
Optical study of electrochromic moving fronts for the investigation of ion transport in conducting polymers†
Sahika
Inal
,
George G.
Malliaras
and
Jonathan
Rivnay‡
*
Department of Bioelectronics, École Nationale Supérieure des Mines, CMP-EMSE, MOC, 13541 Gardanne, France
First published on 11th March 2016
Abstract
Conducting polymers, which can simultaneously support transport of ions in addition to electronic charges, form the basis for electrochemical devices that transduce ionic signals into electronic signals, and vice versa. Understanding and controlling mixed conduction is, however, challenging due to a lack of methodology to simultaneously probe ion (and hole) injection and/or transport, as well as the morphology and film microstructure through which ions traverse. Here, we present a straightforward technique that takes advantage of the electrochromic nature of conducting polymers in order to deduce links between conjugated polymer aggregates, molecular (dis)order, and ion penetration. The morphology of poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) films and, subsequently, ionic transport are tuned by the addition of the co-solvent ethylene glycol in the dispersions. Time lapse UV-VIS spectra of these films, acquired during their redox-cycle, provide valuable insight into the pathways of ions inside the conducting polymer and the nature of aggregates therein. The structure–property relationships gained using such techniques promise to guide the design of polymeric active materials for devices that rely on mixed conduction.
Introduction
Organic electronic materials have become commonplace in a variety of applications ranging from transparent conductors to light emitting/energy harvesting optoelectronic devices due to their electronic conductivity, synthetic tunability, solution processability, and low temperature processing.1 Recently, interest in these materials has increased in response to emerging technologies that rely on their reversible electrochemical activity such as electrochromic displays, biosensors, supercapacitors, and actuators.2–7 These applications share the common requirement of mixed conduction in the material, i.e. electronic transport in the conducting phase as well as ion injection from an electrolyte, resulting in its electrochemical (de)doping. Compared to inorganic films, the free volume associated with weak intermolecular interactions and the absence of a physical barrier facilitate penetration of hydrated ions and their transport in the films.8Organic electronic materials that allow for ion transport are often conjugated polyelectrolytes or complexes of conducting polymers with electronically insulating, but polar, hygroscopic components.9–14 Such a conjugated polymer:polyelectrolyte composite is the water-dispersible conducting polymer poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS), which has been a popular choice in a number of electrochemical stimulation, charge storage and biosensing applications. The bulk interaction of hydrated ions with PEDOT:PSS chains leads to low impedance electrodes15 and high transconductance electrochemical transistors.16 While such applications successfully take advantage of ionic carriers, the nature of ion transport in materials such as PEDOT:PSS is not well understood.17 This is partly due the simultaneous presence of electronic transport which renders typical methods for quantification of ionic transport unusable.11 On the other hand, the electrochromic nature of conducting polymers, i.e. the change in optical transmission upon (de)doping due to injection (extraction) of ions from (into) an electrolyte, has been a means to capture the transient nature of ion motion.18–20 Stavrinidou et al. developed an experimental configuration where holes and ions move in the same direction, enabling straightforward extraction of ion drift mobility in conjugated polymer films.21 The one-dimensional (1-D) geometry of this experiment permits direct observation of a moving front as hydrated ions drift into an electrically percolating network of conjugated species. This technique was subsequently used to quantify the effect of polyanion chemistry in PEDOT-based inks,22 and of protein-based bio-additives on the hydration and ionic mobility in vapor-polymerized PEDOT films.23 Despite the literature emphasizing the benefits of ion transport and/or providing ion mobility values of conducting polymer films, there is a lack of characterization tools for the investigation of the relationship between ionic transport and film microstructure/morphology. It is, however, crucial to understand this relationship in order to tailor the conduction pathways for both ions and electrons. The ability to control such transport pathways will help to improve the performance of existing devices and develop novel device architectures that rely on mixed conduction.
In this work, we investigate in situ optical absorption properties of PEDOT:PSS films during the course of the lateral moving front experiment. It is well-known that the UV-VIS spectrum of a conjugated system can reveal subtleties of the local degree of ordering in its film.24–26 Thus, by monitoring changes in the absorbance characteristics during a redox cycle, we extract information not only about kinetics of ion injection and motion, but also about the local ordering of the conjugated polymer film. We modify the morphology of PEDOT:PSS films with a co-solvent additive, i.e. ethylene glycol, and compare the evolution of the optical absorption of these films at multiple distances from the conducting polymer/electrolyte interface. We explicitly demonstrate agreement between the microscopy-based approach of Stavrinidou et al.,21 and the spectroscopic approach herein for a number of conducting polymer formulations. The spectroscopic data are analyzed using Spano's well-established model for thiophene-based polymers,27,28 demonstrating the effect of aggregate content (modulated by the EG content in the dispersion) on ionic transport. This technique proves to be robust for a number of PEDOT:PSS formulations, and therefore provides a reliable means to understand ion motion in conjugated polymers – an important aspect of future mixed conductor materials design.
Experimental
Device fabrication
Planar junction devices used for electrochromic moving front experiments were fabricated as previously described.21 Briefly, PEDOT:PSS (Clevios PH-1000 from Heraeus Holding GmbH.), dodecyl benzene sulfonic acid (DBSA; 0.002 vol%) and ethylene glycol (EG; 0, 5, 50% by volume of dispersion as noted in the text/figures) are mixed, sonicated and filtered before spin casting on 2 μm thick parylene-C (SCS Labcoater 2) coated 1 × 3 inch glass substrates. A 100 nm gold electrode is vapor deposited on one end of the film, followed by the deposition of a 40 μm-thick SU-8 film patterned on top of the PEDOT:PSS, serving as an ion barrier. Pattering of the ion barrier created an opening for the planar electrolyte/conducting polymer film interface. A polydimethylsiloxane (PDMS) rim was placed on top of the SU-8 well to confine the electrolyte (10 mM KCl in DI water). The length of PEDOT:PSS film between the electrolyte and the Au contact was 3.2 cm.Moving front experiments
Dedoping by K+ ions was achieved by applying a +2 V bias to the Ag/AgCl electrode that is immersed in the electrolyte with respect to the Au electrode. The device was then short-circuited to return to its doped state (this defined one cycle). The de-doping was found to be reversible and the film returned to its initial conduction state upon removal of the applied bias. To ensure sufficient hydration and stable cycling of the films, at least three dedoping cycles were applied before acquiring data for analysis. Voltage was applied and current was measured using a Keithley 2612A source-measure unit connected to a computer running custom LabView software. Microscope-based measurements were performed using an inverted Carl Zeiss Axio-Observer Z1. For the spectroscopic version of the moving front experiments, i.e. spectro-temporal moving front, samples were mounted in a custom 3D printed holder which aligns fiber optic cables directly across from each other in order to measure the absorption spectrum of the film at a position X from the edge of the electrolyte/polymer interface. One fiber led to the white light source (Hamamatsu) and the other to a spectrometer comprising the detector unit (Ocean Optics USB4000). The optical fiber core is 400 μm in diameter. Upon biasing the sample, UV-VIS spectra were recorded at one second intervals using Ocean Optics OceanView software. All analysis was performed with custom MATLAB software.Results and discussion
The time course of the optical spectra due to the electrochromic moving front is compared at two positions, X, from the electrolyte/film interface (Fig. 1A, left). The complete moving front device structure is assembled and mounted on a custom holder allowing the light path from a white light source/fiber optic to pass through the film at the desired location. Fig. 1B and D show the time-lapse UV-VIS spectra of a PEDOT:PSS film recorded in 1 s intervals at X = 1 mm and 4 mm, respectively. Both spectra demonstrate the transient change in the absorbance properties of the film (400 nm ≤ λ ≤ 850 nm) as the de-doping front progresses. The changes in the spectra associated with the drift of the injected cations (0 s ≤ t ≤ 90 s) are further represented in a time–wavelength color plot (Fig. 1C and E). Note that as the front passes the point of observation (X), the spectrum exhibits a complete saturation, indicating that a maximum level of de-doping occurs. Moreover, the spectra acquired at different distances from the polymer/electrolyte interface confirm the 1-D motion of cations inside the film towards the Au electrode: the de-doping ions take a longer time (t ∼ 40 s) to arrive at the X = 4 mm position (Fig. 1E), compared to X = 1 mm (t ∼ 10 s) (Fig. 1C). The shape of the color plot in Fig. 1E suggests a diffusive spread of ionic carriers, as the moving front undergoes 1-D drift. | ||
| Fig. 1 Spectroscopic moving front experiment. (A) Configuration of the spectro-temporal moving front experiment using a white light source/fiber spectrometer (left), and spatio-temporal moving front experiment using a microscope objective (right), at a specified distance X from the edge of the film–electrolyte interface, to track the motion of injected ions. In all cases, the time of application of bias is t = 0 s. The absorbance spectra and the corresponding time–wavelength color plot for X ≈ 1 mm (B and C), and X ≈ 4 mm (D and E), for a PEDOT:PSS film (0 vol% EG). B and D show the absorbance spectra from t = 1 s (blue) to t = 90 s (red) using the same color scheme. (F) Normalized absorbance intensity of the film at a distance X as the de-doping front propagates, validating the agreement between the spectro-temporal (red symbols) and traditional spatio-temporal moving front experiment (black symbols) (X values as noted). The normalized absorbance intensity profiles of the same film obtained from microscopy studies are shown as an inset (blue: t = 5 s, red: t = 45 s). | ||
In order to validate the spectroscopy studies in the context of previous moving front studies,21 we monitored the change in the transmitted light intensity through the same films with an optical microscope (Fig. 1A, right). In this case, propagation of the de-doping front over time was analyzed through bright-field images21 (see Fig. S1 for the time lapse images, ESI†). The results obtained via this spatio-temporal moving front technique were then compared with those acquired with the spectrally-resolved approach introduced above. Fig. 1F shows the direct comparison of these two techniques investigating the same phenomenon in the very same films. The normalized absorption intensity of the film at a distance X as measured by the microscope objective is plotted (black symbols), along with the integrated intensity of the optical spectra (red). Note that the UV-VIS spectra were convoluted with the sensitivity spectrum of the microscope camera. The change in absorption intensity over time measured by the two techniques is in good agreement at both X positions, with the largest error arising from the placement of the fiber light source at the desired position. This validates the use of the spectro-temporal moving front technique to complement or supplant the microscopy-based method. In fact, while the latter allows for the estimation of ion mobility, it yields information only about the spatial extent of the ion/redox-active site interaction.
In order to demonstrate the utility of the moving front technique for various materials/formulations, we investigate films cast from PEDOT:PSS dispersions that contain 5% and 50% of ethylene glycol (EG) by volume. It is well known that polar, high boiling co-solvents affect the structure/morphology of PEDOT:PSS films, which consequently influence the electronic transport properties.29,30 EG, which is often employed as a conductivity enhancer in the dispersion, aids in molecular organization of the film and mesoscale phase separation of the PEDOT:PSS-rich and PSS-rich domains.31–36Fig. 2 shows the absorbance spectra of the three PEDOT:PSS films during the transit of the de-doping front (1 s ≤ t ≤ 25 s), as well as the corresponding time–wavelength color plots. The time-dependent changes in the spectra are further processed to show the effective derivative, i.e., the difference in successive absorbance spectra that are 1 s apart (Fig. 2, lower panels). The time course of the optical absorption at X = 1 mm reveals that while all samples show a similar early-time progression (1 s ≤ t ≤ 10 s), the absorption increase lingers for an additional ca. 5 s and 10 s for films cast from 5 vol% EG and 50 vol% EG containing dispersions, respectively (see color plots of the derivative spectra). The shape of the spectral color plots suggests a slower complete electrochromic modulation with the increase in EG concentration. In all samples, the moving front first manifests itself as an increase in polaron absorption (λ > 800 nm), presumably as the bipolaron absorption in the NIR subsides.37,38 The polaron absorption is replaced by π → π* transitions as cations de-dope PEDOT chromophores. Visible absorption is recovered first by disordered/amorphous chromophores, (λ ∼ 600 nm), but quickly evolves towards absorption contributed by PEDOT aggregates. The latter is associated with low energy-vibronic features observed at ca. 685 nm (A00 transition) and at 625 nm (A01 transition).24 Notably, during the final 5–10 seconds before reaching saturation, the spectra of EG-containing films show a shift in dominant absorption increase from the A00 to A01 transition; best observed in the derivative spectra in Fig. 2, bottom.
 | ||
| Fig. 2 Spectroscopic analysis of electrochromic moving fronts for various PEDOT:PSS formulations. Raw, An–A0 data and analysis of spectro-temporal moving front experiments for 0 vol% (left), 5 vol% (middle), and 50 vol% (right) EG content in PEDOT:PSS formulations. n represents the nth spectrum after application of bias; since spectra are recorded every second, n = t. Top: Absorbance spectra (An–A0) of each film from t = 1 s (blue) to t = 25 s (red). Bottom: Associated color plots of the absorbance and its derivative (An+1–An)/dt (with dt = 1 s) as a function of time. | ||
Given the subtle differences in the UV-VIS spectra during the time course of the moving front experiment, a more quantitative approach is sought by using analysis based on the Spano model.27,28,39 This model enables a quantitative analysis of the absorption spectra of linear H-aggregates, formed in the ordered domains of conjugated polymer chains, in relation to the film morphology. Spano's analytical model is frequently utilized for the conjugated polymer poly(3-hexylthiophene), P3HT, and analogues with variant side chains.24,25,40 Since PEDOT:PSS films, comprising aggregated thiophene-based chains, show similar spectral features as compared to P3HT (Fig. 2), we employ the Spano model for H-aggregates to the absorption spectra of PEDOT:PSS films acquired during the transit of de-doping ions. The parameters obtained from this analysis hint at the relative variation in aggregate absorption, aggregate fraction, and the molecular arrangements of photophysical aggregates during the redox cycle. This provides valuable information regarding the physical pathways of drifting ions inside PEDOT:PSS films, which we affect by varying the content of EG in the dispersion.35
The Spano model allows us to isolate the changes in absorbance spectrum due to aggregates. Fig. S2 (ESI†) shows the absorption spectra (An–A0) recorded at X ∼ 4 mm for the pristine PEDOT:PSS film and the film cast with 50 vol% EG. The fits to these spectra are performed as in ref. 25, detailed in the ESI,† and shown for a number of cases in Fig. S3. Fig. 3 summarizes the results of the fits to the Spano model. By quantifying the time dependent aggregate absorption in the films, we track the ions as they de-dope the PEDOT aggregates which are present in regions of the film differing in morphology and composition. The spectro-temporal moving front technique is thereafter applicable to any mixed conductor, the only requirement being that the ion transport is less effective than electronic transport, i.e., electronic transport does not limit the electrochemical (de)doping.41 The two films vary significantly in the evolution of aggregate absorption with time (Fig. 3A). For the film cast with 50 vol% EG in the dispersion, two distinct rates are involved in the build-up of the visible spectrum (Fig. 3A, dashed lines): we attribute the initial fast rise in the absorption to relatively disordered PEDOT chains that are mixed in or adjacent to the PSS-rich phase, while the lagging absorption arises due to chains in dense PEDOT:PSS-rich domains which are formed when co-solvents such as EG are employed.32,34,36 Such domains are comprised of more closely packed aggregates.42 The lingering of this absorption contribution at longer time points (before saturation) is also directly observable in the time–wavelength analysis in Fig. 2.
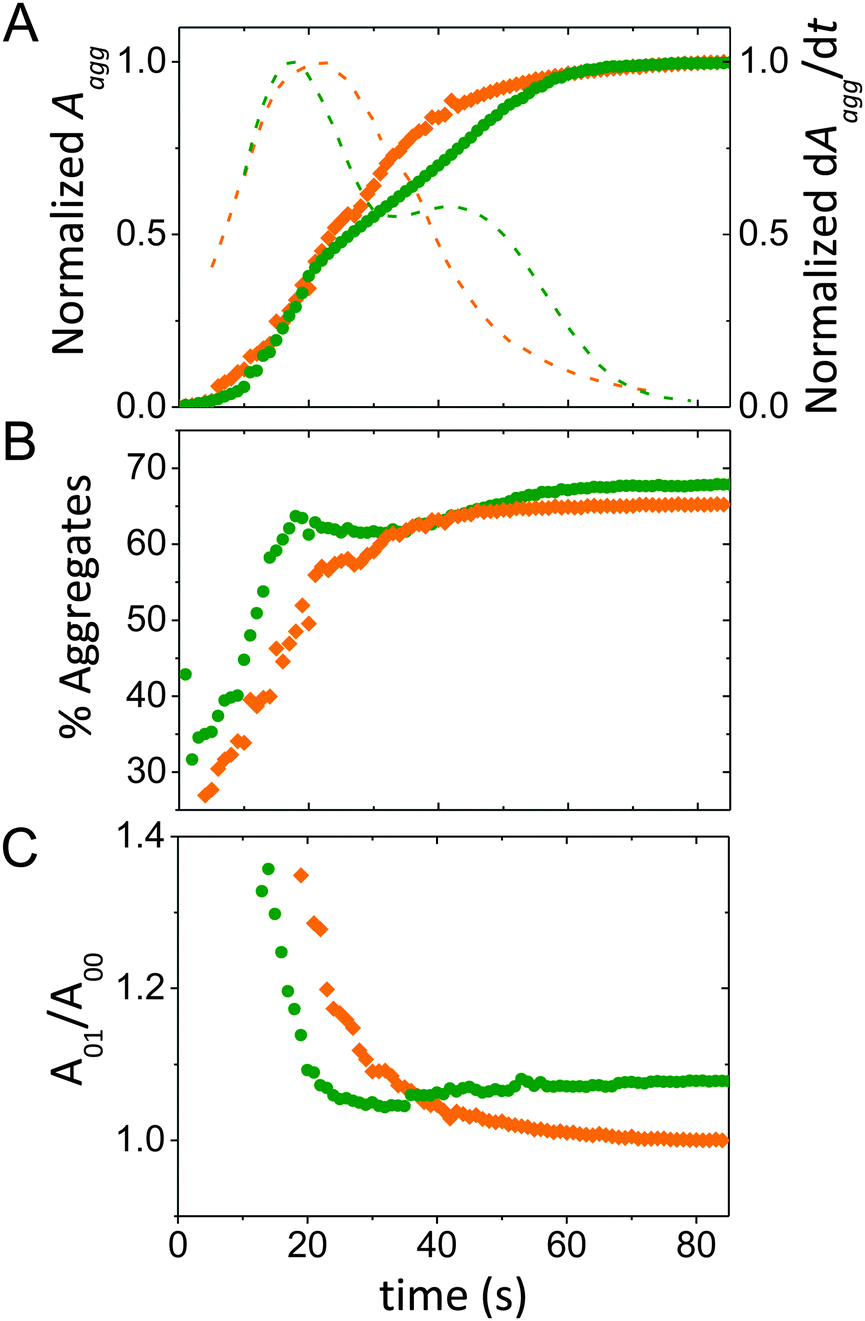 | ||
| Fig. 3 Analysis of absorbance spectra during the course of a moving front experiment. Data from a pristine PEDOT:PSS film is shown in orange and from a PEDOT:PSS film prepared from 50 vol% EG containing dispersion is in green. (A) The normalized absorption of PEDOT aggregates as a function of time at X = 4 mm from the electrolyte/film interface. The normalized rate of increase in aggregate absorption (dotted lines) is on the right axis. (B) The estimated aggregate fraction in the films, and (C) the ratio of the A01 to A00 with time, as the moving front passes. | ||
By quantifying the amorphous contribution to the spectra, the time course of the aggregate fraction in the film can be better compared (Fig. 3B). At early time points (t < 20 s), the increase in aggregate fraction is similar for both films. However, with 50 vol% EG, it takes ca. 25 s longer to reach saturation in the aggregate fraction profile due to the final ca. 7% of aggregates. While aggregated PEDOT is thought to be necessary for enhanced electronic transport, in this case, it also seems to slow down the penetration of ions. On the other hand, although EG is known to enhance π–π scattering within PEDOT-rich regions,32,33,36 the saturated absorption of the film cast from EG-containing dispersion reveals only ∼6% larger aggregate fraction compared to the film cast without EG. This might be due to the presence of relatively short PEDOT chains existing in such dispersions,43 which would contribute to the vibronic signature of photophysical aggregates, but not to scattering due to π-stacking.
The subtleties of molecular (dis)order can be further investigated by quantifying the shape and amplitude ratios of the vibronic peaks. According to the Spano model, when intrachain order, i.e. planarity of monomers within chain segments is low, optical spectra resemble H-aggregates where the A01 transition is more intense than A00.39,44 Within an H-aggregate model, the ratio of the 0–0 and 0–1 vibronic peaks can be used to estimate the magnitude of the interchain coupling.26,27 As the de-doping front proceeds, we see a decrease in the ratio of the A01 to A00 aggregate peaks, (A01/A00), more abrupt with the EG content, meaning that interchain coupling in the aggregates diminishes with time (Fig. 3C). This finding might indicate that at longer time points, the de-doping ions interact with longer and/or more planarized PEDOT chains. It is interesting to note that A01/A00 shows a different time-dependent variation and final state when comparing the two films. A higher final value of A01/A00 for the film cast with EG content indicates stronger intermolecular coupling than the pristine film, i.e. a closer interchain distance and tighter π-stacking, evidenced by Palumbiny et al. through X-ray studies.42 Here, it must be noted that Spano's H-aggregate model assumes a certain molecular packing/order of the chromophores that, while supported to some extent with prior structural studies, has not been thoroughly validated for PEDOT:PSS films. Moreover, since this experiment probes a changing population of PEDOT chromophores in response to drifting ion interaction, one alternate possibility could be that the assumption of aggregate type (H vs. J) varies with time as the front progresses, which would necessitate application of a more complex model.39 We thus highlight that the experimental configuration and data collection methodology presented herein can be applied to any number of appropriate models provided their reasonable agreement with the observed molecular arrangements.
Conclusions
We have put forth a method that offers a link between the molecular packing/microstructure of electrochromic films and ion motion therein. We monitored the electrochromic moving front in order to deduce the local environment of K+ ions (as they dedope the PEDOT chromophores). Three formulations of PEDOT:PSS, with varying EG content in dispersion, were targeted to study films that are known to differ in morphology. By fitting the absorption profiles of these films to the Spano model, we find that the increase in aggregate absorption lingers at longer times, slowing down the overall ion penetration/motion within the polymer film. This is observed only in the presence of EG in the casting dispersion, known as an enhancer of electronic conductivity. Thus, to design an ideal mixed ion-electron conductor, aggregation should be sufficient for efficient electronic transport, but its extent should be controlled since dense, aggregated regions hinder effective ion penetration and slow the complete electrochemical de-doping. The moving front experiments combined with microstructural/morphological probes such as X-ray scattering will help building a complete picture of the nature of mixed conduction in organic electronic systems. Furthermore, these studies are performed in situ during device biasing, mimicking the operation of state-of-the-art devices that rely on mixed conduction, e.g., actuators and electrochemical transistors. The techniques presented here will, thereby, contribute to the rational design of new polymeric ionic/electronic mixed conductors for emerging applications.Acknowledgements
J. R. and S. I. acknowledge funding from the Fondation Recherche Medicale and Fondation Francaise pour la Recherche sur l'Epilepsie, respectively. S. I. thanks R. Lehmann for assistance in data analysis. The authors thank P. Pingel for fruitful discussions on the Spano model.References
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS.
- P. Lin, F. Yan, J. Yu, H. L. W. Chan and M. Yang, Adv. Mater., 2010, 22, 3655–3660 CrossRef CAS PubMed.
- E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS.
- C. S. Haines, M. D. Lima, N. Li, G. M. Spinks, J. Foroughi, J. D. W. Madden, S. H. Kim, S. Fang, M. Jung de Andrade, F. Göktepe, Ö. Göktepe, S. M. Mirvakili, S. Naficy, X. Lepró, J. Oh, M. E. Kozlov, S. J. Kim, X. Xu, B. J. Swedlove, G. G. Wallace and R. H. Baughman, Science, 2014, 343, 868–872 CrossRef CAS PubMed.
- A. Malti, J. Edberg, H. Granberg, Z. U. Khan, J. W. Andreasen, X. Liu, D. Zhao, H. Zhang, Y. Yao, J. W. Brill, I. Engquist, M. Fahlman, L. Wågberg, X. Crispin and M. Berggren, Adv. Sci., 2016, 3, 1500305 Search PubMed.
- D. T. Simon, S. Kurup, K. C. Larsson, R. Hori, K. Tybrandt, M. Goiny, E. W. H. Jager, M. Berggren, B. Canlon and A. Richter-Dahlfors, Nat. Mater., 2009, 8, 742–746 CrossRef CAS PubMed.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- S. Sax, G. Mauthner, T. Piok, S. Pradhan, U. Scherf and E. J. W. List, Org. Electron., 2007, 8, 791–795 CrossRef CAS.
- S. Inal, J. Rivnay, P. Leleux, M. Ferro, M. Ramuz, J. C. Brendel, M. M. Schmidt, M. Thelakkat and G. G. Malliaras, Adv. Mater., 2014, 26, 7450–7455 CrossRef CAS PubMed.
- D. Tordera, M. Kuik, Z. D. Rengert, E. Bandiello, H. J. Bolink, G. C. Bazan and T.-Q. Nguyen, J. Am. Chem. Soc., 2014, 136, 8500–8503 CrossRef CAS PubMed.
- P. S. David, G. R. Stephen and C. L. Mark, Iontronics, CRC Press, 2010, pp. 43–83 Search PubMed.
- S. Ghosh and O. Inganäs, Electrochem. Solid-State Lett., 2000, 3, 213–215 CrossRef CAS.
- C. K. Baker, Y. J. Qiu and J. R. Reynolds, J. Phys. Chem., 1991, 95, 4446–4452 CrossRef CAS.
- X. Cui and D. C. Martin, Sens. Actuators, B, 2003, 89, 92–102 CrossRef CAS.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J.-M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, e1400251 Search PubMed.
- J. M. Leger, Adv. Mater., 2008, 20, 837–841 CrossRef CAS.
- X. Wang, B. Shapiro and E. Smela, Adv. Mater., 2004, 16, 1605–1609 CrossRef CAS.
- T. Johansson, N.-K. Persson and O. Inganäs, J. Electrochem. Soc., 2004, 151, E119–E124 CrossRef CAS.
- K. Aoki, T. Aramoto and Y. Hoshino, J. Electroanal. Chem., 1992, 340, 127–135 CrossRef CAS.
- E. Stavrinidou, P. Leleux, H. Rajaona, D. Khodagholy, J. Rivnay, M. Lindau, S. Sanaur and G. G. Malliaras, Adv. Mater., 2013, 25, 4488–4493 CrossRef CAS PubMed.
- S. Inal, J. Rivnay, A. I. Hofmann, I. Uguz, M. Mumtaz, D. Katsigiannopoulos, C. Brochon, E. Cloutet, G. Hadziioannou and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 147–151 CrossRef CAS.
- E. Stavrinidou, O. Winther-Jensen, B. S. Shekibi, V. Armel, J. Rivnay, E. Ismailova, S. Sanaur, G. G. Malliaras and B. Winther-Jensen, Phys. Chem. Chem. Phys., 2014, 16, 2275–2279 RSC.
- S. T. Turner, P. Pingel, R. Steyrleuthner, E. J. W. Crossland, S. Ludwigs and D. Neher, Adv. Funct. Mater., 2011, 21, 4640–4652 CrossRef CAS.
- D. T. Duong, M. F. Toney and A. Salleo, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 205205 CrossRef.
- J. Clark, C. Silva, R. Friend and F. Spano, Phys. Rev. Lett., 2007, 98, 206406 CrossRef PubMed.
- F. C. Spano, J. Chem. Phys., 2005, 122, 234701 CrossRef PubMed.
- F. C. Spano, Chem. Phys., 2006, 325, 22–35 CrossRef CAS.
- J. Huang, P. F. Miller, J. S. Wilson, A. J. de Mello, J. C. de Mello and D. D. C. Bradley, Adv. Funct. Mater., 2005, 15, 290–296 CrossRef CAS.
- H. Shi, C. Liu, Q. Jiang and J. Xu, Adv. Electron. Mater., 2015, 1 DOI:10.1002/aelm.201500017.
- J. Ouyang, C. W. Chu, F. C. Chen, Q. Xu and Y. Yang, Adv. Funct. Mater., 2005, 15, 203–208 CrossRef CAS.
- C. M. Palumbiny, C. Heller, C. J. Schaffer, V. Körstgens, G. Santoro, S. V. Roth and P. Müller-Buschbaum, J. Phys. Chem. C, 2014, 118, 13598–13606 CAS.
- T. Takano, H. Masunaga, A. Fujiwara, H. Okuzaki and T. Sasaki, Macromolecules, 2012, 45, 3859–3865 CrossRef CAS.
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Müller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076–1081 CrossRef CAS.
- D. Alemu Mengistie, P.-C. Wang and C.-W. Chu, J. Mater. Chem. A, 2013, 1, 9907–9915 CAS.
- Q. Wei, M. Mukaida, Y. Naitoh and T. Ishida, Adv. Mater., 2013, 25, 2831–2836 CrossRef CAS PubMed.
- J. Ouyang, ACS Appl. Mater. Interfaces, 2013, 5, 13082–13088 CAS.
- A. O. Patil, A. J. Heeger and F. Wudl, Chem. Rev., 1988, 88, 183–200 CrossRef CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- P. Pingel, A. Zen, R. D. Abellón, F. C. Grozema, L. D. A. Siebbeles and D. Neher, Adv. Funct. Mater., 2010, 20, 2286–2295 CrossRef CAS.
- E. Stavrinidou, P. Leleux, H. Rajaona, M. Fiocchi, S. Sanaur and G. G. Malliaras, J. Appl. Phys., 2013, 113, 244501 CrossRef.
- C. M. Palumbiny, F. Liu, T. P. Russell, A. Hexemer, C. Wang and P. Müller-Buschbaum, Adv. Mater., 2015, 27, 3391–3397 CrossRef CAS PubMed.
- J. Zhang, H. Ellis, L. Yang, E. M. J. Johansson, G. Boschloo, N. Vlachopoulos, A. Hagfeldt, J. Bergquist and D. Shevchenko, Anal. Chem., 2015, 87, 3942–3948 CrossRef CAS PubMed.
- T. P. Martin, A. J. Wise, E. Busby, J. Gao, J. D. Roehling, M. J. Ford, D. S. Larsen, A. J. Moulé and J. K. Grey, J. Phys. Chem. B, 2013, 117, 4478–4487 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tc04354a |
| ‡ Present address: PARC, a Xerox company, 3333 Coyote Hill Rd. Palo Alto, CA 94304, USA. E-mail: jonathan.rivnay@parc.com |
| This journal is © The Royal Society of Chemistry 2016 |