
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design and characterization of heteroleptic phosphorescent iridium(III) complexes for highly efficient deep-blue OLEDs†
Yansong
Feng
a,
Xuming
Zhuang
a,
Dongxia
Zhu
b,
Yu
Liu
*a,
Yue
Wang
a and
Martin R.
Bryce
*c
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: yuliu@jlu.edu.cn
bInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Renmin Road 5268, Changchun, Jilin 130024, P. R. China
cDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 19th October 2016
Abstract
Two new deep-blue iridium(III) complexes, (dfpypy)2IrFptz (Ir1) and (Medfpypy)2IrFptz (Ir2), comprising difluoro-bipyridyl (dfpypy) derivatives as cyclometaling ligands and a chelated pyridyl-triazole (Fptz) ancillary ligand are reported. The bipyridyl ligands lead to a significantly increased HOMO–LUMO gap and a hypsochromic shift of the phosphorescence compared to phenylpyridyl analogs. Density function theory (DFT) calculations and electrochemical measurements for Ir1 and Ir2 support their genuine blue phosphorescent emission. The combination of ancillary and cyclometalating ligands in Ir1 and Ir2 significantly influences the molecular orbitals of both complexes, leading to clearly distinct electron density distributions of the HOMO and LUMO compared with other blue-emitting Ir(III) derivatives. Both complexes Ir1 and Ir2 show deep-blue emission with λmax values in the region of 435–465 nm with high PLQYs and short excited-state lifetimes. The phosphorescent organic light emitting diodes (PhOLEDs) based on Ir1 and Ir2 achieve remarkably high EL performance with low efficiency roll-off at high luminance. The bluest color (CIEx,y 0.14, 0.11) and the highest EL efficiency were achieved in the device based on Ir2 (Device 2), where the peak EQE/PE of 13.0%/11.2 lm W−1 together with the corresponding values of 12.6%/8.8 lm W−1 and 10.1%/5.0 lm W−1 at the practical luminances of 100 and 1000 cd m−2 respectively, strongly compete with those of any deep-blue fluorescent and/or phosphorescent OLEDs with similar CIE coordinates previously reported.
Introduction
Since the pioneering report by Baldo et al.,1 phosphorescent organic light-emitting devices (PhOLEDs) based on metal complexes have been intensively studied because internal quantum efficiencies (IQEs) approaching 100% can be achieved by harvesting both singlet and triplet excitons for light emission. The external quantum efficiencies (EQEs) of red and green PhOLEDs,2 together with the sky-blue (y coordinate >0.20 for the Commission Internationale de L'Eclairage coordinates, CIEx,y)3 and the quasi-blue (0.15 ≤ y ≤ 0.20) PhOLEDs,4 reach 20–25% which is ca. four times higher than those of conventional fluorescent OLEDs. Thus they have already been commercialized or are competing with the conventional fluorescent materials in OLEDs for displays and solid-state lighting. However, although much progress has been made in the development of new blue phosphorescent emitters, there are only a few reports of deep-blue PhOLEDs with CIEx and y coordinates both ≤0.15. Notably, devices with excellent blue chromaticity, that is, with x and y values simultaneously close to 0.15 and 0.10, respectively,5 showed EQE values of less than 8%, which are only slightly enhanced compared to the highest level of the conventional fluorescent blue OLEDs with similar CIE coordinates.6Chou et al. prepared two iridium complexes using pyridyl azolates and benzyl phenylphosphine ligands,7 which are among the most efficient true-blue PhOLEDs documented to date with CIEx,y of ∼(0.15, 0.11) and the peak EQEs/PEs (power efficiencies) of ∼11%/8 lm W−1, respectively. Nevertheless, they exhibited relatively high turn-on voltages of ≥4.4 V as well as significant efficiency roll-off (∼8% and 3 lm W−1 at the practical luminance of 100 cd m−2). Forrest and coworkers have very recently reported that PhOLEDs of an Ir complex of N-heterocyclic carbene ligands can attain EQEmax of 10.1 ± 0.2% at very low luminance and EQE 9.0 ± 0.1% at 1000 cd m−2 with CIEx,y 0.16, 0.09.8 Thus, it remains very important to develop blue phosphors for realizing very efficient deep-blue PhOLEDs that meet the requirements for practical applications.
Generally, the effective molecular design concept for deep-blue phosphors is to introduce electron-withdrawing or electron-donating groups at selective positions on the cyclometalated (C^N) ligands. For example, fluorine substitution leads to the 4′,6′-difluorophenylpyridinato ligand in the well-known sky-blue iridium complex FIrpic,2b,3a,c,9 resulting in a larger band gap and blue shifted emission.10 Alternatively, fluorine-substituted 2,3′-bipyridine derivatives, where the additional nitrogen atom has a similar or even stronger effect of lowering the HOMO (highest occupied molecular orbital) level (as opposed to introducing the electron-withdrawing groups at the para position on the phenyl ring),11 have shown potential as the cyclometalating ligands for efficient deep-blue iridium complexes. It is also well known that the blue color can be tuned through structural variations of the ancillary ligand.12 For example, chelated pyridyl-triazole derivatives, which usually give very low total synthesis yields of ≤10% for the resulting complexes, afford stable iridium complexes that show remarkably shorter emission maxima than that of FIrpic, due to their blue-shifting effect compared to picolinate and other ancillary liands.4c,5b,10a,11c,12b
The aim of the present work was to develop new phosphors and to fabricate deep-blue PhOLEDs with enhanced efficiency. To achieve this, we designed and characterized two new heteroleptic Ir(III) complexes (Ir1 and Ir2) containing the unique combination of 2′,6′-difluoro-2,3′-bipyridyl ligands (dfpypy or Medfpypy) and a 5-(2′-pyridyl)-3-trifluoromethyl-1,2,4-triazole ancillary ligand (Fptz). We demonstrate that both Ir1 and Ir2 display true-blue phosphorescence in their solution and film states, and they serve in the emitting layer (EML) of PhOLEDs that show deep-blue emission with very high EL efficiencies. The Ir2-based device (Device 2) represents the deepest blue emission with CIEx,y of (0.14, 0.11) near to the ideal value of (0.14, 0.08). The peak EQE/PE values are 13%/11.2 lm W−1, which maintain very high levels of 12.6%/8.8 lm W−1 and 10.1%/5.0 lm W−1 at luminances of 100 and 1000 cd m−2, respectively. To the best of our knowledge, these EL efficiency values strongly compete with, and even exceed, those previously reported for PhOLEDs with similar CIE coordinates.5–7,13a Additionally, an effective synthetic route for Fptz13b,c provides high yields of Ir1 and Ir2 (>70%), facilitating the low cost of both complexes in the quest for deep-blue PhOLEDs in the future.
Experimental section
General information
All reagents and solvents obtained from commercial suppliers were used without further purification. Anhydrous tetrahydrofuran was distilled with sodium benzophenone ketyl under nitrogen atmosphere and degassed by the freeze–pump–thaw method. All glassware, syringes, magnetic stirring bars and needles were dried in a convection oven for at least 4 h. Reactions were monitored with thin layer chromatography (TLC). Commercial TLC plates (silica gel 60 F254, Merck Co.) were visualized under UV light at 254 and 365 nm. Silica column chromatography used silica gel 60 G (particle size 5–40 μm, Merck Co.). 1H NMR spectra were recorded on a 500 MHz Bruker Avance spectrometer using CDCl3 as solvent and tetramethylsilane (TMS) as an internal standard (δ = 0.00 ppm). GC/MS data were recorded on a Thermo Fisher ITQ1100 instrument. UV-vis spectra were measured on a Shimadzu UV-2550 spectrophotometer. Fluorescence spectra were recorded using a Shimadzu RF-5301 PC spectrometer and a Maya2000Pro optical fiber spectrophotometer. Solid state PL efficiencies were measured using an integrating sphere (C-701, Labsphere Inc.), with a 365 nm Ocean Optics LLS-LED as the excitation source, and the light was introduced into the integrating sphere through an optical fiber. Single crystal X-ray diffraction intensity data were collected on a Rigaku RAXIS-PRID diffractometer using the ω-scan mode with graphite-monochromator Mo Kα radiation. DSC experiments were recorded on a NETZSCH DSC 204 instrument at a scanning rate of 10 K min−1. Thermogravimetric analysis (TGA) was performed on a TA Q500 thermogravimeter by measuring the weight loss while heating at a rate of 10 °C min−1 from 25 to 800 °C under nitrogen. Electrochemical measurements were performed with a BAS 100W Bioanalytical electrochemical work station, using Pt working electrode, platinum wire as auxiliary electrode, and a porous glass wick Ag/Ag+ as reference electrode. The voltammograms were referenced to the ferrocene/ferrocenium couple and the scan rate was 100 mV s−1.Fabrication of the OLEDs and EL measurements
The ITO (indium-tin oxide) coated glass substrates (20 Ω square−1) were first cleaned in detergent solution, ethanol, acetone, isopropanol and deionized water. All organics were purified by gradient sublimation and thermally evaporated at a rate of 1.0 Å s−1 at a pressure of ca. 3.5 × 10−4 Pa. A LiF layer (1 nm) was deposited at a rate of 0.2 Å s−1. The Al cathode was deposited at a rate of 10 Å s−1; the active area of the diode segments was 2 × 2.5 mm2. EL spectra and brightness–current density–voltage characteristics were measured by combining a Spectrascan PR-650 spectrophotometer with a computer-controlled direct current power supply Keithley model 2400 voltage–current source under ambient conditions at room temperature.Results and discussion
Synthesis, single crystal structures and theoretical calculations
The chemical structures of Ir1 and Ir2 are shown in Scheme 1. The two cyclometalating ligands, dfpypy and Medfpypy, were synthesized by Suzuki coupling following the reported method.11a The corresponding μ-chloro-bridged dimers [(dfpypy)2Ir(μ-Cl)]213 and [(Medfpypy)2Ir(μ-Cl)]211c were prepared by the reaction of dfpypy and Medfpypy with IrCl3·3H2O in a mixture of 2-ethoxyethanol and water. Here, it is worth noting that overall yields of >70% were obtained for both new iridium complexes Ir1 and Ir2, which represent the most effective syntheses for deep-blue phosphors reported in literature.5b,11c | ||
| Scheme 1 The synthesis and chemical structures of Ir1 and Ir2. | ||
The molecular structures of Ir1 and Ir2 in single crystals were determined by X-ray diffraction. As shown in Fig. 1a and Table S1 in the ESI,† both complexes exhibit distorted octahedral geometries around the iridium center with the classical C,C-cis and N,N-trans configurations. The bond lengths and angles are summarized in Table S2 (ESI†). The Ir–C bond lengths, ranging from 1.981 to 2.008 Å, and Ir–N (in dfpypy) bond lengths, ranging from 2.040 to 2.055 Å, are comparable to those observed in several blue-emitting Ir(III) complexes (Ir–C = 1.997–2.005 Å for Ir(dfpypy)3; Ir–C = 1.992–2.009 Å for Ir(dfpypy)2(pic); Ir–C = 2.009–2.013 Å for Ir(dfpypy)2(fppz); Ir–N = 2.116–2.136 Å for Ir(dfpypy)2(pic); Ir–N = 2.044–2.048 Å for Ir(dfpypy)2(fppz)).11
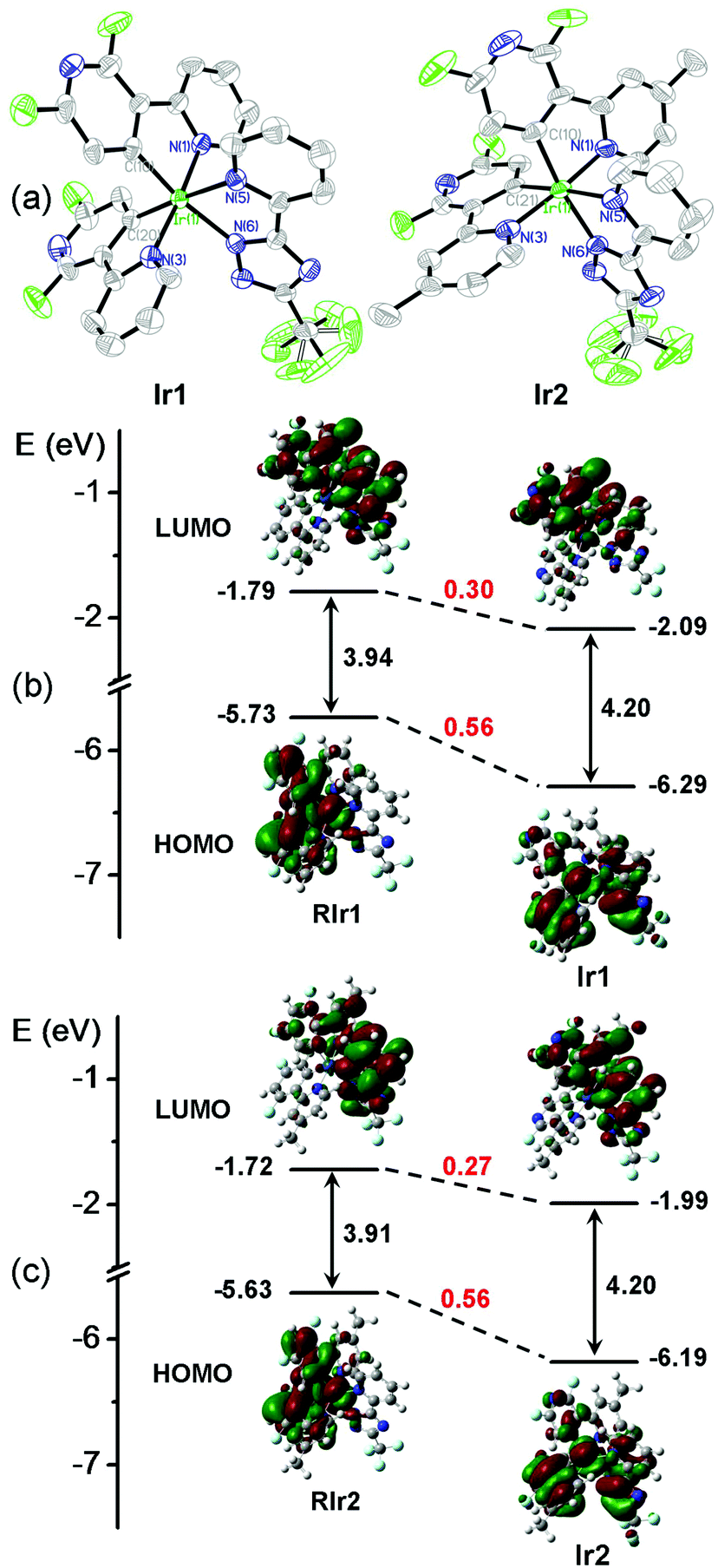 | ||
| Fig. 1 (a) ORTEP plots of the X-ray molecular structures of Ir1 and Ir2. Hydrogen atoms are omitted for clarity. (b and c) Molecular orbital diagrams for Ir1/Ir2 and two reference complexes (RIr1/RIr2) with selected isodensity frontier molecular orbitals mainly involved in the electronic transitions. All the DFT energy values are given in electron volts (eV). The structures of RIr1 and RIr2 are shown in Fig. S3 (ESI†). | ||
Density functional theory (DFT) calculations were performed in order to estimate the energy levels and electron density distributions of the orbitals of Ir1 and Ir2 together with two reported reference complexes RIr1 and RIr2,11c,12b which have two difluorophenylpyridinato-N,C2 main ligands (dfppy and Medfppy) instead of dfpypy and Medfpypy, respectively (see Fig. S3 in ESI†). Fig. 1b and c show the optimized structures and a schematic representation of the energy levels of the four complexes. The HOMOs of RIr1 and RIr2 are mostly localized on both dfppy ligands together with the iridium atom, whereas the HOMOs of Ir1 and Ir2 are distributed over one of the two dfpypy ligands and the metal d orbitals, also with a significant contribution from the ancillary ligand orbital. This clear difference results in much lower HOMO levels of Ir1 and Ir2 (−6.29 and −6.19 eV, respectively) compared to RIr1 and RIr2 (−5.73 and −5.63 eV, respectively). On the other hand, the LUMOs are distributed similarly in all four complexes over part of one of the main ligands as well as the ancillary ligand. Thus, although Ir1 and Ir2 show lower LUMO levels of −2.10 and −1.99 eV compared to RIr1 and RIr2 (−1.79 and −1.72 eV) respectively, the difference of the LUMO energy between Ir1 and Ir2 and the corresponding reference complexes is lower than for the HOMO. These data lead to the larger HOMO–LUMO energy gaps of ∼4.2 eV for Ir1 and Ir2, compared to ∼3.9 eV for RIr1 and RIr2. These data confirm that the bipyridyl cyclometalating ligand of Ir1 and Ir2 is beneficial for achieving the desired blue shift in the phosphorescent emission compared to the phenylpyridyl analogs. The improvement in the purity of blue emission from Ir1 and Ir2 is experimentally confirmed by the photophysical and electrochemical studies described below.
Photophysical and electrochemical properties
The UV-vis absorption and phosphorescence spectra of Ir1 and Ir2 are shown in Fig. 2a, measured in CH2Cl2 solution at room temperature (RT). Both complexes display strong absorption bands at approximately 253 and 255 nm assigned to intraligand π–π* transitions.14 The lowest-energy absorption band at λmax 330–355 nm can be attributed to the spin-allowed singlet metal-to-ligand charge-transfer (1MLCT) transitions. Moreover, the spin-forbidden triplet 3MLCT or 3LC transitions (or both) appear as a weak long-tail absorption at the range of 410–430 nm (Fig. 2a inset), implying that the excitation energy for the triplet state (T1) of both complexes is ∼2.9 eV.14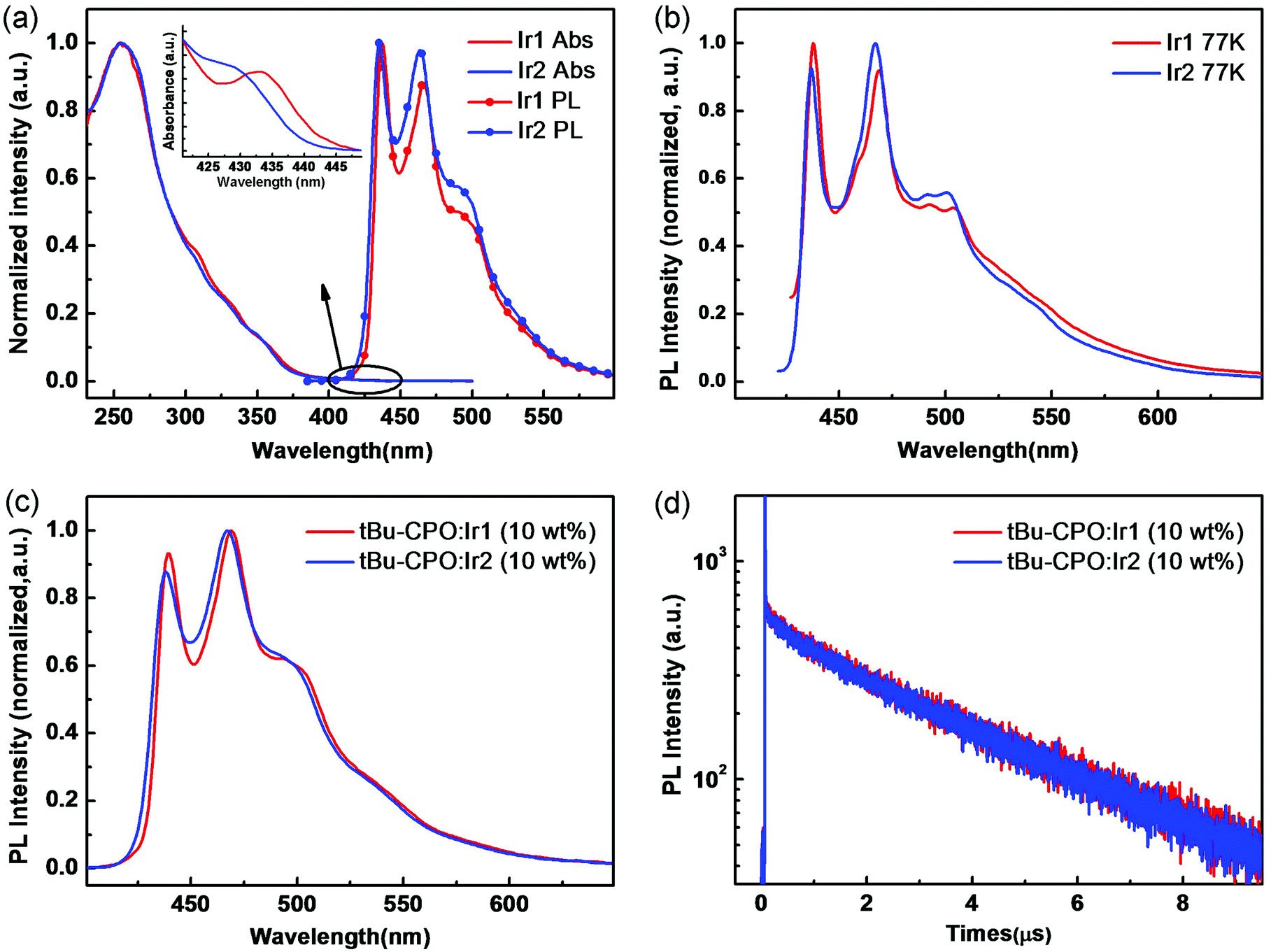 | ||
| Fig. 2 UV-vis absorption and PL spectra in CH2Cl2 at room temperature (a) and PL spectra in THF at low temperature (77 K) (b) of Ir1 and Ir2. PL spectra (c) and luminescence decay profiles (d) of tBu-CPO films doped with 10 wt% Ir1 and Ir2. Emission data were obtained in degassed solutions. The structure of tBu-CPO is shown in Fig. S3 (ESI†). | ||
Both Ir1 and Ir2 show similar intense blue phosphorescence in solution (RT) with emission at λmax 437, 466 nm (RT)/438, 468 nm (77 K) for Ir1 and 435, 464 nm (RT)/437, 467 nm (77 K) for Ir2 (Fig. 2b), which are strongly blue shifted compared to RIr1 and RIr2 (∼460, 490 nm)11c,12b in agreement with the DFT calculations above. The photoluminescence (PL) emission and the transient PL properties of thin films using t-BuCPO15 as host doped with complexes of Ir1 and Ir2 (10 wt%) were measured. Both films displayed strong and structured phosphorescence spectra with similar emission maxima in the deep blue 430–470 nm region (Fig. 2c) to those in solution. The PL quantum yields (PLQYs) were 0.65 ± 0.03 and 0.70 ± 0.03, by employing an integrating sphere. The excited-state lifetimes of Ir1 and Ir2 are monoexponential and are 2.97 μs and 3.01 μs, respectively, consistent with their emission originating from a triplet excited state. The PL spectra and transient PL of the doped films are shown in Fig. 2c and d.
The solution electrochemical properties of Ir1 and Ir2 were investigated by cyclic voltammetry (CV), and both complexes exhibit reversible reduction and oxidation behaviour as shown in Fig. S6 (ESI†). The respective HOMO and LUMO energy levels were experimentally determined from the redox curves relative to the vacuum level, and are −6.27 and −2.57 eV for Ir1, −6.24 and −2.56 eV for Ir2. These data show that Ir1 and Ir2 have larger HOMO–LUMO gaps than RIr1 and RIr2 with the HOMO/LUMO values of ∼−5.6/−2.9 eV.11c Therefore, the electrochemical, photophysical and computational data are entirely consistent and demonstrate that the novel combination of main and ancillary ligands in Ir1 and Ir2, where cyclometalating C^N bipyridine ligands are employed instead of phenylpyridine ligands, leads to a distinctively different HOMO/LUMO distribution compared to previous blue-emitting Ir(III) complexes10,11 and explains the observed deep blue emission.
Characterization of deep-blue phosphorescent OLEDs
To explore the electroluminescence (EL) characteristics of Ir1 and Ir2, OLEDs based on a wide energy-gap host molecule t-BuCPO15 were fabricated by vacuum evaporation processes. The PhOLEDs have the following configuration: ITO/TAPC (30 nm)/mCP (10 nm)/EML (30 nm)/TPBi (30 nm)/LiF (1 nm)/Al (200 nm). In each device, TAPC (1,1-bis{4-[N,N-di(p-tolyl)amino]phenyl}cyclohexane) served as a hole-transporting layer, mCP (1,3-bis(carbazol-9-yl)benzene) as an electron-blocking layer (EBL) to prevent the formation of an exciplex between TAPC and the EMLs, TPBi (1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene) as an electron-transporting layer (ETL), with ITO (indium-tin oxide) anode and LiF/Al cathode. The devices using doped t-BuCPO: Ir1 or Ir2 (10 wt%) films as the EMLs, are denoted as Device 1 and Device 2, respectively. As shown in Fig. 3a, there are slight barriers (0.2–0.3 eV) against hole injection from mCP into the EMLs based on t-BuCPO, and against electron injection from TPBi into the EMLs. Furthermore, t-BuCPO was chosen as the host due to its sufficiently high T1 of ∼3.2 eV to sensitize the emission of Ir1 or Ir2, together with its rather deep HOMO energy level and its favorable bipolar donor–acceptor structure.15 As a result, both holes and electrons can conduct smoothly and are confined effectively within the EMLs, ensuring that highly efficient exciton-formation/-transfer can occur even at high current densities.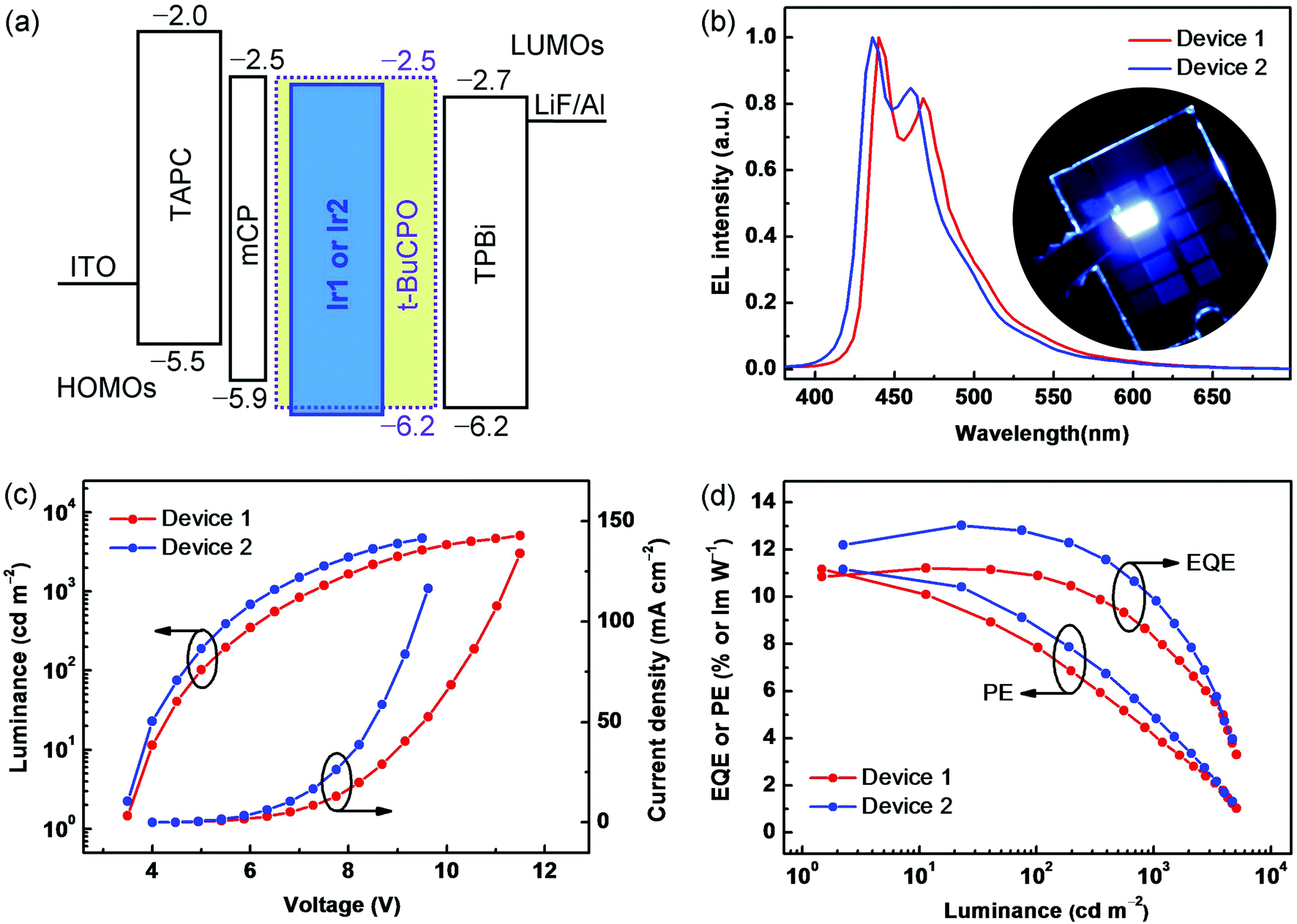 | ||
| Fig. 3 (a) Energy diagram of the materials used in the OLEDs. (b) EL spectra of Devices 1 and 2. Inset: A photograph of Device 2 illuminating at 1000 cd m−2. (c) Current density–voltage–luminance (J–V–L) curves of Devices 1 and 2. (d) Power efficiency (PE)–luminance (L)–external quantum efficiency (EQE) curves of Devices 1 and 2. | ||
The devices showed stable EL spectra (Fig. 3b) within a range of driving voltages (4–8 V) (Fig. S8, ESI†), and no host emission is observed in the EL spectra, which also exhibit good color durability after continuous open-condition operation for 2 hours. Deep-blue EL with two λELmax bands at 430–440 nm and 460–470 nm at a current density of 5 mA cm−2 (∼500 cd m−2) resemble the PL spectra of the complexes. Owing to the blue-region emitting peaks of both devices, together with their full width at half maximum (FWHM) of less than 50 nm, the similar CIE coordinates of (0.15, 0.13) and (0.14, 0.11) were obtained. Fig. 3b shows a photograph of Device 2 demonstrating the observed color purity. It is interesting to note that the relative intensities at the shortest wavelength bands of Ir1 and Ir2 are higher in the EL than in the PL spectra due to optical microcavity effects,16 leading to the desired deep-blue EL. The current density–voltage–luminance (J–V–L) and EL efficiency–luminance (PE/EQE–L) characteristics are shown in Fig. 3c and d, respectively, and the EL data are summarized in Table 1. Devices 1 and 2 exhibit rather low turn-on voltages of ∼3.5 V, indicating also that these HTL–EML–ETL systems possess the predominant factor facilitating both hole and electron injection and transport,8,17 which also dominated the rapidly increasing J–V and L–V curves after the onset, where the driving voltages are as low as 5.0/4.7 V and 7.3/6.4 V at the practical luminances of 100 and 1000 cd m−2, respectively. Furthermore, the peak EQEs/PEs of 11.2%/11.1 lm W−1 and 13.0%/11.2 lm W−1 with remarkably low roll-off were obtained in Devices 1 and 2, respectively. Device 2 shows the bluest color and the higher EQE/PE in terms not only of the peak values above, but also by maintaining such high levels as 12.6%/8.8 lm W−1 and 10.1%/5.0 lm W−1 at the luminances of 100 and 1000 cd m−2, respectively, which are needed for practical displays and/or solid-state lighting. Here, the slight improved EL performance of Ir2 compared to Ir1 indicates that the methyl substituent could partly suppress the aggregation of the emitting core in the solid thin films, and thereby favour enhanced device EL efficiency.18 To the best of our knowledge, these EL efficiencies strongly compete with, and even exceed, any previously reported values for deep-blue PhOLEDs with similar CIEx,y values.5–8
| V on [V] | L max [cd m−2] | η ext [%] | η p [lm W−1] | CIE(x,y)c | |
|---|---|---|---|---|---|
| a Applied voltage required to reach a luminance of 1 cd m−2. b The efficiencies listed are the maximum values, and the values at 100 and 1000 cd m−2, respectively. c Recorded at 1000 cd m−2. | |||||
| 1 | 3.5 | 5080 | 11.2, 10.8, 8.3 | 11.1, 7.9, 4.2 | 0.15, 0.13 |
| 2 | 3.5 | 4710 | 13.0, 12.6, 10.1 | 11.2, 8.8, 5.0 | 0.14, 0.11 |
For comparison, we note that deep-blue OLEDs using a thermally-activated delayed fluorescence (TADF) organic (metal-free) emitter achieve 20% EQE at 100 cd m−2 with roll-off to ca. 12% at 1000 cd m−2.19 However, the TADF-OLEDs' CIEx,y coordinates (0.15, 0.13) are less blue than our Device 2, and the complicated TADF device architecture with seven separate organic layers could limit practical applications.
Conclusions
In summary, a straightforward synthetic route is reported for the high-yielding synthesis of two new heteroleptic phosphorescent complexes Ir1 and Ir2 consisting of cyclometalating ligands based on difluorobipyridyl derivatives, in combination with a chelated pyridyl-triazole ancillary ligand. This rational materials design has successfully achieved a very wide HOMO–LUMO gap. As a result, both phosphors show deep-blue emission with λmax values in the 435–465 nm range, high PLQYs and short excited-state lifetimes. In particular, the PhOLEDs based on Ir1 and Ir2 realized high performance deep-blue EL. The deepest blue color (CIEx,y 0.14, 0.11) and the highest EL efficiency were achieved in the device based on Ir2 (Device 2), where the peak EQE/PE (13.0%/11.2 lm W−1) together with the corresponding values at the practical luminances of 100 cd m−2 (12.6%/8.8 lm W−1) and 1000 cd m−2 (10.1%/5.0 lm W−1) strongly compete with any previously reported deep-blue fluorescent and/or phosphorescent OLEDs with similar CIE coordinates. This work represents a significant advance in the design and characterization of new transition metal complexes which display outstanding efficiency in deep-blue OLEDs.Acknowledgements
This work was supported by National Basic Research Program of China (973 Program, 2013CB834805), Natural Science Foundation of China (91333201, 21221063, 51373062, 51473028) and the key scientific and technological project of Jilin province (20150204011GX). The work at Durham University was supported by EPSRC (Engineering and Physical Sciences Research Council) grant EP/K039423/1.Notes and references
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS; (b) M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS; (c) M. Ikai, S. Tokito, Y. Sakamoto, T. Suzuki and Y. Taga, Appl. Phys. Lett., 2001, 79, 156–158 CrossRef CAS.
- (a) W. Soichi, I. Nobuhiro and K. Junji, Jpn. J. Appl. Phys., 2007, 46, 1186–1188 CrossRef; (b) S.-J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125–2130 CrossRef CAS; (c) Y. Tao, Q. Wang, C. Yang, C. Zhong, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 2923–2929 CrossRef CAS; (d) S. Lee, K.-H. Kim, D. Limbach, Y.-S. Park and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 4105–4110 CrossRef CAS; (e) G. Li, D. Zhu, T. Peng, Y. Liu, Y. Wang and M. R. Bryce, Adv. Funct. Mater., 2014, 24, 7420–7426 CrossRef CAS; (f) Y. Feng, P. Li, X. Zhuang, K. Ye, T. Peng, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 12544–12547 RSC.
- (a) H. Shin, S. Lee, K.-H. Kim, C.-K. Moon, S.-J. Yoo, J.-H. Lee and J.-J. Kim, Adv. Mater., 2014, 26, 4730–4734 CrossRef CAS PubMed; (b) K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062–5066 CrossRef CAS PubMed; (c) J.-H. Lee, S.-H. Cheng, S.-J. Yoo, H. Shin, J.-H. Chang, C.-I. Wu, K.-T. Wong and J.-J. Kim, Adv. Funct. Mater., 2015, 25, 361–366 CrossRef CAS.
- (a) S. O. Jeon, S. E. Jang, H. S. Son and J. Y. Lee, Adv. Mater., 2011, 23, 1436–1441 CrossRef CAS PubMed; (b) M. S. Park, D. H. Choi, B. S. Lee and J. Y. Lee, J. Mater. Chem., 2012, 22, 3099–3104 RSC; (c) J.-B. Kim, S.-H. Han, K. Yang, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, Chem. Commun., 2015, 51, 58–61 RSC.
- (a) C.-H. Hsieh, F.-I. Wu, C.-H. Fan, M.-J. Huang, K.-Y. Lu, P.-Y. Chou, Y.-H. O. Yang, S.-H. Wu, I. C. Chen, S.-H. Chou, K.-T. Wong and C.-H. Cheng, Chem. – Eur. J., 2011, 17, 9180–9187 CrossRef CAS PubMed; (b) S. Lee, S.-O. Kim, H. Shin, H.-J. Yun, K. Yang, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14321–14328 CrossRef CAS PubMed.
- (a) J. N. Moorthy, P. Venkatakrishnan, D.-F. Huang and T. J. Chow, Chem. Commun., 2008, 2146–2148 RSC; (b) T. Peng, K. Ye, Y. Liu, L. Wang, Y. Wu and Y. Wang, Org. Electron., 2011, 12, 1914–1919 CrossRef CAS; (c) W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS; (d) R. Kim, S. Lee, K.-H. Kim, Y.-J. Lee, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, Chem. Commun., 2013, 49, 4664–4666 RSC.
- Y.-C. Chiu, J.-Y. Hung, Y. Chi, C.-C. Chen, C.-H. Chang, C.-C. Wu, Y.-M. Cheng, Y.-C. Yu, G.-H. Lee and P.-T. Chou, Adv. Mater., 2009, 21, 2221–2225 CrossRef CAS.
- J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92–98 CrossRef CAS PubMed.
- (a) S. Tokito, T. Iijima, Y. Suzuri, H. Kita, T. Tsuzuki and F. Sato, Appl. Phys. Lett., 2003, 83, 569–571 CrossRef CAS; (b) R. J. Holmes, S. R. Forrest, Y.-J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422–2424 CrossRef CAS; (c) L. Xiao, S.-J. Su, Y. Agata, H. Lan and J. Kido, Adv. Mater., 2009, 21, 1271–1274 CrossRef CAS; (d) E. Baranoff and B. F. E. Curchod, Dalton Trans., 2015, 44, 8318–8329 RSC.
- (a) S. Takizawa, H. Echizen, J. Nishida, T. Tsuzuki, S. Tokito and Y. Yamashita, Chem. Lett., 2006, 35, 748–749 CrossRef CAS; (b) S. O. Jeon, S. E. Jang, H. S. Son and J. Y. Lee, Adv. Mater., 2011, 23, 1436–1441 CrossRef CAS PubMed; (c) K. S. Yook and J. Y. Lee, Org. Electron., 2011, 12, 1711–1715 CrossRef CAS.
- (a) S. J. Lee, K.-M. Park, K. Yang and Y. Kang, Inorg. Chem., 2009, 48, 1030–1037 CrossRef CAS PubMed; (b) C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS; (c) H. J. Park, J. N. Kim, H.-J. Yoo, K.-R. Wee, S. O. Kang, D. W. Cho and U. C. Yoon, J. Org. Chem., 2013, 78, 8054–8064 CrossRef CAS PubMed; (d) Y. Kang, Y.-L. Chang, J.-S. Lu, S.-B. Ko, Y. Rao, M. Varlan, Z.-H. Lu and S. Wang, J. Mater. Chem. C, 2013, 1, 441–450 RSC; (e) F. Kessler, Y. Watanabe, H. Sasabe, H. Katagiri, M. K. Nazeeruddin, M. Grätzel and J. Kido, J. Mater. Chem. C, 2013, 1, 1070–1075 RSC.
- (a) R. J. Holmes, B. W. D'Andrade, S. R. Forrest, X. Ren, J. Li and M. E. Thompson, Appl. Phys. Lett., 2003, 83, 3818–3820 CrossRef CAS; (b) S. J. Yeh, M. F. Wu, C. T. Chen, Y. H. Song, Y. Chi, M. H. Ho, S. F. Hsu and C. H. Chen, Adv. Mater., 2005, 17, 285–289 CrossRef CAS; (c) L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed; (d) K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed.
- (a) H. Oh, K.-M. Park, H. Hwang, S. Oh, J. H. Lee, J.-S. Lu, S. Wang and Y. Kang, Organometallics, 2013, 32, 6427–6436 CrossRef CAS; (b) H. Xu, Y. Yue, L. Qu, Y. Hao, H. Wang, L. Chen and B. Xu, Dyes Pigm., 2013, 99, 67–73 CrossRef CAS; (c) X. Zhang, J. K. Rueter, Y. Chen, M. Moorjani, M. C. Lanier, E. Lin, R. S. Gross, J. E. Tellew, J. P. Williams, S. M. Lechner, S. Markison, T. Joswig, S. Malany, M. Santos, J. C. Castro-Palomino, M. I. Crespo, M. Prat, S. Gual, J.-L. Diaz, J. Saunders and D. H. Slee, Bioorg. Med. Chem. Lett., 2008, 18, 1778–1783 CrossRef CAS PubMed.
- (a) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed; (b) P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634–1641 CrossRef CAS.
- A. Wada, T. Yasuda, Q. Zhang, Y. S. Yang, I. Takasu, S. Enomoto and C. Adachi, J. Mater. Chem. C, 2013, 1, 2404–2407 RSC.
- (a) V. Bulović, V. B. Khalfin, G. Gu, P. E. Burrows, D. Z. Garbuzov and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3730–3740 CrossRef; (b) Y. Zhang, J. Lee and S. R. Forrest, Nat. Commun., 2014, 5, 5008 CrossRef CAS PubMed.
- (a) F.-M. Hsu, C.-H. Chien, C.-F. Shu, C.-H. Lai, C.-C. Hsieh, K.-W. Wang and P.-T. Chou, Adv. Funct. Mater., 2009, 19, 2834–2843 CrossRef CAS; (b) H. Sasabe, Y. Seino, M. Kimura and J. Kido, Chem. Mater., 2012, 24, 1404–1406 CrossRef CAS.
- K. Q. Ye, J. Wang, H. Sun, Y. Liu, Z. C. Mu, F. Li, S. M. Jiang, J. Y. Zhang, H. X. Zhang, Y. Wang and C.-M. Che, J. Phys. Chem. B, 2005, 109, 8008–8016 CrossRef CAS PubMed.
- I. Lee and J. Y. Lee, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details; differential scanning calorimetric (DSC) and thermogravimetric analysis (TGA) data; X-ray crystallographic data files (CIF), crystal data and structure refinement parameters; cyclic voltammograms for Ir1 and Ir2. CCDC 1052634 and 1052689. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6tc04119a |
| This journal is © The Royal Society of Chemistry 2016 |