
Quantitative prediction of morphology and electron transport in crystal and disordered organic semiconductors†
Abstract
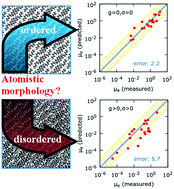
The morphologies and electron mobilities for 20 single-crystal and 21 thin-film organic n-type semiconductors are predicted using a multi-mode methodology previously applied by our group for p-type materials [I. Yavuz, et al., J. Am. Chem. Soc., 2015, 137, 2856–2866]. The calculations simulate Marcus charge-hopping with a kinetic Monte Carlo method using the VOTCA package of Andrienko et al. The calculations assume perfect order for single crystal morphologies, but structural disorder is incorporated into thin-film calculations using molecular dynamics to simulate the energetic disorder of thin-film morphologies. Predicted electron mobilities for both morphologies are typically within one order of magnitude.
 Please wait while we load your content...
Please wait while we load your content...

