
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the aggregation-induced enhanced emission behavior and self-assembly of phosphole-lipids†
Zisu
Wang
,
Benjamin S.
Gelfand
,
Pengcheng
Dong
,
Simon
Trudel
and
Thomas
Baumgartner
*
Department of Chemistry and Centre for Advanced Solar Materials, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada. E-mail: thomas.baumgartner@ucalgary.ca
First published on 18th December 2015
Abstract
Herein, we report on the synthesis, self-assembly, as well as the photophysical properties of a novel series of P-benzylated phosphole-lipids. In the context of this structure–property study we have systematically altered the number, position, and length of the alkyl chains in the 3-,4-, and 5-position of the benzyl group. Both the self-assembly and photophysical properties of the compounds were found to correlate strongly with the alkyl chain length and chain arrangement of the mesogenic moiety.
Introduction
Organic materials that contain main group elements, such as P,1a–d B,1e,f and Si,1g are experiencing a surge in interest. This is due to the unique structural and electronic properties these elements can impart on the organic scaffold of the material.Among structural motifs commonly found in organo-main group molecules, phospholes are a unique class of compounds.1a Distinct from the analogous pyrroles, phospholes are partially aromatic, as the phosphorus lone pair has only little overlap with the π orbital of the backbone.1a The limited aromaticity of the molecule actually comes from the interaction of the σ* orbital of the P–R single bond and the π* orbital of the conjugated scaffold (Fig. 1). Due to these unique interactions, chemical modification of the phosphole lone pair can have a considerable effect on the optoelectronic properties of the resulting compounds. By adjusting the substituent ‘E’ on the lone pair, the emission properties of the compounds can be easily tuned without the need for modification of the backbone.2
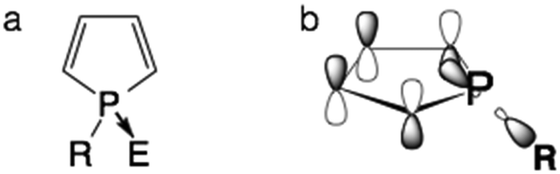 | ||
| Fig. 1 The unique electronics of phospholes. (a) General bonding patterns of phospholes; (b) σ*–π* interaction in phospholes. | ||
Over more than a decade, our group has been involved with the development of functional conjugated organophosphorus species and we have established various highly desirable optoelectronic features for the materials.3 To expand the general utility of such species in a materials context, we have recently reported on a series of self-assembled dithienophosphole-based materials. These ‘phosphole-lipids’ exhibit liquid crystalline properties over a wide temperature range (Fig. 2).4a Some of the phosphole-lipids also display gelation properties in a wide range of solvents at low concentrations.2b The self-assembly features of phosphole-lipids are the results of a balancing act between intermolecular π–π interactions of the phosphole backbones, the intermolecular ionic interactions and the thermal disorder imparted by the flexible alkyl chains. As can be seen in Fig. 2, with larger π conjugation in its phosphole backbone, the increased intermolecular π–π interactions in 1a led to a more crystalline soft crystal phase unlike the liquid crystal phase found in the smaller DTP. Therefore, we became interested in further exploring this delicate balance between the driving forces and tuning the self-assembly feature of our phosphole-lipid system in more detail.
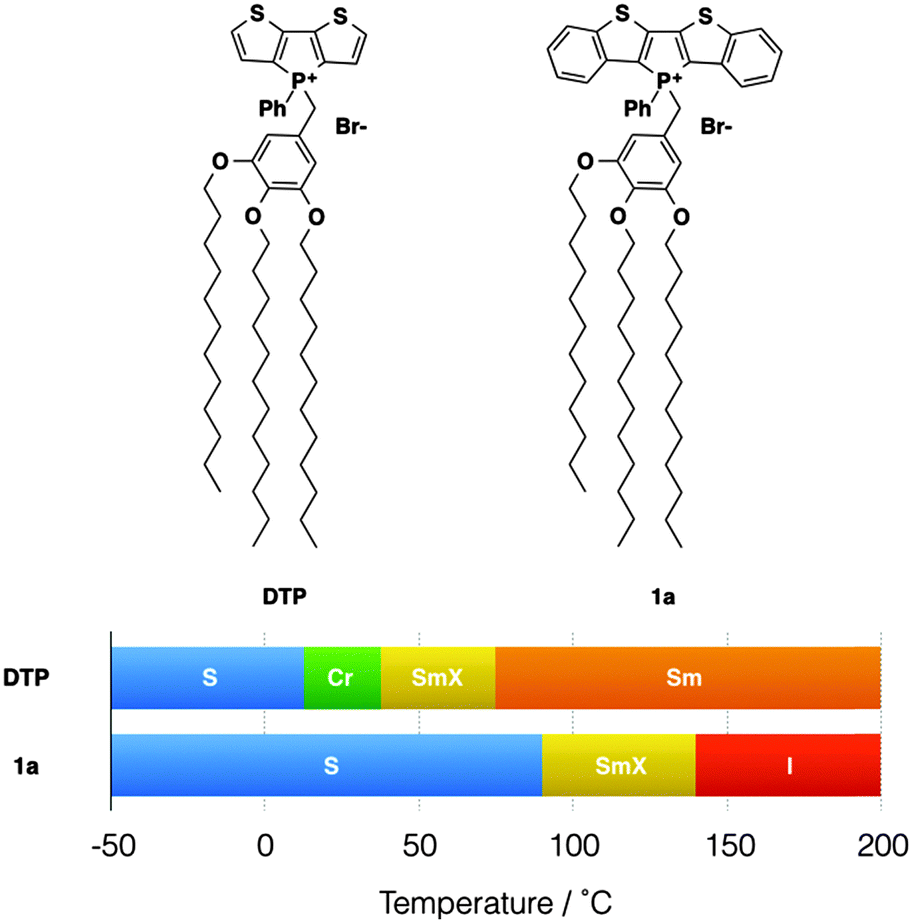 | ||
| Fig. 2 Phosphole-lipid liquid crystals. S, “solid”; Cr, crystalline state; SmX, soft crystal; Sm, smectic phase; I, isotropic liquid. | ||
In phosphole-lipids, quaternization at the phosphorus lone pair affords the ionic feature of the compounds, which is a critical directing force for the self-assembly process. At the same time, due to the aforementioned σ*–π* hyperconjugation, the benzyl moiety also heavily participates in the optical transitions of the molecule. As such, the rotational flexibility of the benzyl moiety also impacts the optoelectronic behavior of the compounds both in solution and in the solid state.4a,c We were further able to confirm that stimuli-responsive features of phosphole-lipids such as aggregation-induced enhanced emission (AIEE) and mechanochromism also stem from this particular role of the benzyl moiety.4
AIEE is the phenomenon where the aggregated-state fluorescence quantum yield of the compound exceeds that of the compound in solution.5 Contrary to the more common aggregation-caused quenching (ACQ) of fluorescence that is found in organic chromophores, AIEE-enabled materials hold more appeal in application settings such as OLED displays. As well, due to the increased fluorescence of the compounds in the solid state, these materials provide interesting opportunities in studying the photoluminescence process in the solid state.5 Tetraphenylethylene-containing compounds and phenyl-appended siloles are two of the most studied AIEE systems for their excellent response to concentration variations not only in solution, but also in the solid state (Fig. 3).5 The AIEE in both systems stems from the free/restricted intramolecular motion of conjugated peripheral substituents.
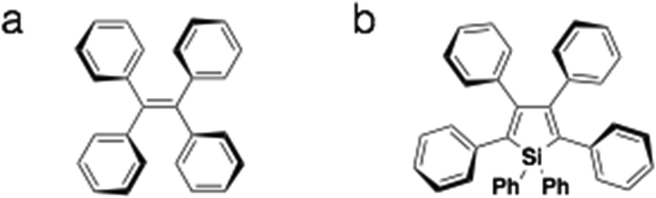 | ||
| Fig. 3 Examples of common AIEE chromophores. (a) Tetraphenylethylene; (b) hexaphenylsilole. | ||
While the general principle of AIEE can be understood with relative ease, the effect of self-organization on AIEE still needs to be better defined. To this end, the well defined self-assembly features of the phosphole-lipid system enable us to gain a glimpse into the relationship between self-assembly and AIEE.
Herein, we report on our efforts in further exploring and enhancing both the self-assembly and the AIEE properties of phosphole-lipids. In the context of this study, the di(benzothieno)phosphole-based lipid was chosen because of the promising packing features of the π-extended head group.4
Results and discussion
Synthesis
To gain deeper insights into the underlying parameters required for the self-assembly and ultimately the AIEE of the phosphole-lipid system, a variety of target compounds were chosen for this study, in which we have selectively altered the number and position of the benzyl alkyl chains as well as their length (Scheme 1).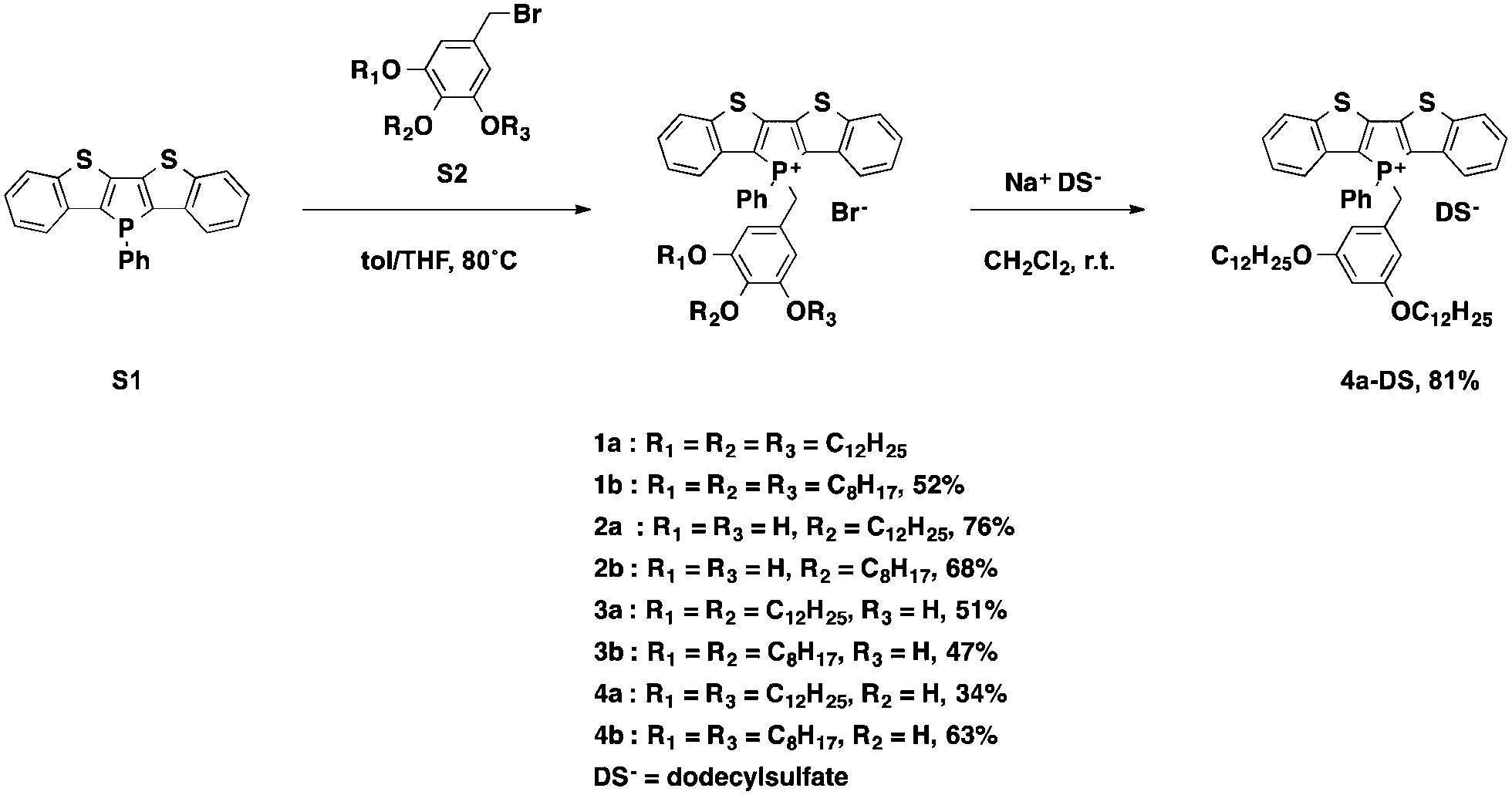 | ||
| Scheme 1 Synthesis of phosphole-lipids. Quaternization of S1 with various benzyl bromides (S2) afforded the desired phosphole-lipids 1–4. The bromide anion was exchanged with dodecylsulfate via metathesis. | ||
The dodecyl-functionalized species (2a, 3a and 4a) were aimed at paralleling and expanding our original studies4 in order to see if a different placement of the dodecyl chains led to distinct self-assembly behavior and/or altered electronics. To this end, we expected to pinpoint and systematically study the effect of chain arrangements on the resulting solid-state properties. The octyl-functionalized species (1b, 2b, 3b and 4b) were targeted to study the effects of a shifted balance with regard the potential intra- and intermolecular interactions via van-der-Waals and π-stacking interactions.
The di(benzothieno)phosphole S1 was synthesized following our reported procedure.6 The various alkoxybenzyl bromides S2 were obtained in high yields by following the standard procedure reported by Zhao et al.7 Quaternization of the phosphorus center afforded the target compounds in modest to good yields (Scheme 1).
To also test the counteranion as effective handle for tuning the self-assembly properties of phosphole-lipids, a model study was conducted with the cation of 4a and the dodecylsulfate anion. Dodecylsulfate was chosen because its ability to promote amphiphilic properties8 and its expected compatibility with the dodecyl chains already present in the cationic portion of 4a. The exchange of the bromide for dodecylsulfate was accomplished by repeated aqueous extraction of an equimolar solution of the phosphole-lipid 4a and sodium dodecylsulfate (Na+DS−) in dichloromethane to yield the anion-exchanged species 4a-DS in 81% yield.
Photophysical properties
| Compound | λ abs (nm) | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) εa εa |
λ
emsolution (nm) |
ϕ
PLsolution
|
λ
emsolid-state (nm) |
ϕ
PLsolid-state
|
AIEE responsed (%) |
|---|---|---|---|---|---|---|---|
|
a Molar absorption coefficient (M−1 cm−1).
b Relative, measured against a quinine sulfate standard adjusted to the same absorbance, λex = 365 nm.
c Absolute, determined using an integrating sphere, λex = 365 nm.
d Obtained by subtracting ϕPLsolution from ϕPLsolid-state. Negative value indicates aggregation quenching, positive value indicates AIEE.
e Not available due to low signal to noise ratio.
|
|||||||
| 1b | 350, 415 | 4.02, 3.88 | n/ae | 0.00 | n/ae | 0.00 | 0 |
| 2a | 350, 416 | 4.12, 3.99 | 514 | 0.26 | 526 | 0.03 | −23 |
| 2b | 350, 415 | 4.11, 3.96 | 512 | 0.26 | 529 | 0.05 | −21 |
| 3a | 347, 413 | 3.98, 3.86 | n/ae | 0.00 | n/ae | 0.00 | 0 |
| 3b | 348, 414 | 4.07, 3.96 | n/ae | 0.00 | 529 | 0.09 | 9 |
| 4a | 347, 417 | 4.05, 3.90 | 524 | 0.13 | 517 | 0.33 | 20 |
| 4b | 349, 416 | 4.07, 3.92 | 522 | 0.10 | 518 | 0.19 | 9 |
| 4a-DS | 348, 417 | 4.04, 3.90 | 524 | 0.17 | 529 | 0.10 | −7 |
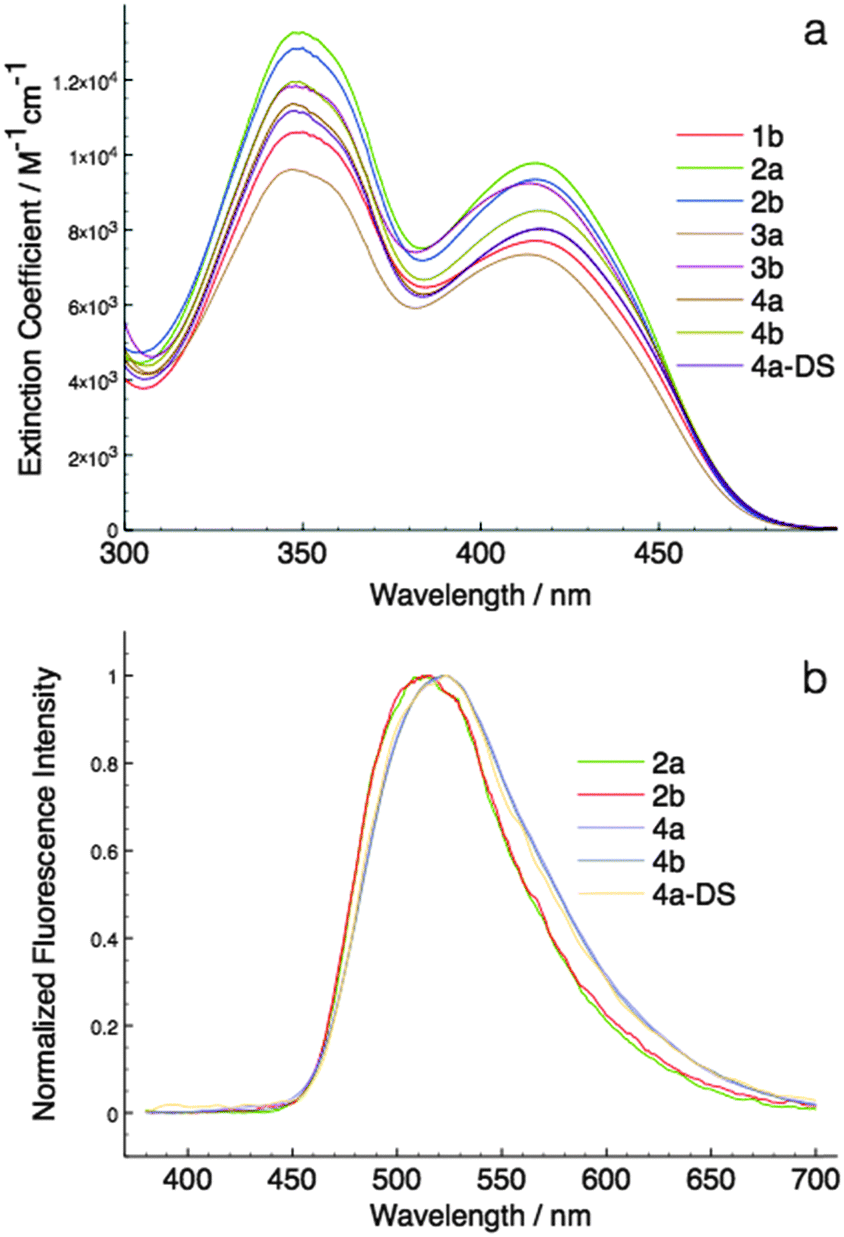 | ||
| Fig. 4 Solution photophysics of phosphole-lipids. (a) UV-Vis absorption spectra of the phosphole-lipids; (b) fluorescence spectra of phosphole-lipids (low emission species was omitted for clarity due to low signal to noise ratio). | ||
Theoretical calculations
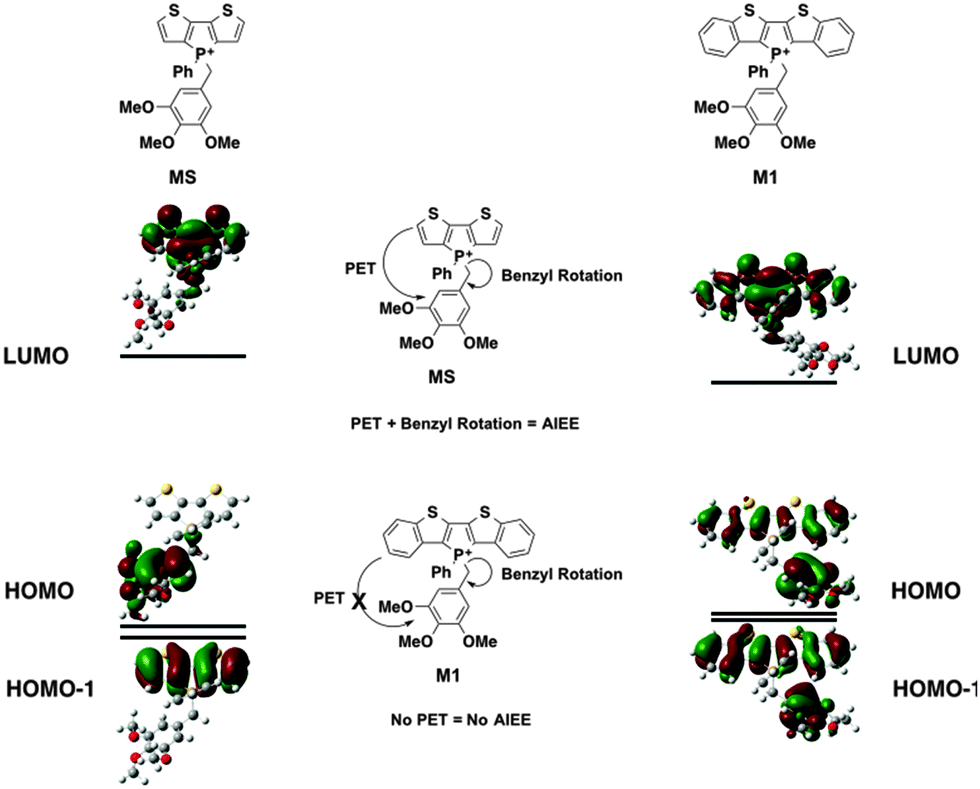 | ||
| Fig. 5 Mechanism of AIEE in phosphole-lipids. | ||
In M1, however, significant overlap exists between the FMOs of the species (Fig. 5). The PET process is therefore suppressed in this case, which leads to the lack of AIEE observed in the previously reported 1a species.
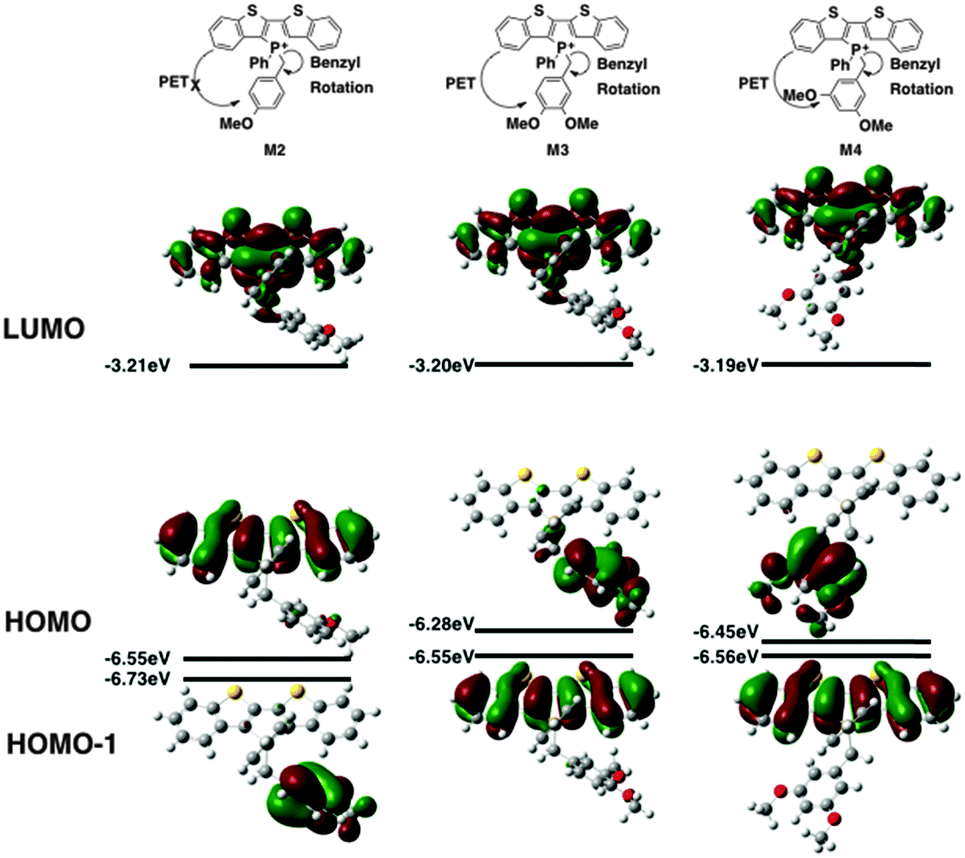 | ||
| Fig. 6 Frontier orbitals of model phosphole-lipids. | ||
Contrary to the observed photophysics, M3 and M4 are electronically quite similar to each other according to the calculations. The LUMO (primarily the π* orbital of the conjugated phosphole head group), HOMO (primarily the π orbital of the benzyl moiety) and HOMO−1 (primarily the π orbital of the phosphole head group) all have the same distribution in both species. A minor stabilization of the HOMO exists in M4, which can be attributed to the weaker π-donating nature of the meta-alkoxy substituent. Similar to MS, M3 and M4 are both PET-enabled making them strong candidates of AIEE features. Combining this result with the solution fluorescence observations, we can tentatively draw the conclusion that the significantly enhanced emissions of the 4 compounds do not originate from electronics but rather from different self-assembly in solution.
M2, on the other hand, deviates significantly from the other species in this study (Fig. 6). Both its HOMO and LUMO reside mostly on the π backbone of the conjugated phosphole head group. In addition, while spatial separation exists between HOMO−1 and LUMO in M2, the weak donating character of a single alkoxy substituent leads to a significant stabilization of HOMO−1, creating a considerable energy gap between HOMO and HOMO−1. As a result of the energy difference, the LUMO → HOMO−1 fluorescence transition is suppressed. Consequently, the compounds should behave much like other non-AIEE chromophores where the solution fluorescence is high. This prediction fits rather well with the observed high fluorescence of the compounds in solution.
Since the AIEE mechanism relies on the delicate energy balance between various orbitals, the conformation of the alkoxy substituents on the benzyl moiety was also investigated. The optimized structure (M4) placed the alkoxy group in plane with the benzene ring, which is quite reasonable as this conformation permits the conjugation between the p orbitals of the oxygen and the benzene ring. This conformation is also observed in the single crystal X-ray data of compound 4a (vide infra). DFT calculations were also conducted based on the X-ray crystallographic data of 4a (Fig. 7, M4-XRD with the dodecyl chains truncated to methyl for efficiency). With the endo-conformation of the benzyl substituent, there exists significant orbital communication between the benzyl substituent and the phosphole backbone. As a result, both the HOMO and the HOMO−1 were slightly stabilized compared to the optimized structure. This enlargement of the HOMO–LUMO gap was reflected in the slightly blue-shifted fluorescence of 4a in the solid state as compared to the solution (Δλ = 7 nm). To our surprise, a profound effect was observed, when the alkoxy group was rotated 90° with respect to the benzene plane, suppressing the lone pair π-conjugation (Fig. 7, M4-alkoxy rotated). The lack of π donation results in a significant stabilization (−0.58 eV) of the benzyl π orbital, which leads to the promotion of the phosphole π orbital to become the HOMO. In this configuration, the AIEE should be suppressed. However, considering the possibility of somewhat free rotation of the alkoxy groups in solution (at least to some extent), would explain the observed AIEE, but also accounts in part the residual fluorescence of compounds 4a,b in solution. This finding also indirectly supports the distinct emission features of the 3-series in solution, as the ortho-relationship of the two alkoxy chains seems to restrict their free rotation, in contrast to the 3,5-arrangement in 4a,b.
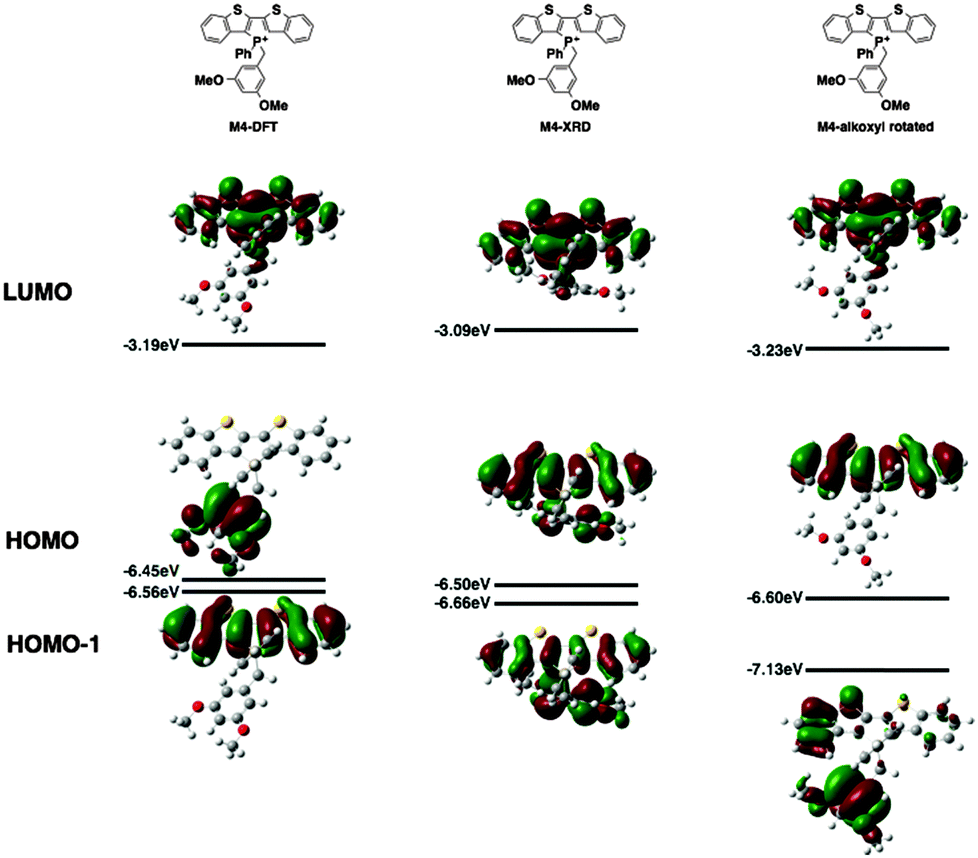 | ||
| Fig. 7 DFT calculations of M4 as optimized (left), based on X-ray data (middle) and M4 with the alkoxy groups rotated (right). | ||
| Calculated absorption bands | Nature of the transition | |
|---|---|---|
| a Contribution of the particular transition to the absorption band. b f = oscillator strength of the transition as calculated by TD-DFT. | ||
| M1 |
HOMO → LUMO (95%)a
HOMO−1 → LUMO (4%) (436 nm, fb = 0.2821) |
πphos–π*phos
πbenzyl–π*phos |
|
HOMO−3 → LUMO (88%)a
HOMO−4 → LUMO (10%) (358 nm, fb = 0.1636) |
πphos–π*phos | |
|
HOMO−4 → LUMO (88%)a
HOMO−3 → LUMO (10%) (352 nm, fb = 0.1226) |
πphos–π*phos | |
| M2 |
HOMO → LUMO (99%)a
(441 nm, fb = 0.2657) |
πphos–π*phos |
|
HOMO−2 → LUMO (97%)
(360 nm, f = 0.1252) |
πphos–π*phos
πbenzyl–π*phos |
|
|
HOMO−3 → LUMO (97%)
(358 nm, f = 0.182) |
πphos–π*phos | |
| M3 |
HOMO−1 → LUMO (99%)
(439 nm, f = 0.2889) |
πphos–π*phos |
|
HOMO−2 → LUMO (98%)
(364 nm, f = 0.077) |
πphos–π*phos
πbenzyl–π*phos |
|
|
HOMO−3 → LUMO (97%)
(357 nm, f = 0.1814) |
πphos–π*phos | |
|
HOMO−4 → LUMO
(347 nm, f = 0.04) |
πphos–π*phos
πbenzyl–π*phos |
|
| M4 |
HOMO−1 → LUMO (99%)
(435 nm, f = 0.2818) |
πphos–π*phos |
|
HOMO−2 → LUMO (94%)
HOMO−3 → LUMO (3%) (363 nm, f = 0.1355) |
πphos–π*phos
πbenzyl–π*phos |
|
|
HOMO−2 → LUMO (2%)
HOMO−3 → LUMO (93%) HOMO−4 → LUMO (2%) (357 nm, f = 0.1849) |
πphos–π*phos
πbenzyl–π*phos |
|
As can be seen in Fig. 6, the HOMO–LUMO energy gaps appear to be quite varied. However, the UV-Vis spectra of the compounds were rather similar to each other (Fig. 4a). This discrepancy can be easily resolved by considering the TD-DFT calculation results. According to TD-DFT, both the low energy (∼415 nm) and the high-energy (∼350 nm) absorption bands are dominated by the π–π* transition of the phosphole head group. Benzyl involvements were rather limited in the excitation processes. Since the di(benzothieno)phosphole head groups are the same in all compounds, it is no surprise that the absorption spectra are similar in all cases.
Solvatochromism
To substantiate the PET process in the new phosphole-lipids, solvatochromism studies were carried out for two representative species 2a and 4a. These compounds were chosen for their contrasting FMO arrangements (Fig. 6, M2 and M4).Since the intramolecular PET process relies on the polar charge-separated excited state, polar solvents are more capable of stabilizing the excited state. Therefore it follows that, with a polar solvent, a bathochromic shift of the absorption peaks should be observed compared to a nonpolar solvent if the PET is at play in the relaxation process.
As can be seen in Fig. 8, 4a indeed demonstrates significant solvatochromism where the growth of the low-energy charge-transfer bands is accompanied by the increase of solvent polarity. On the contrary, 2a had limited response to solvent polarity. As such, the solvatochromism of 4a in conjunction with the DFT results provides evidence of the presence of PET in phosphole-lipids with suitable FMO arrangements.
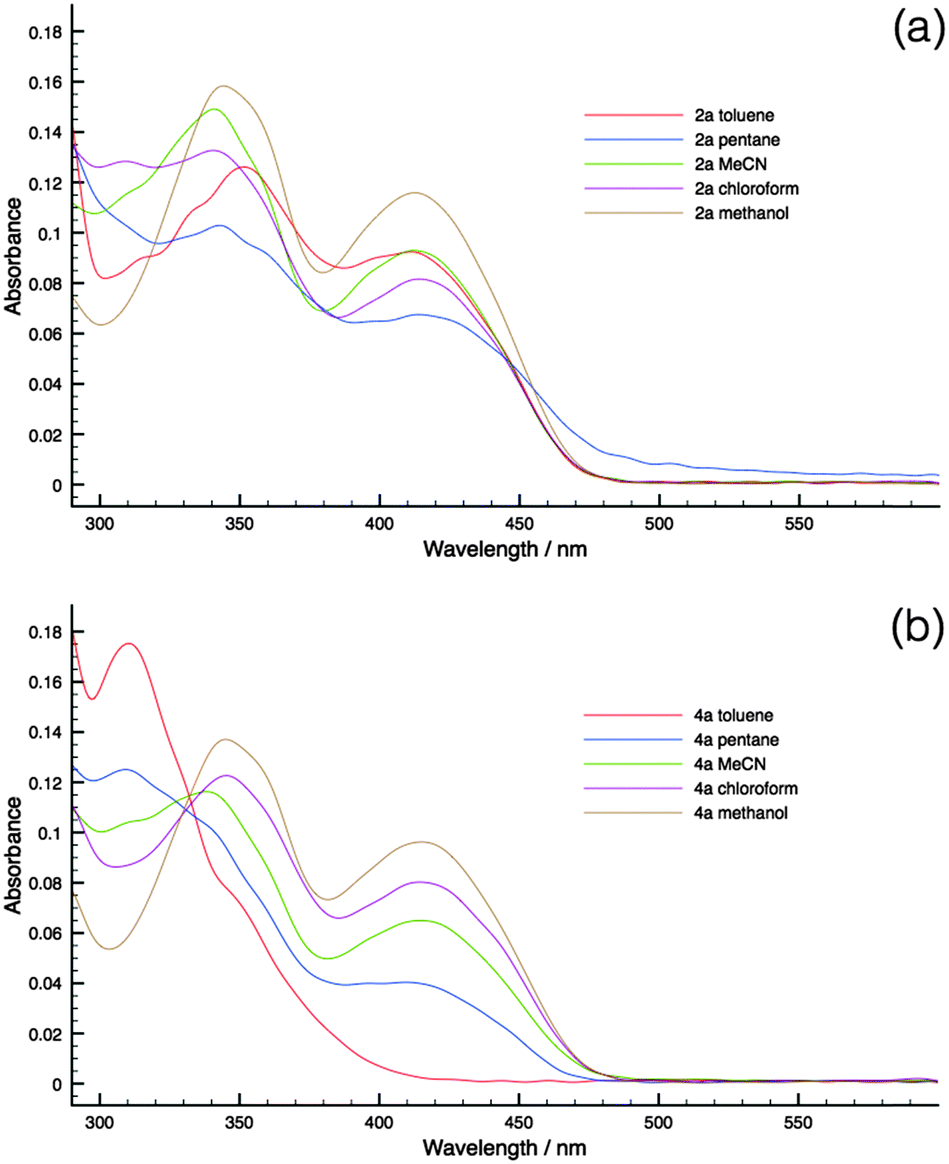 | ||
| Fig. 8 Solvatochromism of 2a (top) and 4a (bottom). All experiments were conducted at 10−5 M to mitigate effect of aggregation. (a) Absorption of 2a in various solvents; (b) absorption of 4a in various solvents, note the disappearance of the high-energy band at 300 nm and the appearance of the low energy bands at 350 nm and 420 nm, respectively. | ||
Self-organization and AIEE
As discussed above, the AIEE of phosphole-lipids relies heavily on the intramolecular rotation of the benzyl moiety. Therefore, self-organization of the molecules both in solution and in the solid state plays an important role in tuning the AIEE properties of the compounds. To better understand and further explore and leverage this important relationship, the self-organization of the phosphole-lipids was studied with an array of methods.As can be seen in Table 3, the 31P NMR signal shows distinct shifts as a function of both concentration and species investigated. As concentration differences lead to conformational changes at the benzylic methylene,4 the shift of the 31P NMR signals was correspondingly assigned to conformational variations.
| Compound | δ 31P (10−1 M)/ppm | δ 31P (10−3 M)/ppm | Δδ31P/ppb |
|---|---|---|---|
| 2a | 19.6769 | 19.9097 | 233 |
| 3a | 20.4754 | 20.8383 | 363 |
| 4a | 20.5358 | 20.6126 | 77 |
With a shift of only 77 ppb, 4a (3,5-C12 chains) experiences the least changes. This small response to a relatively large concentration range (10−3 – 10−1 M) indicates a more rigid conformational environment around the phosphorus center, where the rotation of the benzylic methylene group is restricted. This rigidity of 4a in solution was also confirmed by the fluorescence quantum yield of the compound in solution. While theory predicted 4a to have limited fluorescence intensity in solution, in actuality the compound exhibited significant fluorescence in solution (ϕPL = 13%). Compound 4b, the octyl analogue of 4a displays comparable fluorescence (ϕPL = 10%). The rigidity of the molecule along with the free rotation of the alkoxy substituent (vide supra) combined, give the 4-series of compounds increased solution fluorescence as compared to other phosphole-lipids.
By contrast, the 31P NMR signal of 3a (3,4-C12 chains) experiences a shift of 360 ppb, indicating a considerably more flexible conformational environment. In this case, this flexibility was mirrored by the non-emissive nature of the compound in solution; the octyl analogue 3b was also non-emissive in solution.
While 2a (4-C12 chain) was comparable to 3a in terms of 31P NMR shift (232 ppb), the solution fluorescence of the compound was quite intense (ϕPL = 26%) and that of 2b (4-C8 chain) was equally strong. According to the theoretical prediction, the PET process was suppressed in this compound. As such, the high fluorescent quantum yields of the 2-series confirm the result of the DFT-calculated results.
While 31P NMR spectroscopy revealed the intramolecular interactions of the compounds in solution, 1H NMR spectroscopy was able to provide key information on the intermolecular π–π interactions of the compounds at various concentrations. The benzo-proton Ha was selected for examination due to its potential for response to intermolecular π–π interactions (Fig. 9). At low concentrations, the molecules appear more monomeric with little intermolecular interactions. With increased concentration, increased intermolecular π-interactions between two phosphole head groups occur (Fig. 9b). Since in almost all phosphole systems π stacking almost always exists in a slip-stack fashion, proton Ha should experience the most change in its chemical environment with increased π–π interaction.10
 | ||
| Fig. 9 (a) Proton assignments on the benzo moiety; (b) the proposed dimerization process of phosphole-lipids. Note the position of proton Ha. | ||
The results of the concentration-dependent 1H NMR spectroscopy studies are summarized below. As can be seen in Table 4, 4a displayed the least amount of changes to concentration variation. This is in line with the 31P NMR spectroscopy studies and confirms the rigidity of the compounds in solution. Even at rather high concentration (10−1 M), 4a still exhibited significant monomeric characteristics. The concentration response of 3a clearly contrasts with 4a. This prominent response of 3a also confirms the 31P NMR results and indicates a highly flexible system, where the rotation of the benzyl group can easily accommodate the dimerization of the compounds. Compound 2a also appears to be more flexible than 4a, albeit to a lesser degree than 3a.
| Compound | δ 1H (10−1 M)/ppm | δ 1H (10−3 M)/ppm | Δδ1H/ppb |
|---|---|---|---|
| 2a | 8.1214 | 8.1411 | 20 |
| 3a | 8.1066 | 8.1819 | 75 |
| 4a | 8.2226 | 8.2080 | 15 |
The results of concentration-dependent 31P and 1H NMR studies demonstrate empirically that the attachment pattern of the alkyl chains has considerable impact on the solution self-assembly process. The 3,5-bisalkyl arrangement (4a) displays significant rigidity and resistance to intermolecular interactions in solution, while the 3,4-bisalkyl and 4-alkyl arrangement appear to be rather flexible in their scaffold. Despite the fact this difference in rigidity being reflected by the solution emission properties of these compounds, the newly synthesized phosphole-lipids were also studied in the solid state to better understand the mechanism for these self-assembly processes.
As can be seen in Table 5, the self-assembly features of the newly synthesized phosphole-lipids range from the highly crystalline 4-series of compounds to the highly isotropic 1b species. As a result, the fluorescence of the compounds in the solid state also varies significantly according to the differences in morphology. In general, crystalline compounds display higher fluorescence intensity due to the close packing in the crystalline phase. When crystallinity decreases, so does solid-state fluorescence.
Compound 1b, when newly prepared, appeared to be a highly viscous liquid at room temperature, however, which solidified after several months. While the eventual solid displayed certain degrees of long-range order, as evident by PXRD (see ESI†), it is its liquidity that dominates the self-assembly of the compound on a more accessible time scale. This highly disordered feature was also found in another short-chain-decorated phosphole species in an earlier study.11 With truncated chain length and enlarged conjugated head group, both the shape anisotropy of the molecule and charge density decrease. Since these forces are integral in the ordering of the compounds in aggregation, decreased shape anisotropy and charge density are likely the cause for the high liquidity of 1b. Like its previously studied cousin 1a, the species was essentially non-emissive in the aggregated state (ϕPL = 0.17%).
While 2a was amorphous in nature, 2b displayed some soft crystal/liquid crystal features as evident by it PXRD results. The low solid-state fluorescence of the compounds is, however, more likely due to ACQ rather than the AIEE effect. This conclusion is based on the particular FMOs arrangements where the PET effect was suppressed (Fig. 6, M2).
While also being amorphous solids, akin to 2a, films of the 3-series can be obtained by drop casting and were studied by scanning electron microscopy (SEM, Fig. 10); films formed by both 3a and 3b appear to be of similar thickness (∼20 μm, see ESI†). In both cases, amorphous defects were found evenly distributed over the entire surface. However, whereas the film of 3a appeared to be relatively homogeneous, rectangular crystallites can be found embedded through out the surface of the film of 3b. The increased crystallinity of 3b was reflected by increased solid-state fluorescence quantum yield of 3b (ϕPL = 9%) as compared to 3a (ϕPL = 0%). However, due to the diminutive size of the crystallites, PXRD of the film did not support any increased crystallinity for 3b.
 | ||
| Fig. 10 Scanning electron microscopy of 3-series of phosphole-lipids. (a) Phosphole-lipids 3a and 3b; (b) crystallites on the surface of 3b film; (c) amorphous surface of 3a film; (d) amorphous surface of 3b film with crystallites on the surface. | ||
Contrary to the amorphous nature of the 2- and 3-series, the crystallinity of the 4-series was confirmed by the relatively sharp melting and crystallization transitions on DSC, large crystalline domains on POM and the prominent sharp peaks in PXRD graphs (see ESI†). This significantly increased crystallinity of the 4-series is the likely reason for its considerable solid-state fluorescence (Table 5). In fact, 4a and 4b proved to be two of the most highly emissive phosphole-lipid species observed to date (4aϕPL = 33%, 4bϕPL = 19%). Furthermore, we were able to obtain single crystals of 4a by slow evaporation of solvent mixture (dichloromethane and hexane) at room temperature (Fig. 11).
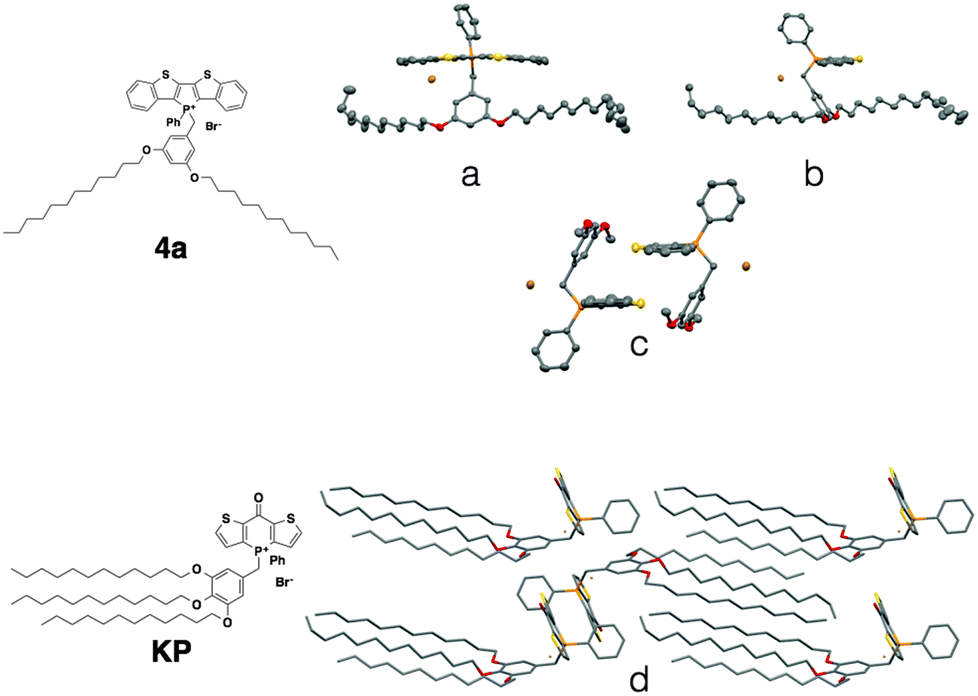 | ||
| Fig. 11 Single crystal XRD data of phosphole-lipid (4a) and phosphinine-lipid (KP). (a) and (b) solid-state structures of 4a as monomers; (c) the intermolecular packing of 4a (dodecyl chains are truncated for clarity); (d) solid-state packing of KP.9 | ||
Due to the thermal disorder generally imparted by the highly flexible dodecyl chains, reports of single crystal X-ray crystallographic data of compounds possessing multiple dodecyl substituents are few and far between. Therefore, the single crystal X-ray data of 4a afforded us a unique opportunity in understanding the self-assembly feature of the phosphole-lipid system.‡
One of the most significant features of 4a in the crystalline state is the anti-parallel arrangement of the two dodecyl chains. Contrary to our expectation that the chains would lie parallel to each other and form a linear molecule with polar and non-polar regions on each end, the two dodecyl chains in crystalline 4a adopt a ‘split’ stance with the chains pointing away from each other (Fig. 11a). In a recent study, we also reported on the single crystal X-ray data of a ‘phosphinine lipid’ KP (Fig. 11, KP).12 By comparing 4a and KP with two different chain attachment patterns (3,5- and 3,4,5-), we can better understand the peculiar geometry of the 3,5-chain arrangement in 4a. While the dodecyl chains in KP were aligned parallel to each other in the solid state (Fig. 11d), the dodecyl chains in 4a are arranged antiparallel to each other (Fig. 11a). This difference in chain arrangement can be attributed to the lack of van-der-Waals interchain interaction in 4a. The presence of the 4-dodecyl substituents in KP very likely acts as a ‘glue’ to pull the other two chains together resulting in a parallel arrangement. This particular arrangement of the dodecyl chain in crystalline 4a can also shed light on the self-assembly processes of the compound in solution (Fig. 11b, Tables 2 and 3). Without an alkyl substituent in the 4-position, the 3- and 5-dodecyl chains are more or less free floating in solution. From the crystallographic data, it can be deduced that the dodecyl chains would be found primarily on the periphery of the molecule, creating a barrier preventing the approach of another molecule (Fig. 12). It is reasonable to believe this is the basis for the apparent reluctance of 4a to aggregate in solution.
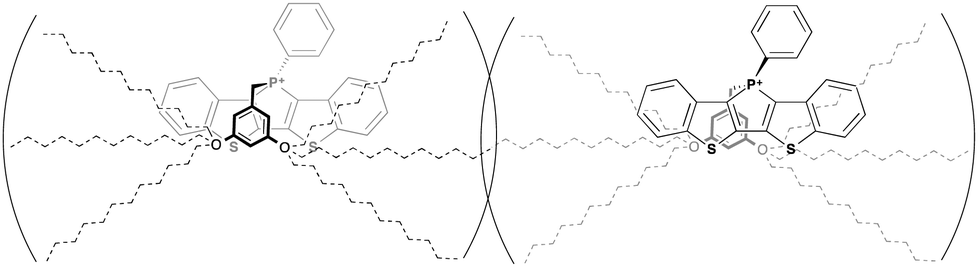 | ||
| Fig. 12 Proposed mechanism for intermolecular repulsion of 4a in solution. | ||
Another important feature of 4a is the absence of π–π interactions in 4a (Fig. 11c), which is commonly found in related phosphole compounds. In most of the other non-lipid phospholes,6,10 including closely related ionic model compound M1,4c π–π interactions in the solid state are predominantly of the intermolecular variety where the phosphole π-scaffolds arrange in a face-to-face fashion. In 4a however, intermolecular π-stacking interactions between the phosphole scaffolds are almost completely suppressed (Fig. 11c). Instead of the π–π dimers commonly observed in other dithienophosphole systems, 4a appears to remain monomeric, even in the crystalline state. This remarkable ‘rigidity’ of the molecule was also mirrored in the concentration-dependent 31P NMR studies discussed previously. Although the corresponding 4b also appears crystalline, crystals of sufficient quality for single-crystal X-ray crystallography could not be obtained.
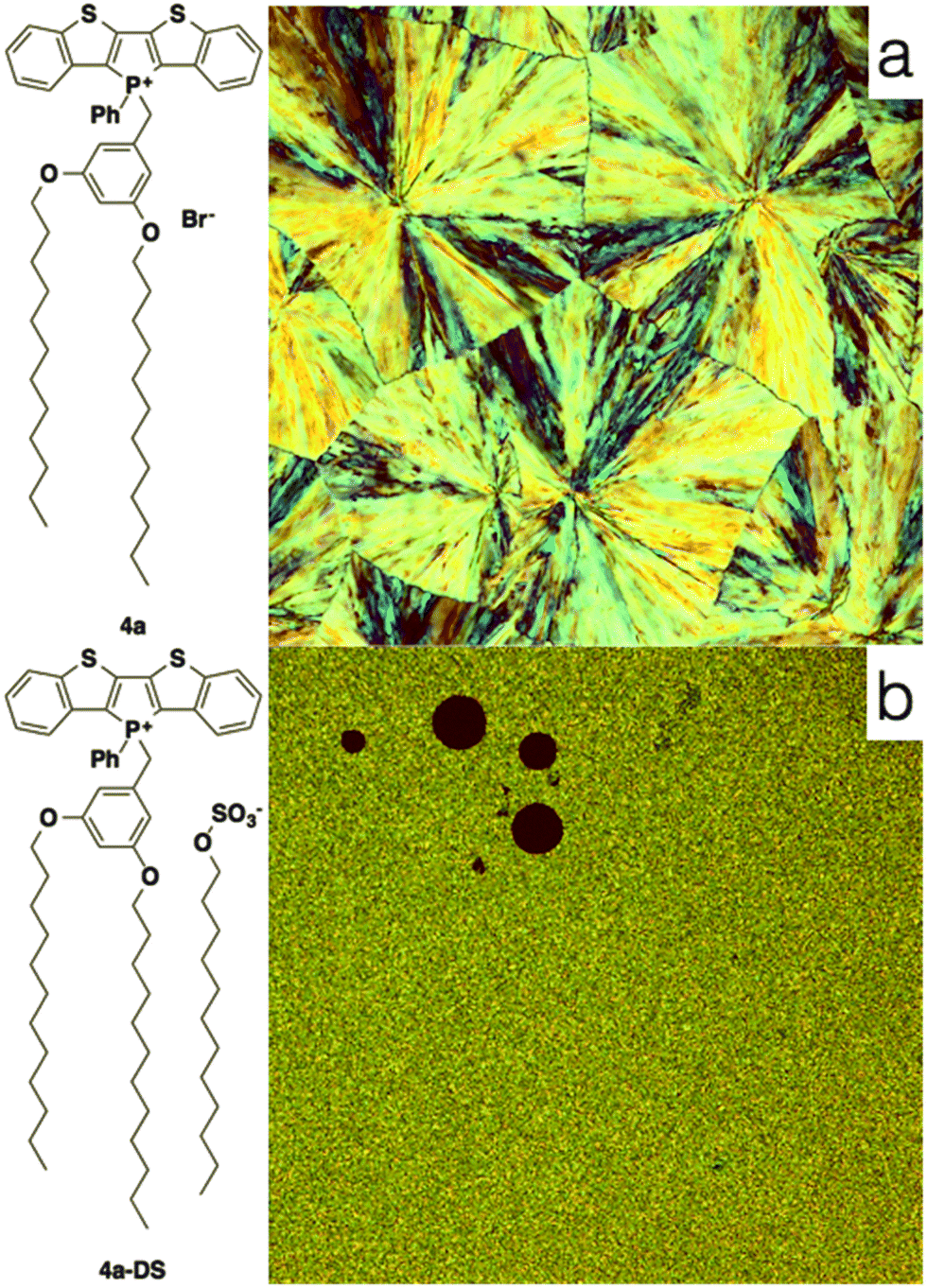 | ||
| Fig. 13 Polarized optical microscopy of 4a (a) and 4a-DS (b). Note the differences in domain size between the two micrograph. | ||
Conclusions
In this study, we successfully demonstrated the tuning of AIEE features of di(benzothieno)phosphole-lipids by self-assembly. Theoretical calculations proved to be an important and reliable tool for predicting and rationalization the opto-electronic properties of the phosphole-lipids. By varying attachment patterns of the alkyl chains on the benzyl moiety, we were able to achieve both crystalline and amorphous self-assembly of the molecules in the solid state. When regarding the theoretical, photophysical and self-assembly results of all the newly synthesized phosphole-lipids, it can be seen that the self-assembly and related AIEE of phosphole-lipids are based on the delicate balance of all the forces involved in the process, such as ionic interaction, van der Waals interactions, π stacking, and sterics. Within the phosphole-lipid system, the benzyl moiety plays an important role in both the self-assembly and the photophysics. In terms of photophysics, by varying the number and electronic nature of the substituent, the orbital energies of the benzyl moiety can be tuned relative independently of the phosphole head groups. By adjusting the donor strength of the benzyl moiety, we can turn on/off the AIEE of the compounds. In terms of the self-assembly, the chain length and attachment patterns determine the self-assembly feature of the compounds. The 4-position on the benzyl moiety, in particular, can be used as a switch for crystalline(4a)/amorphous(3a) features and, moreover, to restrict the potential for free rotation of the neighboring alkoxy groups, which has a considerable effect on the AIEE of the system. While the effect of the chain length is more complicated, it can be said that by varying the chain length we can introduce crystallinity (3avs.3b) or liquidity (1avs.1b) and the related photophysical properties. Finally, the dodecylsulfate anion provided us a convenient avenue for tuning the self-assembly feature of our system without drastically altering the intrinsic energy of the chromophores involved. Through this study, we can now begin to assemble the toolbox for rationally designing and building self-assembled phosphole-lipid chromophores with targeted properties.Acknowledgements
Financial support by NSERC of Canada and the Canada Foundation for Innovation (CFI) is gratefully acknowledged. We also thank Alberta Innovates – Technology Futures for a graduate scholarship (Z. W.). Thanks to Profs. T. Sutherland and C. C. Ling for helpful discussion and the use of the polarized optical microscope.Notes and references
- (a) T. Baumgartner and R. Reáu, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (b) T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed; (c) C. Romero-Nieto and T. Baumgartner, Synlett, 2013, 920–937 CrossRef CAS; (d) M. Stolar and T. Baumgartner, Chem. – Asian J., 2014, 9, 1212–1225 CrossRef CAS PubMed; (e) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (f) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef; (g) S. Yamaguchi and K. Tamao, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and T. Appeloig, John Wiley & Sons, Chichester, 2001, vol. 3, pp. 641–694 Search PubMed.
- (a) T. Baumgartner, T. Neumann and B. Wirges, Angew. Chem., Int. Ed., 2004, 43, 6197–6201 CrossRef CAS PubMed; (b) Y. Dienes, M. Eggenstein, T. Neumann, U. Englert and T. Baumgartner, Dalton Trans., 2006, 1424–1433 RSC; (c) S. Durben, Y. Dienes and T. Baumgartner, Org. Lett., 2006, 8, 5893–5896 CrossRef CAS PubMed; (d) Y. Ren and T. Baumgartner, J. Am. Chem. Soc., 2011, 133, 1328–1340 CrossRef CAS PubMed.
- (a) Y. Ren and T. Baumgartner, Dalton Trans., 2012, 41, 7792–7800 RSC; (b) Z. Wang and T. Baumgartner, Chem. Rec., 2015, 15, 199–217 CrossRef CAS PubMed.
- (a) Y. Ren, W. H. Kan, M. A. Henderson, P. G. Bomben, C. P. Berlinguette, V. Thangadurai and T. Baumgartner, J. Am. Chem. Soc., 2011, 133, 17014–17026 CrossRef CAS PubMed; (b) Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner, Angew. Chem., Int. Ed., 2012, 51, 3964–3968 CrossRef CAS PubMed; (c) Y. Ren and T. Baumgartner, Inorg. Chem., 2012, 51, 2669–2678 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (c) Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- Y. Dienes, M. Eggenstein, T. Kárpáti, T. C. Sutherland, L. Nyulászi and T. Baumgartner, Chem. – Eur. J., 2008, 14, 9878–9889 CrossRef CAS PubMed.
- L.-C. Lee and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 5579–5582 CrossRef CAS PubMed.
- (a) A. C. Nieuwkerk, A. T. M. Marcelis, A. Koudijs and E. J. R. Sudhölter, Liebigs Ann., 1997, 8, 1719–1724 CrossRef; (b) M. R. Bozorgmehr, M. Saberi and H. Chegini, J. Mol. Liq., 2014, 199, 184–189 CrossRef CAS.
- M. J. Frisch, et al., Gaussian09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed, (see ESI† for full reference).
- (a) T. Baumgartner, W. Bergmans, T. Kárpáti, T. Neumann, M. Nieger and L. Nyulászi, Chem. – Eur. J., 2005, 11, 4687–4699 CrossRef CAS PubMed; (b) Y. Ren, Y. Dienes, S. Hettel, M. Parvez, B. Hoge and T. Baumgartner, Organometallics, 2009, 28, 734–740 CrossRef CAS; (c) S. Durben and T. Baumgartner, Angew. Chem., Int. Ed., 2011, 50, 7948–7952 CrossRef CAS PubMed; (d) X. He, J.-B. Lin, W. H. Kan, P. Dong, S. Trudel and T. Baumgartner, Adv. Funct. Mater., 2014, 24, 897–906 CrossRef CAS.
- Y. Ren, F. Biegger and T. Baumgartner, J. Phys. Chem. C, 2013, 117, 4748–4758 CAS.
- X. He, J.-B. Lin, W. H. Kan and T. Baumgartner, Angew. Chem., Int. Ed., 2013, 52, 8990–8994 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1431111. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5tc03311j |
‡ Crystal data and structure refinement for 4a (C53H70BrO2PS2): Mr = 914.09, temperature = 173(2) K, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.5341(3), b = 14.4282(4), c = 18.4781(4) Å, α = 85.0625(17), β = 77.5714(16), γ = 80.3446(13)°, V = 2443.82(12) Å3, Z = 2, ρcalc = 1.242 g cm−3, μ = 0.998 mm−1, F(000) = 972.0, crystal size = 0.070 × 0.050 × 0.020 mm3, λ = 0.71073 Å, 2Θ range for data collection = 4.892° to 55.124°, index ranges = −12 ≤ h ≤ 12, −18 ≤ k ≤ 18, −23 ≤ l ≤ 24, 20775 measured reflections, 11196 [Rint = 0.0484] independent reflections, GoF on F2 = 1.117, R1 = 0.0682, wR2 = 0.1164 [I ≥ 2σ(I)], R1 = 0.1021, wR2 = 0.1309 [for all data], largest difference peak and hole 0.79 and −0.40 e Å−3. , a = 9.5341(3), b = 14.4282(4), c = 18.4781(4) Å, α = 85.0625(17), β = 77.5714(16), γ = 80.3446(13)°, V = 2443.82(12) Å3, Z = 2, ρcalc = 1.242 g cm−3, μ = 0.998 mm−1, F(000) = 972.0, crystal size = 0.070 × 0.050 × 0.020 mm3, λ = 0.71073 Å, 2Θ range for data collection = 4.892° to 55.124°, index ranges = −12 ≤ h ≤ 12, −18 ≤ k ≤ 18, −23 ≤ l ≤ 24, 20775 measured reflections, 11196 [Rint = 0.0484] independent reflections, GoF on F2 = 1.117, R1 = 0.0682, wR2 = 0.1164 [I ≥ 2σ(I)], R1 = 0.1021, wR2 = 0.1309 [for all data], largest difference peak and hole 0.79 and −0.40 e Å−3. |
| This journal is © The Royal Society of Chemistry 2016 |