC.
- P. J. Fraker, L. E. King, D. Lill-Elghanian and W. G. Telford, Methods Cell Biol., 1995, 46, 57–76 CrossRef CAS PubMed.
- T. Tanaka, D. Halicka, F. Traganos and Z. Darzynkiewicz, in Chromatin Protocols, ed. S. P. Chellappan, Humana Press, 2009, pp. 161–168 Search PubMed.
- G. P. Watters, D. J. Smart, J. S. Harvey and C. A. Austin, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2009, 679, 50–58 CrossRef CAS PubMed.
- V. Valdiglesias, B. Laffon, E. Pásaro and J. Méndez, J. Toxicol. Environ. Health, Part A, 2011, 74, 980–992 CrossRef CAS PubMed.
- M. Nüsse, W. Beisker, J. Kramer, B. M. Miller, G. A. Schreiber, S. Viaggi, E. M. Weller and J. M. Wessels, Methods in Cell Biology, 1994, 42, 149–158 Search PubMed.
- S. L. Avlasevich, S. M. Bryce, S. E. Cairns and S. D. Dertinger, Environ. Mol. Mutagen., 2006, 47, 56–66 CrossRef CAS PubMed.
- N. P. Singh, M. T. McCoy, R. R. Tice and E. L. Schneider, Exp. Cell Res., 1988, 175, 184–191 CrossRef CAS PubMed.
- B. Laffon, E. Pásaro and J. Méndez, Toxicol. Lett., 2002, 126, 61–68 CrossRef CAS PubMed.
- Z. Magdolenova, Y. Lorenzo, A. Collins and M. Dusinska, J. Toxicol. Environ. Health, Part A, 2012, 75, 800–806 CrossRef CAS PubMed.
- B. Laffon, V. Valdiglesias, E. Pásaro and J. Méndez, Biol. Trace Elem. Res., 2010, 133, 12–19 CrossRef CAS PubMed.
- H. L. Karlsson, S. Di Bucchianico, A. R. Collins and M. Dusinska, Environ. Mol. Mutagen., 2014, 56, 82–96 CrossRef PubMed.
- P. Gould, Nano Today, 2006, 1, 34–39 Search PubMed.
- L. B. Thomsen, T. Linemann, K. M. Pondman, J. Lichota, K. S. Kim, R. J. Pieters, G. M. Visser and T. Moos, ACS Chem. Neurosci., 2013, 4, 1352–1360 CrossRef CAS PubMed.
- Z. Chen, J.-J. Yin, Y.-T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, ACS Nano, 2012, 6, 4001–4012 CrossRef CAS PubMed.
- B. H. Kenzaoui, C. C. Bernasconi, H. Hofmann and L. Juillerat-Jeanneret, Nanomed., 2012, 7, 39–53 CrossRef CAS PubMed.
- J.-J. Xiang, J.-Q. Tang, S.-G. Zhu, X.-M. Nie, H.-B. Lu, S.-R. Shen, X.-L. Li, K. Tang, M. Zhou and G.-Y. Li, J. Gene Med., 2003, 5, 803–817 CrossRef CAS PubMed.
- A. G. Kolhatkar, A. C. Jamison, D. Litvinov, R. C. Willson and T. R. Lee, Int. J. Mol. Sci., 2013, 14, 15977–16009 CrossRef PubMed.
- S. Larumbe, C. Gómez-Polo, J. I. Pérez-Landazábal and J. M. Pastor, J. Phys.: Condens. Matter, 2012, 24, 266007 CrossRef CAS PubMed.
- S. Rittikulsittichai, B. Singhana, W. W. Bryan, S. Sarangi, A. C. Jamison, A. Brazdeikis and T. R. Lee, RSC Adv., 2013, 3, 7838–7849 RSC.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 CrossRef CAS PubMed.
- D. J. Barbosa, J. P. Capela, M. de L. Bastos and F. Carvalho, Toxicol. Res., 2015, 4, 801–842 RSC.
- H. Xie, L. Hu and G. Li, Chin. Med. J., 2010, 123, 1086–1092 CAS.
- S. J. H. Soenen and M. De Cuyper, Contrast Media Mol. Imaging, 2009, 4, 207–219 CrossRef CAS PubMed.
- M. A. Malvindi, V. De Matteis, A. Galeone, V. Brunetti, G. C. Anyfantis, A. Athanassiou, R. Cingolani and P. P. Pompa, PLoS One, 2014, 9, e85835 Search PubMed.
- M. Lévy, F. Lagarde, V.-A. Maraloiu, M.-G. Blanchin, F. Gendron, C. Wilhelm and F. Gazeau, Nanotechnology, 2010, 21, 395103 CrossRef PubMed.
- E. Mahon, D. R. Hristov and K. A. Dawson, Chem. Commun., 2012, 48, 7970–7972 RSC.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- Y. Ibuki and T. Toyooka, Methods Mol. Biol., 2012, 926, 157–166 CAS.
- A. Kunzmann, B. Andersson, C. Vogt, N. Feliu, F. Ye, S. Gabrielsson, M. S. Toprak, T. Buerki-Thurnherr, S. Laurent, M. Vahter, H. Krug, M. Muhammed, A. Scheynius and B. Fadeel, Toxicol. Appl. Pharmacol., 2011, 253, 81–93 CrossRef CAS PubMed.
- A. Krais, L. Wortmann, L. Hermanns, N. Feliu, M. Vahter, S. Stucky, S. Mathur and B. Fadeel, Nanomedicine, 2014, 10, 1421–1431 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- D. O. Morgan, The Cell Cycle: Principles of Control, New Science Press in association with Oxford University Press, London, 2007 Search PubMed.
- M. Mahmoudi, K. Azadmanesh, M. A. Shokrgozar, W. S. Journeay and S. Laurent, Chem. Rev., 2011, 111, 3407–3432 CrossRef CAS PubMed.
- F. Namvar, H. S. Rahman, R. Mohamad, J. Baharara, M. Mahdavi, E. Amini, M. S. Chartrand and S. K. Yeap, Int. J. Nanomed., 2014, 9, 2479–2488 CrossRef PubMed.
- D. Couto, R. Sousa, L. Andrade, M. Leander, M. A. Lopez-Quintela, J. Rivas, P. Freitas, M. Lima, G. Porto, B. Porto, F. Carvalho and E. Fernandes, Toxicol. Lett., 2015, 234, 67–73 CrossRef CAS PubMed.
- H. A. Jeng and J. Swanson, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2006, 41, 2699–2711 CrossRef CAS PubMed.
- V. Valdiglesias, S. Giunta, M. Fenech, M. Neri and S. Bonassi, Mutat. Res., Rev. Mutat. Res., 2013, 753, 24–40 CrossRef CAS PubMed.
- C. Garcia-Canton, A. Anadón and C. Meredith, Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA, 2012, 26, 1075–1086 CrossRef CAS PubMed.
- T. Nikolova, M. Dvorak, F. Jung, I. Adam, E. Krämer, A. Gerhold-Ay and B. Kaina, Toxicol. Sci., 2014, 140, 103–117 CrossRef CAS PubMed.
- M. Sánchez-Flores, E. Pásaro, S. Bonassi, B. Laffon and V. Valdiglesias, Toxicol. Sci., 2015, 144, 406–413 CrossRef PubMed.
- V. Valdiglesias, C. Costa, G. Kiliç, S. Costa, E. Pásaro, B. Laffon and J. P. Teixeira, Environ. Int., 2013, 55, 92–100 CrossRef CAS PubMed.
- V. Valdiglesias, C. Costa, V. Sharma, G. Kiliç, E. Pásaro, J. P. Teixeira, A. Dhawan and B. Laffon, Food Chem. Toxicol., 2013, 57, 352–361 CrossRef CAS PubMed.
- Y. Li, J. Liu, Y. Zhong, D. Zhang, Z. Wang, L. Wang, Y. An, M. Lin, Y. Gao and J. Zhang, Int. J. Nanomedicine, 2011, 2805 CrossRef CAS PubMed.
- V. Shah, O. Taratula, O. B. Garbuzenko, M. L. Patil, R. Savla, M. Zhang and T. Minko, Curr. Drug Discovery Technol., 2013, 10, 8–15 CAS.
- T. Zhang, L. Qian, M. Tang, Y. Xue, L. Kong, S. Zhang and Y. Pu, J. Nanosci. Nanotechnol., 2012, 12, 2866–2873 CrossRef CAS PubMed.
- D. Chen, Q. Tang, X. Li, X. Zhou, J. Zang, W. Xue, J. Xiang and C. Guo, Int. J. Nanomed., 2012, 7, 4973–4982 CrossRef CAS PubMed.
- W. Wu, B. Chen, J. Cheng, J. Wang, W. Xu, L. Liu, G. Xia, H. Wei, X. Wang, M. Yang, L. Yang, Y. Zhang, C. Xu and J. Li, Int. J. Nanomed., 2010, 5, 1079–1084 Search PubMed.
- Z. Magdolenova, A. Collins, A. Kumar, A. Dhawan, V. Stone and M. Dusinska, Nanotoxicology, 2014, 8, 233–278 CrossRef CAS PubMed.
- H. L. Karlsson, Anal. Bioanal. Chem., 2010, 398, 651–666 CrossRef CAS PubMed.
- V. Stone, H. Johnston and R. P. F. Schins, Crit. Rev. Toxicol., 2009, 39, 613–626 CrossRef CAS PubMed.
- G. L. Eichhorn and Y. A. Shin, J. Am. Chem. Soc., 1968, 90, 7323–7328 CrossRef CAS PubMed.
- S. Toyokuni and J. L. Sagripanti, Free Radical Res., 1999, 31, 123–128 CrossRef CAS PubMed.
- A. Azqueta and A. R. Collins, Arch. Toxicol., 2013, 87, 949–968 CrossRef CAS PubMed.
- S. C. Hong, J. H. Lee, J. Lee, H. Y. Kim, J. Y. Park, J. Cho, J. Lee and D.-W. Han, Int. J. Nanomed., 2011, 6, 3219–3231 CAS.
- K. Bhattacharya, M. Davoren, J. Boertz, R. P. Schins, E. Hoffmann and E. Dopp, Part. Fibre Toxicol., 2009, 6, 17 CrossRef PubMed.
- P. Jaruga and M. Dizdaroglu, Nucleic Acids Res., 1996, 24, 1389–1394 CrossRef CAS PubMed.
- G. M. Benhusein, E. Mutch, S. Aburawi and F. M. Williams, Libyan J. Med., 2010, 5 Search PubMed.
- D. R. Lloyd and D. H. Phillips, Mutat. Res., 1999, 424, 23–36 CrossRef CAS PubMed.
- M. Frankenberg-Schwager, Radiother. Oncol., 1989, 14, 307–320 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |