
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel hybrid nanofibrous strategy to target progenitor cells for cost-effective in situ angiogenesis†
N.
Sachot
ab,
O.
Castaño
 *abc,
H.
Oliveira
d,
J.
Martí-Muñoz
ab,
A.
Roguska
e,
J.
Amedee
d,
M.
Lewandowska
f,
J. A.
Planell
ab and
E.
Engel
abg
*abc,
H.
Oliveira
d,
J.
Martí-Muñoz
ab,
A.
Roguska
e,
J.
Amedee
d,
M.
Lewandowska
f,
J. A.
Planell
ab and
E.
Engel
abg
aBiomaterials for Regenerative Therapies, Institute for Bioengineering of Catalonia (IBEC), 08028 Barcelona, Spain. E-mail: ocastano@ibecbarcelona.eu
bCIBER en Bioingeniería, Biomateriales y Nanomedicina, CIBER-BBN, 50018 Zaragoza, Spain
cMaterials Science and Physical Chemistry, Universitat de Barcelona (UB), 08028 Barcelona, Spain
dInserm U1026, Tissue Bioengineering, University of Bordeaux, 33076 Bordeaux, France
eInstitute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland
fFaculty of Materials Science and Engineering, Warsaw University of Technology, 02-507 Warsaw, Poland
gMaterials Science and Metallurgy, Technical University of Catalonia (UPC), 08028 Barcelona, Spain
First published on 22nd September 2016
Abstract
Although the impact of composites based on Ti-doped calcium phosphate glasses is low compared with that of bioglass, they have been already shown to possess great potential for bone tissue engineering. Composites made of polylactic acid (PLA) and a microparticle glass of 5TiO2–44.5CaO–44.5P2O5–6Na2O (G5) molar ratio have already demonstrated in situ osteo- and angiogenesis-triggering abilities. As many of the hybrid materials currently developed usually promote osteogenesis but still lack the ability to induce vascularization, a G5/PLA combination is a cost-effective option for obtaining new instructive scaffolds. In this study, nanostructured PLA-ORMOGLASS (organically modified glass) fibers were produced by electrospinning, in order to fabricate extra-cellular matrix (ECM)-like substrates that simultaneously promote bone formation and vascularization. Physical–chemical and surface characterization and tensile tests demonstrated that the obtained scaffolds exhibited homogeneous morphology, higher hydrophilicity and enhanced mechanical properties than pure PLA. In vitro assays with rat mesenchymal stem cells (rMSCs) and rat endothelial progenitor cells (rEPCs) also showed that rMSCs attached and proliferated on the materials influenced by the calcium content in the environment. In vivo assays showed that hybrid composite PLA-ORMOGLASS fibers were able to promote the formation of blood vessels. Thus, these novel fibers are a valid option for the design of functional materials for tissue engineering applications.
Introduction
The development of materials for tissue engineering has long been focused on implants aimed at simply replacing non-functional or damaged organs. But scaffold design has evolved considerably in recent years, and third generation biomaterials have been developed.1,2 These biomaterials are able to stimulate specific cellular responses, being both bioactive and bioabsorbable.3–5 In bone regeneration, many material fabrication approaches have been implemented for the production of functional substrates able to interact and properly integrate with the host tissue and/or trigger cell differentiation towards osteoblastic lineage. But the obtained scaffolds generally do not promote angiogenesis, a vascularization process essential for the formation of fully functional new living tissues.6 Several studies performed by our group have demonstrated that composites made of polylactic acid (PLA) and TiO2–CaO–P2O5–Na2O bioactive inorganic glass have the potential to efficiently promote bone formation and integrate with bone tissues.7–9 More recently, another study pointed to the formation of tube-like structures when endothelial progenitor cells (responsible for the formation of vessels) were cultured on the material.10 The suitable biological performance of these scaffolds was mainly attributed to their stiffness and the ions released from the glass, in particular calcium ions, as well as the intrinsic surface properties of the materials.8,10 The combination of PLA with this bioactive glass seemed, therefore, to be a promising and low-cost combination. Up to now, this material has only been shaped by using the salt leaching-solvent casting method and the rapid prototyping technique.11,12 As not only the chemistry but also the structure of a scaffold influence cellular response, the choice of appropriate scaffold architecture is critical; for this reason, biomimetic scaffolds are being investigated extensively. In particular, numerous studies reported in a review by Jang et al.13 have shown the potential of electrospun mats to support cell adhesion, proliferation and differentiation in bone tissue engineering. Other studies also revealed the importance of developing materials with specific features (composition, for example) and validated the use of Si–Ca–P2 ORMOGLASSES (organically modified glasses) to prepare hybrid materials.14–17 However, the fabrication of hybrids involving other systems (i.e. other constituents) is required in order to demonstrate the usefulness of ORMOGLASSES for tissue engineering, particularly for bone regeneration.The aim of this study was to prepare hybrid composite fibers, by means of electrospinning, containing PLA and a titanium-based ORMOGLASS (previously reported by Sanzana et al.18 as the G5 composition) in order to produce scaffolds with the capacity to sustain both osteo and angiogenesis. Hence, we evaluated the applicability of a sol–gel methodology to the previously developed G5 glass composition and established a new organometallic glass preparation protocol. Then fiber fabrication followed a dispersed blend approach, allowing for the participation of the titanium-based ORMOGLASSES to the final mechanical support of the matrices. The obtained composite fiber mats were characterized and their biological responses were investigated. We hypothesized that 3rd generation PLA–titanium based ORMOGLASS composite scaffolds, fabricated as fibrous mats, would constitute a favourable environmental interface for cellular activity (bioinspired structure)19,20 and that the interactions between the phases could be improved in comparison to the other materials already prepared using these two types of compounds.
Results and discussion
Material morphology
Titanium-based ORMOGLASS particles were prepared by a hydrolysis-tuned sol–gel method and associated with PLA, as the organic matrix, enabling the formation of fibers by electrospinning without polymer degradation. As observed in Fig. 1, titanium-based ORMOGLASS/PLA blend electrospun fibers were efficiently prepared, using two different G5 compositions (OG5, i.e. 10 and 20 (v/v) %), either randomly oriented or aligned, showing a full width at half maximum (FWHM) of 62.8 and 51.5° of misorientation-angle (Fig. S1, ESI†). EDS analysis showed a good concordance with the theoretical mineral composition of the mineral phase in the two blends produced (Table S1, ESI†). Note the versatility of the method, for example, we could increase or decrease the Ti content in order to modify the behaviour of the ormoglasses when immersed in aqueous solutions.21 | ||
| Fig. 1 Morphology of the hybrid fibers (FESEM zoomed pictures). | ||
Finding a stable combination of alkoxides and PLA is a laborious work. Numerous processing conditions influence the fiber deposition and the final fiber aspect (the voltage applied or the tip-collector distance, for example22). The viscosity of the solution is another crucial factor. Without a suitable viscosity (too low or too high), the fibers do not form properly.23 It was thus essential to prepare a blend with a suitable viscosity to ensure fiber deposition. Note that PLA can suffer alcoholysis and chain scission when the ORMOGLASS alkoxides mix is added (data not shown). Once experimental parameters were optimized, even when the addition of the ORMOGLASS gel into the polymeric solution induced a decrease in the viscosity, the viscosity of the solution was in an appropriate range and remained stable over time (Table S2, ESI†). This was the first time that a glass with the G5 composition was processed using the sol–gel method and that PLA-OG5 ORMOGLASS blends were used to obtain extra cellular matrix (ECM)-like fibers. Previous reports only considered the use of G5 glasses in non-fibrous and non-hybrid composite applications.8,12
Water contact angle assessment
ORMOGLASSES enhance the hydrophilicity in hybrid architectures as inorganic bioglasses do. The wettability of all hybrid fibers produced was improved in comparison to pure PLA fibers (Fig. S2, ESI†) in accordance with other hydrophilic composites.7,12 In our case, it can also be noted that aligned fibers had a lower contact angle than the random ones. The positioning of aligned fibrous samples (parallel or perpendicular) influences the evaluation of the water contact angle due to the anisotropic topography of the materials (see Fig. S3, ESI†).Thermal characterization
Polymer chain motion was not restricted by the ORMOGLASS and two different phases were present in contrast to what was previously observed in ormoglass coated PLA.17 Differential scanning calorimetry (DSC) results showed that the thermal properties of the polymer were globally not affected by the incorporation of the ORMOGLASS. In fact, the thermograms of the hybrid fibers associated with the first heating ramp were similar to those of PLA fibers. Moreover, based on the second heating ramps, no significant differences in Tg were observed for the PLA and the hybrid fibers, as expected (Fig. S4, ESI†). Interactions at the nanometric level can be created thanks to the intrinsic advantages of the fabrication method,24 but no strong chemical interactions, able to induce significant modifications in Tg, were formed. Note that Tg in this amorphous PLDLLA (70% LL/30% LD PLA) ranges between 55 and 60 °C and basically depends on the molecular weight.25The higher the ORMOGLASS content, the earlier the degradation of PLA occurred. Thermograms obtained from thermogravimetric analysis revealed that when subjected to high temperatures, the physical–chemical properties of the materials differed in terms of degradation. Degradation of PLA occurred at a lower temperature in the hybrid fibers than that of the pure PLA ones (Fig. 2 and Table S5, ESI†). The ORMOGLASS promoted an earlier PLA degradation, which was even more anticipated when the ORMOGLASS content was increased. Under thermal-oxidative conditions (air), the thermal stability of PLA was thus influenced by the content of the ORMOGLASS. This is in accordance with a study performed by Wen et al., which showed that an increase in titanium content in polymeric networks decreased the degradation temperature of the polymer.26 Other researchers also proved that addition of hydrophilic or organic species (organosilicon compounds) in PLA resulted in a decrease in the decomposition temperature of the polymer.27,28 Moreover, as observed on the derivative graphic and in comparison to the pure PLA fibers, two novel degradation peaks at lower temperatures appeared for the hybrid fibers. This indicated that compounds related to glass additionally degraded before PLA. According to the FTIR vapour phase analysis (performed under N2 flow for this kind of set-up in Fig. S5, ESI†), the infrared spectra associated with these two peaks were assigned to alkylphosphates. The fact that such compounds were identified for the hybrid fibers suggested that not all the phosphate contained in the glass gel had reacted to form the glass network. Carta et al. also attributed these two peaks to unreacted alkylphosphate molecules.29 Knowing that the phosphorous precursor is composed of mono and di-ethyl phosphate species, the non-reaction of these two species would explain that two peaks (i.e. two different compounds) are observed. Liang et al. suggested that such free molecules degraded because they are small and thus able to volatilize easily.30 To enhance the incorporation of the phosphate into the ORMOGLASS, modification of the hydrolysis parameters of the glass (i.e. catalyst used, hydrolysis rate, etc.) will be needed to control this aspect. Finally, it may also be possible that part of the alkylphosphate molecules detected were not all resulting from the signal of unreacted molecules. Perhaps some of them resulted from the chain scission of the phosphate groups located at the end of the glass chain network (backbone).31 These end groups need, in fact, less energy of activation to volatilize in comparison to phosphate groups well incorporated into the inside of the ORMOGLASS network.
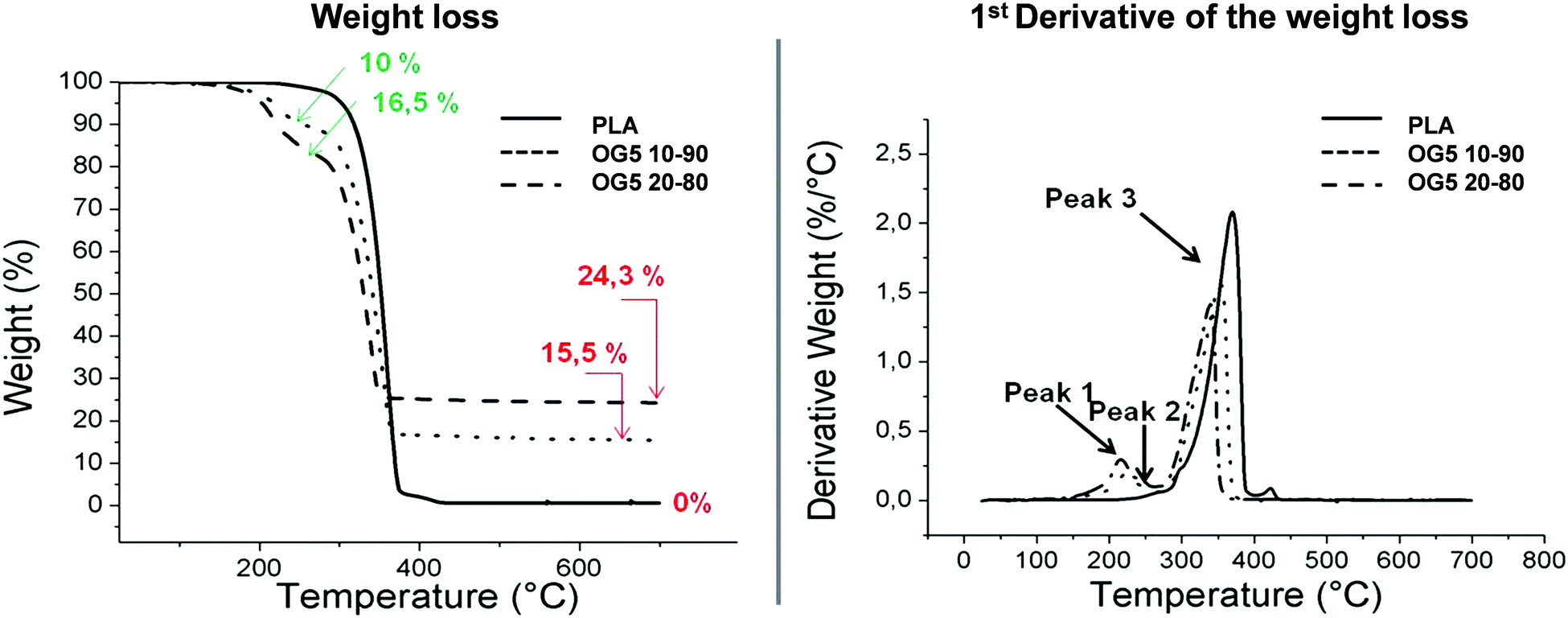 | ||
| Fig. 2 TGA curves of the hybrid fibers and their associated derivative curves (DTA). The percentages in red represent the amount of inorganic ORMOGLASS remaining at the end of the assay. The percentages in green correspond to the weight loss associated with the degradation of alkylphosphate molecules (peaks 1 and 2) in the hybrid fibers. | ||
The weight percent of the OG5 20–80 fibers was not exactly double that of the OG5 10–90 ones as would have been expected according to the v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v ratio defined. Observing the TGA thermograms, i.e. the weight and derivative weight (DTA) graphics, associated with the degradation of the fibers in Fig. 2, we can infer by evaluating the sample weight changes that the final inorganic compound remaining after thermal treatment constituted 15.5% of the sample (in mass) for the OG5 10–90 fibers and 24.3% for the OG5 20–80 ones. However, these values did not represent the exact ORMOGLASS weight of the fibers as two other weight losses attributed to the presence of the ORMOGLASS during the degradation were identified. The percentage of the ORMOGLASS weight contained in the OG5 10–90 fibers was increased to 25.5% and that in the OG5 20–80 ones to 40.8%. These two weight losses that occurred between 150 and 280 °C were related to alkylphosphates, as revealed by the analysis of the vaporized gaseous products and based on studies reported in the literature.29,30 Differences in the degradation temperatures of PLA were also observed for the hybrid fibers (Table S3, ESI†). While PLA fibers had a degradation temperature (max peak) of 360 °C, hybrid fibers exhibited lower values: 355 °C for the OG5 10–90 fibers and 341 °C for the OG5 20–80 fibers.
:v ratio defined. Observing the TGA thermograms, i.e. the weight and derivative weight (DTA) graphics, associated with the degradation of the fibers in Fig. 2, we can infer by evaluating the sample weight changes that the final inorganic compound remaining after thermal treatment constituted 15.5% of the sample (in mass) for the OG5 10–90 fibers and 24.3% for the OG5 20–80 ones. However, these values did not represent the exact ORMOGLASS weight of the fibers as two other weight losses attributed to the presence of the ORMOGLASS during the degradation were identified. The percentage of the ORMOGLASS weight contained in the OG5 10–90 fibers was increased to 25.5% and that in the OG5 20–80 ones to 40.8%. These two weight losses that occurred between 150 and 280 °C were related to alkylphosphates, as revealed by the analysis of the vaporized gaseous products and based on studies reported in the literature.29,30 Differences in the degradation temperatures of PLA were also observed for the hybrid fibers (Table S3, ESI†). While PLA fibers had a degradation temperature (max peak) of 360 °C, hybrid fibers exhibited lower values: 355 °C for the OG5 10–90 fibers and 341 °C for the OG5 20–80 fibers.
Mechanical behaviour
As in inorganic–organic conventional hybrids, alkoxide based ormoglass also increases the mechanical resistance. Tensile tests demonstrated that the approached Young's modulus and the yield strength of the hybrid fibers exhibited higher values than the PLA ones (Fig. S6 and S7, ESI†). This attested that tougher materials were produced. The mechanical behaviours of the hybrids closely depend on the nature of the inorganic phase introduced into the polymeric matrix. As reported by Wen et al., for example,26 hybrids produced by the incorporation of inorganic phases containing metal alkoxides greatly improved the mechanical features of the pure polymeric material. They also showed that these mechanical properties can be tailored by modifying the content of the inorganic glass. Here, similar results were observed. Based on these statements and the fact that no increase in crystallinity was detected by DSC, it is assumed that the use of alkoxide precursors and the sol–gel method is the main reason for the increase of the values in the mechanical properties measured for the hybrids. As a more general observation, it can be seen that aligned fibers exhibited higher mechanical properties. This behaviour is commonly observed when tensile tests are performed parallel to the principal axis of the fiber alignment.32,33BSA protein adsorption
Bovine serum albumin (BSA) adsorbs better in the hybrid formulations due to their higher hydrophilicities, higher calcium release profile and lower electronegative surface potentials. Protein adsorption on the surface of biomaterials is crucial, as this phenomenon constitutes the first real interaction that the material establishes with the biological fluid and contributes to efficient cell adhesion.34,35 Bovine serum albumin shows a higher adsorption on the surface of the hybrid fibers, in comparison with PLA alone (Fig. S8, ESI†). Several explanations can be provided for this observation: the first one considers that the composition of the ORMOGLASS contained a high amount of calcium, and albumin is known as a major calcium-binding protein present in tissues and fluids.36 Thus, hybrid fibers may include a significant amount of binding sites available for the protein. In fact, for pure PLA substrates, previous surface activation or functionalization is usually required to promote albumin adsorption.35 Another reason may be the charge of the hybrid fiber surface. According to Klinger et al., the electrostatic interactions between albumin and titanium surfaces are the main mechanisms involved in protein adsorption. BSA is a negatively charged protein in the pH range in which the assay is conducted. As PLA fibers generally showed a more electronegative potential than the hybrid fibers, BSA had less affinity with the PLA fibers than with the hybrids (Fig. S8 and Table S4, ESI†). This may also be emphasized by the pH of the solution in which the assay is performed. As revealed by the pH measurements, when hybrid fibers are immersed in diverse solutions (medium, simulated body fluid (SBF), water), the pH of the solutions slightly decreased (Fig. 3 and Fig. S9, ESI†). The complementary pH measurements reported in Fig. S10 (ESI†) confirmed that the pH of the solution in which the albumin adsorption assay is performed was lower for the hybrids than for PLA. This means that the electrostatic potential of the hybrid fibers was even more electropositive than that of the PLA fibers under the conditions of the test. Consequently, zeta potential (ZP) and pH together drastically affected the adsorption efficiency (Fig. S11, ESI†). Zhu et al. and Krajewski et al. already described this ZP–pH dependence in the literature.37,38 Finally, the last two factors that may have favoured the adhesion of BSA are wettability and the reactive groups present at the fiber surface.35,39 The low wettability of the PLA fibers compared to the hybrid ones (Fig. S2, ESI†) may have prevented BSA from binding to the surface as a smaller amount can reach the whole surface. It is also hypothesized that suitable reactive groups such as hydroxyl might be present at the fiber surface, providing additional binding sites for the protein.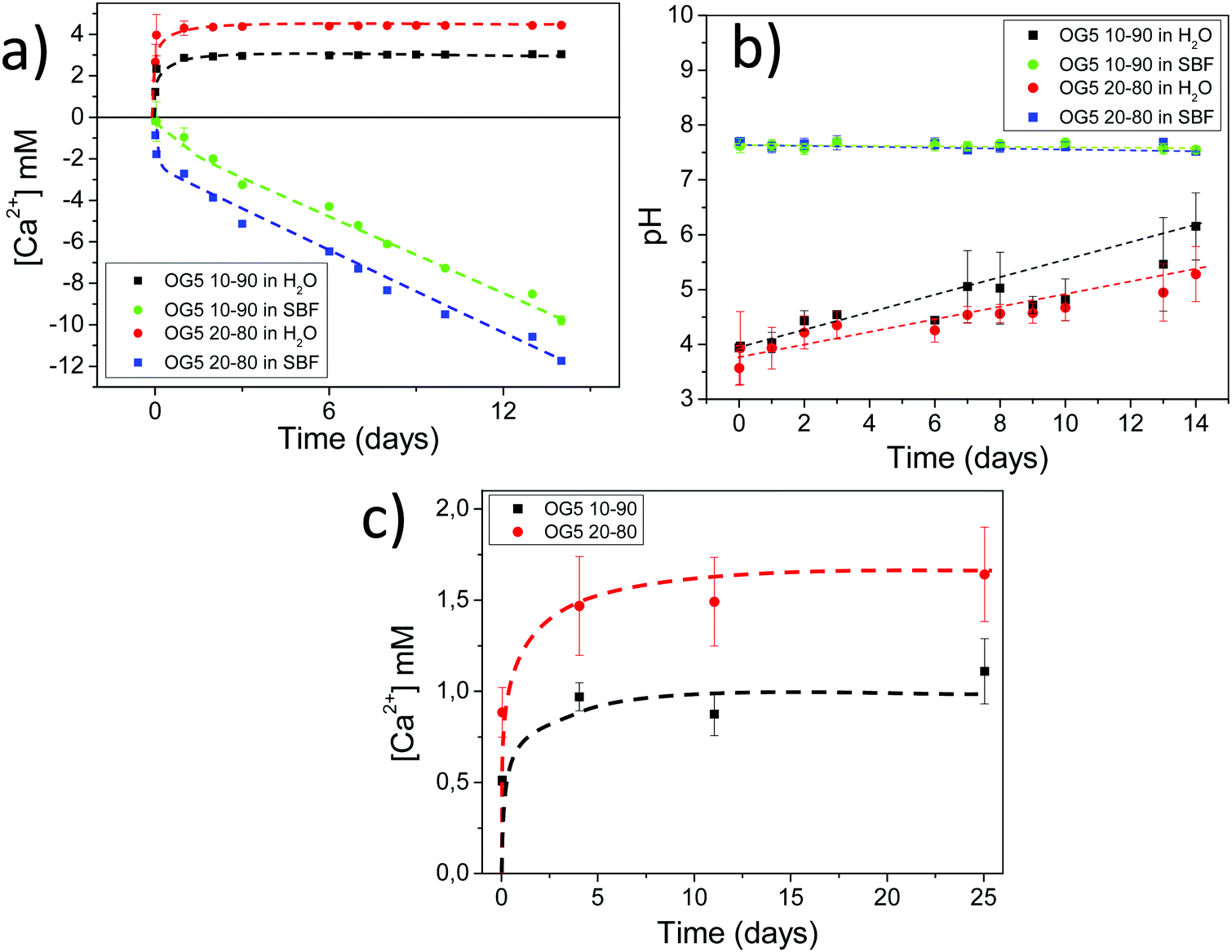 | ||
| Fig. 3 (a) Calcium release and (b) environmental pH of blended fiber sheets both in pure water and SBFF solution. (c) Calcium release of a monolayer of blended fibers deposited on cover glasses in HEPES buffered solution. | ||
Proliferation of rMSCs and EPCs
Proliferation of rMSCs is promoted by the hybrid fibrous scaffolds in contrast to that of rEPCs. Biological response is of paramount importance to the success of scaffold implantation. Scaffolds are designed to act as templates to support adequate cellular function. They should thus provide a suitable environment to direct the activity of cells through their material features (e.g. architecture, chemical composition, wettability).40 When cultured on the hybrid fibers for 1 day, rMSCs showed to attach and spread on the substrate for both fiber compositions (Fig. 4a, b, e and f). At the same time point, rEPCs also adhered on the fiber mats, but showed to spread to a less extent (Fig. 4c, d, g and h). After 10 days, rMSCs proliferated significantly on all fiber types, forming a homogeneous cellular layer (Fig. 5a–h). On the other hand, rEPCs showed a more heterogeneous distribution on the fiber mats, especially on the OG5 10–90 fibers (Fig. 5i–p). Quantification of the cell proliferation confirmed this trend. Based on these results, all hybrid fibers seemed to be suitable substrates to proliferate rMSCs, aligned OG 10–90 mats being the most relevant composition. For rEPCs, however, the cellular response was more limited (Fig. 6). As a general observation, the initial adhesion and proliferation of lower rEPCs could be observed both for the hybrid and pure polymeric mats. Relevant lower adhesion of rEPCs to the fibers (around 50%) and a trend for a higher cell count for the random fibers seem to exist, although not statistically significant. The signalling provided by the tested scaffolds may not be the most suitable to sustain their proliferation.41,42 In previous reports it has been shown that, while calcium enhances rMSC proliferation,43 no significant effect was observed for rEPCs.44 | ||
| Fig. 4 FESEM pictures at low and high magnification of rMSCs and rEPCs at 1 day of culture. Cells are artificially coloured in blue to better visualize the cells on the material and red stars point the round cells (a–d: random; e–h: aligned). | ||
 | ||
| Fig. 5 Pictures obtained thanks to fluorescent staining (microscope images) and cell fixation (FESEM images) of rMSCs (a–h) and rEPC (i–p) after 1 d and 10 d of culture on hybrid fibers. | ||
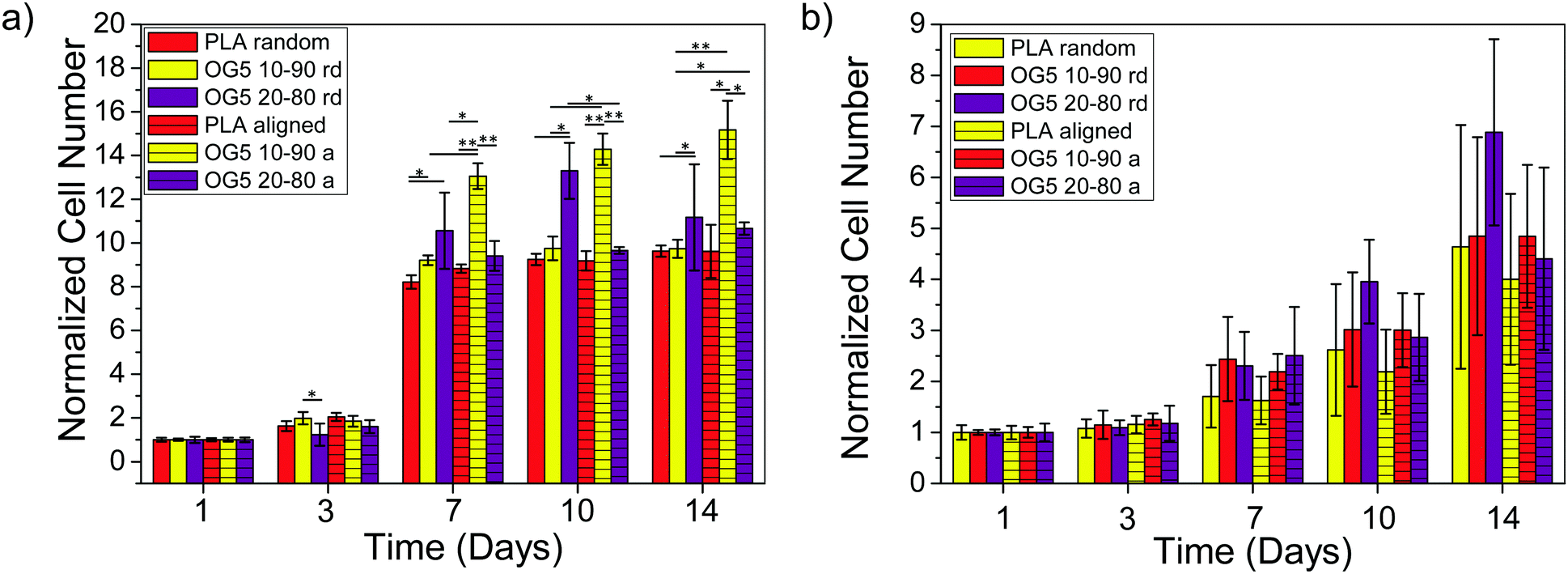 | ||
| Fig. 6 Proliferation quantification of (a) rMSCs and (b) rEPCs cultured on PLA and hybrid fibers – random and aligned (*p < 0.05, **p < 0.001). | ||
It can be additionally noted that cellular adhesion and spreading followed the orientation of the fibers. On random fibers, cells spread randomly, whereas on aligned fibers they aligned along the orientation of the fibers (Fig. 4 and 5). This is a common behaviour, already reported in the literature, which shows how cells modulate their cytoskeleton shape depending on the substrate structure, and how cell–material interactions are influenced by the design of the material.45 On the other hand, electrospun scaffolds are often described as suitable templates for cell infiltration due to their high porosity and high surface area.13 In this in vitro study, cells did not seem to penetrate the scaffolds, according to the FESEM images. This may be explained by the chosen cell types as well as the porosity (small micropores associated with the technique) of the produced scaffolds.46 Even with less tortuosity (aligned fibers), cells did not penetrate efficiently into the membranes. Such behaviour was also reported by Ghasemi-Mobarakeh et al., using an in vitro evaluation with rEPCs, who suggested that the small pore size of nanofibrous scaffolds was the reason for this non-infiltration.47 Several methods can be used to promote cell penetration on electrospun mats, such as selective leaching or a combination of electrospray with co-electrospinning.48,49 For the produced hybrid fibers, an improvement regarding the cell infiltration would be required, if cells are intended to be cultured on the scaffolds in vitro and if infiltration is required for the targeted application.47
In vivo subcutaneous implantation in rats
As a means to evaluate the in vivo response of the developed materials and according to in vitro results, we implanted aligned mats subcutaneously in mice. Histological analysis by HE staining showed that all materials tested induced a mild inflammatory response at both 1 and 4 weeks with a thin fibrous capsule being formed at the interface with the host tissue (Fig. 7, see red arrows at 1-week post implantation). Also, one can observe that the materials containing the ORMOGLASS showed higher infiltration of cells at both time points tested, in relation to pure PLA alone. The abundant cellular infiltration observed after in vivo implantation contrasted with the in vitro results, particularly for the OG5 20–80 fiber formulation (Fig. 7). Nonetheless, previous reports have also shown this discrepant effect. Indeed, Boland et al. has also shown a lack of infiltration under in vitro conditions but abundant cell migration after in vivo implantation.50 It is hypothesized that the external forces applied by the animal activity/motion promoted the migration of the cells towards the centre of the material and aid the cells to push individual fibers from their path during migration. A combination of both in vivo mechanical constraints and inflammatory response plays a role in the cellular penetration, inside the matrices, which due to the controlled release of calcium favours angiogenesis inside the composite matrices. In fact, assays reported in a previous study also demonstrated that cells infiltrated well into fibrous templates under in vivo conditions.15,51 However, it is clear that the infiltration in fibrous mats was lower than in other materials with bigger pore size and interconnectivity.52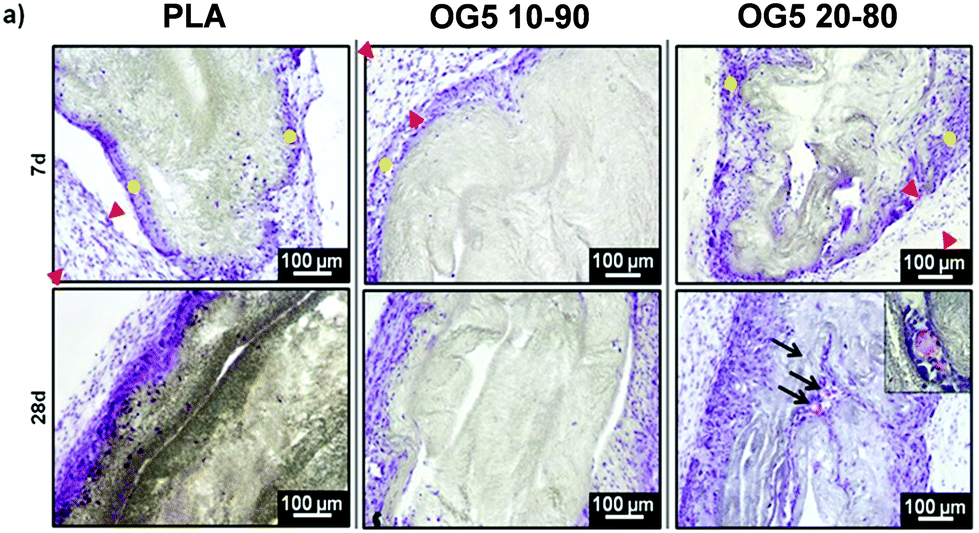 | ||
| Fig. 7 Histological analysis by HE staining after 7 and 28 days of subcutaneous implantation (n = 6). Red arrows denote the presence of fibrous capsules (mild inflammatory response). Yellow dots point to the cells penetrating the scaffolds. Black arrows show the presence of blood vessels. Blood vessels were identified by the staining of red blood cells (inset in the picture related to OG5 20–80 fibers). | ||
The more interesting aspect obtained from this in vivo evaluation was the improved vascularization potential of the hybrid fibers, in relation to PLA alone (see Fig. 7 for hematoxylin (HE) staining and Fig. 8 for platelet endothelial cell adhesion molecule (CD31) staining). Owing to the strong staining eosin by red blood cells and to the morphology of the capillaries, we could observe the existence of vessels inside the materials at both 7 and 28 days post implantation, with higher relevance in the OG5 20–80 fiber material (see black arrows in Fig. 7, at 28 days). Based on these initial results, we then performed CD31 immunohistochemistry on the samples implanted subcutaneously at the described time points (Fig. 8). Upon quantification, we could observe a clear effect regarding the number of vessels formed and the number of ORMOGLASSES in the materials. As observed in Fig. 8a, microvessels could be observed predominantly in the periphery of the material and the density was more pronounced with increased time of implantation. The corresponding quantification (Fig. 8b) shows that at 7 days post-implantation a significant higher density of vessels could be determined for the OG5 20–80 formulation. Again, at 28 days post-implantation both composite formulations demonstrated a significant angiogenic effect in comparison with PLA alone (Fig. 8b). Even though the OG5 10–90 fibers did not seem to improve angiogenesis after one week, a great enhancement in blood vessel density was achieved between the first week and the end of the assay. This suggested that these fibers may be a promising scaffold, but also that they may require more time than the OG5 20–80 ones to induce angiogenesis. This may be attributed to the differences in ORMOGLASS content and the corresponding calcium release profile. As shown above (Fig. 3) OG5 20–80 blended fibers showed a calcium release close to the concentration range at which angiogenesis has been shown to be favoured (3–10 mM [Ca2+]).10,15,44 On the other side, the OG5 10–90 blended fiber formulation showed a lower calcium release kinetics that might explain the delayed angiogenic effect. Note the negative slope in Fig. 3a, typical behaviour when in vitro studies are performed using phosphate buffered solutions in the absence of proteins from the serum. Therefore, experiments were repeated in an organically buffered solution using HEPES, with consistent results. Note, as well, the slightly acid pH resulting from the immersion in pure water in contrast to silicon based ormoglasses previously published by our group,14,15 which can also trigger a higher release rate. Nonetheless, we could demonstrate that these hybrid fibers possess the potential to induce vascularization, due to the improvement of the hydrophilicity and by the release of calcium, shown to enable cell chemotaxis and enhancement in cellular responses.9,10 These hybrid fibers seem thus to be promising according to their bioactivity and biocompatibility. However, further in vivo studies in the bone site are underway to confirm the potential of these biomimetic materials for bone tissue regeneration.
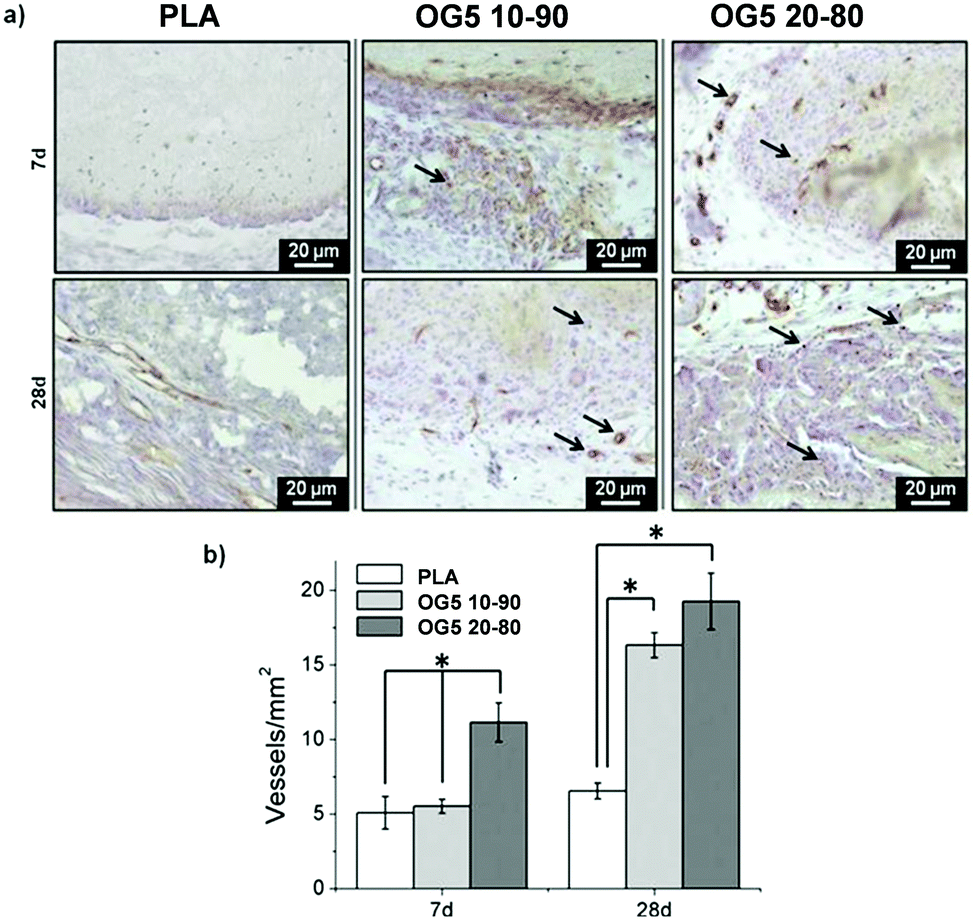 | ||
| Fig. 8 (a) Histological analysis by CD31 immunostaining and (b) blood vessel density quantification of PLA, OG5 10–90 or OG5 20–80 membranes, after 7 and 28 days of subcutaneous implantation (n = 6; *p < 0.05). Black arrows point the presence of endothelial cells and blood vessels. | ||
Other aspects to consider
We have evaluated the initial behaviour of endothelial cells, upon in vitro culture on the matrices, but focused our angiogenesis studies in an in vivo model that is, from our perspective, more relevant in terms of material evaluation. We and others53 considered that the in vitro evaluation of a biomaterial has some limitations. Indeed, endothelial cells are likely to behave differently when in tubular structures and when in contact with other cells, like pericytes and smooth muscle cells and when in response to flow and shear stress. Hence, in vitro assays using endothelial cells will not permit the evaluation of more complex physiological interactions that occur in vivo, including the assessment of indirect effects and the impact of other cellular populations.The production of these titanium-based fibers represents great progress in the development of artificial bioactive matrices. Indeed, up to now, most of the designed biomaterials focused on silica-based glasses, due to the difficulty of preparing phosphate-based glasses containing titanium.54 Hence, only a few reports on this system are found in the literature. Moreover, most of them involve the conventional melt-quenching method.21,55–57
To the best of our knowledge, only Pickup et al. used the sol–gel method to prepare titanium-based glasses in the form of nanostructured powders,58 but the drawback of their approach is that the preparation of their glass requires several weeks; in fact, ageing and drying were performed during 10 weeks at temperatures between 60 °C and 350 °C. For the sol–gel derived glass described in this paper, only three days were necessary to obtain the glass, while using low temperatures (4 °C and ambient temperature). This enabled us also to conserve the organic fragments present in the glass and to work with ORMOGLASSES. Additionally, titanium-based glasses have rarely been used to prepare nanostructured composite materials, being more often investigated as isolated scaffolds.59,60
This is the first time that sol–gel derived glasses, based on titanium, were used to design nanostructured hybrids mimicking an ECM, and with the capacity to sustain angiogenesis. In contrast to usual composites, where the glasses are used as fillers for polymeric matrices (dispersion of microparticles),12,56,61 our approach constitutes a real novelty regarding other titanium-based glasses and composite materials. The preparation of these fibers demonstrated that not only silicon-based glasses15 but also titanium-based ones can be prepared as ORMOGLASS and be used to produce nanofibrous hybrids.
Finally, we want to propose this material as a possible candidate for, but not limited to, clinical applications for guiding bone regeneration. The periosteum is an outer membrane of the bone that provides a high number of pluripotent stem cells, and it has been demonstrated to be crucial as a template for further neovascularization during healing.62–64 An engineered biomimetic membrane promoting efficient vascularization that mimics the periosteum would be an ambitious challenge and a new milestone in biomaterial-based therapies for early stage bone regeneration.65 As this material can be easily transferred to other processing methods such as rapid prototyping, among others, it offers further tissue regeneration potential according to the vascularization requirements of the tissue, such as skin or even osteochondral tissue regeneration.
Conclusions
PLA-titanium based ORMOGLASS composites have been produced as fibrous mats. For the first time, bioactive glass has been successfully obtained by applying the sol–gel method and has been efficiently used to prepare an electrospinnable polymer/glass blend. Electrospinning parameters (blend viscosity, voltage intensity, dispensing rate, tip-collector distance) were correctly set for this novel hybrid system and a continuous fiber deposition was achieved. Fibers, collected as random and aligned structures with two different glass contents, exhibited regular shapes, improved mechanical properties and suitable hydrophilicity. Globally, the addition of ORMOGLASS into the polymeric matrix resulted in the enhancement of the physical–chemical properties of the pure polymeric scaffolds. In addition, the fibers induced promising biological responses. In particular, in vivo implantation demonstrated the potential of the hybrids to induce vascularization. Although further assays are required to assess more precisely the potential of these novel fibers (efficiency of osteogenesis and angiogenesis promotion), this study represents a valuable step forward for the fabrication of biomimetic materials as potential functional in situ cell-instructive scaffolds for vascularized bone regeneration with excellent cost-effectiveness.Experimental
Blend preparation and electrospinning
The preparation of the glass involved a mix of metal alkoxide precursor solutions. Calcium alkoxide precursor solution was obtained as the result of refluxing metallic calcium (Panreac, 98%) in 2-methoxyethanol (Sigma-Aldrich, 99%). Phosphorus alkoxide precursor solution was prepared by refluxing P2O5 (Aldrich, 99.99%) in absolute ethanol (Sigma-Aldrich >99%). The titanium precursor was prepared by dissolving titanium isopropoxide (Alfa Aesar, 97%) in absolute ethanol and the sodium precursor by refluxing metallic sodium (Panreac, 99.8%) in 2-methoxyethanol. The precursor solutions were mixed homogeneously according to the composition of OG5 glass (molar ratio): 44.5P2–44.5Ca–5Ti–6Na2, with strong stirring. Then a basic catalyst (Ti:H2O:NH3:2-propanol (Panreac, 99.7%) catalyst with a molar ratio of 1:60:4.5:100) was added to partially hydrolyze the solution and produce a viscous electrospinnable ORMOGLASS slurry. The hydrolysis was performed at 4 °C and aged for three days. In parallel, polymeric solutions of polylactic acid 3% and 4% w/w (PLA, 70/30 L-lactide/DL-lactide copolymer, Purasorb PLDL 7038) in 2,2,2-trifluoroethanol (TFE, Panreac, 99.8%) were prepared. Then, the ORMOGLASS gel and the polymeric solution were blended according to an ormoglass:polymer ratio of 10:90 (OG5 10–90) and 20:80 (OG5 20–80) (v:v) in order to obtain the electrospinnable blends. The blends, as well as the pure polymeric solution, were finally electrospun using a conventional electrospinning setup with a grounded flat or a rotary collector covered with aluminium. Fibers were then deposited as randomly distributed or aligned fibrous mats. The processing parameters were the following: 8 kV applied voltage, 0.5 ml h−1 blend dispensing rate, 18 cm tip-collector distance and 1000 rpm rotating speed (aligned fibers only). In order to minimize differences in the fiber composition due to the preparation of the ORMOGLASS (mix), each blend was used to produce both types of fiber orientation.
Blend viscosity
In order to assess the effect of the addition of the ORMOGLASS gel to the polymeric solution, viscosity measurements of the blends were performed (vibro-viscometer SV10, AND company, Japan). Continuous measurements were done to make sure that the viscosity of the blends was stable over time and suitable for the electrospinning process.Fiber morphology and composition
Small pieces 2 × 2 mm of the electrospun mats were fixed on a metallic support and coated with carbon. The support was then introduced in a Field Emission Scanning Electron Microscope (FESEM, Nova™-Nano SEM-230; FEI Co., The Netherlands) to assess the arrangement and morphology of the fibers. To evaluate the composition of the fibers, the samples used for FESEM observations were loaded in a SEM device coupled with EDS (Quanta 200 XTE 325/D8395; FEI Co., The Netherlands). The measured compositions were averaged from elemental quantifications of three different points on the samples.Fiber wettability
Contact angle measurements were performed using the sessile drop method to evaluate the wettability of the scaffolds (OCA 20 system, Dataphysics, GmbH, Germany). This consists of depositing an ultra-pure water drop (3 μl) on the material and determining the angle between this drop and the material's surface. The contact angle was determined by using the ImageJ software66 and the plugin. This approach enables the determination of the degree of hydrophilicity of the materials: the lower the contact angle values, the more hydrophilic the material. For the aligned fibers, pictures of the water drop on the fibers were taken perpendicular to the orientation of the fibers. For the random fibers, no specific positioning of the sample was performed, as the contact angle values were not influenced by fiber orientation (Fig. S3, ESI†).Fiber surface charge
The zeta potential (ZP) technique was used to assess the surface charge of the fibers. ZP measurements were performed in a 1 mM KCl electrolyte solution using the “adjustable gap cell” set-up (electrokinetic analyzer SurPASS, Anton Paar Ltd, Austria). The electrolyte was titrated from the basic region (pH ∼ 8) to the acid one (pH ∼ 2.5) by adding HCl 0.1 M (Sigma-Aldrich, 37%) using the device syringe pump. The electrolyte was forced to pass between two thin layers of fibers using a pressure program of maximum 300 mbar. The isoelectric points (IEP) and ZP values at pH = 7.4 were monitored to compare the charge surface changes after the treatments.Thermal properties of the polymer
Differential scanning calorimetry (DSC, Q2000 TA, US) was used to determine the effect of the addition of the ORMOGLASS in the polymeric solution on the thermal properties of the produced fibers. As for FESEM, small pieces of the mats were cut at random areas of the electrospun layer (∼1.2 mg sample). Samples were then placed in aluminium pans and subjected to two heating ramps and one intermediate cooling: samples were first heated from −20 to 180 °C, cooled, and heated again. The heating rate for both ramps was fixed at 10 °C min−1 and nitrogen was used as a purging gas. Crystallization and melting properties of the polymer were assessed by analysing the curve of the first heating ramp. The glass transition (Tg) was determined considering the second heating cycle and using the inflexion point method (TA Universal Analysis software, v4.7A, TA, US).Thermogravimetry
A thermoanalyser (TGA Q5000 TA, US) was used to determine the mass of the inorganic phase contained in the fibers and assess the stability of the polymeric phase (evaluation of decomposition temperatures). Samples of ∼10 mg were placed on platinum pans and heated from room temperature (25 °C) up to 700 °C, at a heating rate of 10 °C min−1, in air. TGA coupled with FTIR (Nicolet 8700 Thermo Scientific, US) was also operated to identify the gaseous by-products that decomposed and/or vaporized during the degradation of fibers. In this case, measurements were performed under the same alternative conditions as simple TGA but in nitrogen. However, only the OG5 20:80 fibers were selected for this assay. As they had a higher ORMOGLASS content, it was assumed that an improved infrared signal, related to eventual degradation products from the ORMOGLASS, could be obtained (in comparison to fibers containing a smaller quantity of ORMOGLASS). TGA curves were analysed using the TA Universal Analysis software (v4.7A) and FTIR spectra acquired using the OMNIC 8.2 software (Thermo Scientific, US).
Mechanical properties
Fiber strips (1 cm width), cut from the fiber mats, were clamped by the grips of the tensile device (Tytron 250 Microforce Testing System, MTS, US) and elongated using a force load of 50 N. The MultiPurpose TestWare® software was used to acquire the strain–stress data curves. The Young's modulus was calculated as the slope of the linear region of the curve. The thickness of the fiber layer was measured using the ImageJ66 software using FESEM pictures of the strips in the transversal view. The initial sample length (distance grip to grip) was set to 2 cm. For the aligned fibers, strain was applied in the same axis as the fiber orientation.Protein adsorption
A protein adsorption test was performed to evaluate the ability of the fibers (PLA and hybrids) to support protein anchorage. As such, 1 cm × 1 cm pieces were cut from the fibrous layers and immersed for 1 hour in deionized water at room temperature. Afterwards, fibers were transferred to a 24 well-plate and 1 ml of phosphate buffer saline (PBS) solution containing 0.5 mg of bovine albumin serum (BSA, Sigma-Aldrich) was added to each well and incubated for 30 min at 37 °C. Then the membranes were gently rinsed three times with deionized water and allowed to dry at room temperature. The adsorption of the protein was qualitatively assessed by FTIR (Nicolet 8700 Thermo Scientific, US).pH measurements and calcium release evaluation
The pH and Ca2+ release of the samples were evaluated by immersion in distilled ultra-high pure water, containing 0.02 M KCl (Sigma >99%) for constant ionic strength, in c-SBF67 or in 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES 20 mM, Sigma, >99.5%) buffered solutions. Discrete measurements, at different time points, were performed and the solution was renewed at each time point. Measurements in water and in SBF at 37 °C were performed using a Crison GLP22 + pH-meter (Crison, Spain), a Crison pH microelectrode, a Crison Ca2+ selective electrode and an Ag/AgCl reference electrode.Measurements of calcium release, in organically buffered solutions, were equally performed using fiber monolayer sheets electrospun on 10 mm diameter cover glasses placed in 48 well plates. The slides were immersed in a solution of 2.5 mM CaCl2 and 0.01 M HEPES at pH = 7.4 ± 0.1. The samples were then incubated at 37 °C and the buffer replaced at different time point, for 1 month. The calcium concentration was measured by spectrophotometry using a chromophore (O-cresolphthalein complexone, Sigma-Aldrick 99%). The reason for the use of organically buffered solutions is the trend of calcium to precipitate in contact with phosphates from c-SBF in the form of brushite or hydroxyapatite as can be observed in the negative slope of calcium release rates of hybrid mats in Fig. 3a.
When performing preliminary tests for in vitro assays, it has been noted that the culture medium used turned yellow instead of retaining pink, its usual color. This indicated that the pH changed once the fibers were immersed in culture medium. The possibility to control these changes was evaluated by immersing the fibers in four different solutions: deionized water, SBF, culture medium (advanced Dulbecco's modified Eagle medium, adv. DMEM, Gibco®) and culture medium supplemented with 20 mM of HEPES at pH 7.4 that was enough to solve this aspect.
In vitro biological assays
Before cell seeding, 15 × 15 mm squares were cut from the electrospun fibrous layers and fitted in 24 well plates, sterilized for 15 min under UV radiation, pre-incubated in complete medium for 2 hours at 37 °C and rinsed with PBS. Cells were then seeded at a density of 10000 cells per disc in 24 well plates using 500 μl medium per well.
The cell number was normalized to day 1 data to avoid the different cell attachment response in the different materials that masked the calcium dose effect.
In vivo assays
As a means to evaluate the material biocompatibility and capacity to sustain angiogenesis we performed their subcutaneous implantation in a murine model. The procedures and animal handling followed the principles of Laboratory Animal Care described by the National Society for Medical Research, and approved by the Animal Care and Experiment Committee of the University of Bordeaux Segalen. Experiments were carried out in accredited animal facilities according to European recommendations for laboratory animal care (directive 86/609 CEE of 24/11/86). Swiss Strain mice (female, 3 months) were used for this study. Mice were housed in individually ventilated cages in a 12-hour light-dark cycle at 21–23 °C and 40–60% humidity, and allowed free access to irradiated standard rodent diet and sterilized water. Round shape membranes, cut from the electrospun layers with a diameter of 1 cm, were sterilized by UV, incubated for 24 h in PBS and then implanted in the dorsum of the mice (subcutaneous implantation – see Fig. S12, ESI†). The animals were anesthetized using isoflurane and one small incision was made, with scissors, on each side of the dorsum. Each animal received one specimen of the aligned fiber type (PLA, OG5 20–80 or OG5 10–90 membranes) at the left and right dorsal sites. Three animals were used for each fiber type for histological assays (n = 6). The incision was closed using chirurgical staples. After one and four weeks, the animals were euthanized and implanted membranes were extirpated for histological analysis. The membranes and the tissues surrounding the implanted membranes were placed in 4% buffered formaldehyde solution for 24 h, at 4 °C. Then they were immersed for 3 days at 4 °C in a sucrose solution (30%, Sigma-Aldrich, 99.5%) and afterwards, immersed for 2 h in a sucrose solution (30%) containing 7% of porcine gelatin (type A, Sigma-Aldrich), at 37 °C. The samples embedded in gelatin were snap frozen using liquid nitrogen, and 8 μm sections were cut using a cryosection device (Leika, Germany). Hematoxylin and Eosin staining (HE) was performed in order to assess developed inflammation (formation of fibrous capsules), cell penetration into the materials and angiogenesis. CD31 immunostaining was additionally performed to confirm the presence of endothelial cells and to quantify more precisely the degree of angiogenesis. Briefly, samples were hydrated in PBS for 5 minutes, endogenous peroxidase was quenched using 3% (v/v) hydrogen peroxide for 5 min at room temperature (RT) and then blocked with 2% goat serum, in PBS, for 30 min at RT. Primary antibody against CD31 (ab28364, Abcam, UK) was used at 1:250 in 1% (w/v) BSA in PBS, and incubated overnight at 4 °C. After two washes with PBS, the peroxidase VECTASTAIN ABC kit (VectorLabs, USA) was used according to the manufacturer's instructions. Specific staining was obtained using the 3,3′-diaminobenzidine staining solution (VectorLabs, USA). Counterstaining was performed using Mayer's haematoxylin. Samples were then mounted using Clearmount® medium (Invitrogen, USA). Sample imaging was performed using a microscope (Nikon Eclipse 80i) equipped with a digital camera (Nikon Dxm 1200C). Six tissue slices, at three different sample positions, were evaluated per time point and condition. For vessel quantification the total number of microvessels was determined and normalized to the area of the material.
Statistical analysis
Using the Graphpad Prism 5.0 software, the D'Agostino and Pearson omnibus normality test was used in order to verify if data conformed to a Gaussian distribution. Statistically significant differences between the groups were analyzed by the non-parametric Kruskal–Wallis test, followed by a Dunn post-test. The results were considered statistically significant for p < 0.05 and highly statistically significant for p < 0.001.Acknowledgements
The authors thank the European Commission (European ERANET project PI11/03030, NANGIOFRAC), the Spanish Ministry of Economy and Competitiveness (Project MAT2011-29778-C02-01) and the National Centre for Research and Development (contract No. 04/Euronanomed/2012) for funding. O. Castaño also acknowledges the MINECO for the “Ramon y Cajal” and I3 programme. N. Sachot thanks AGAUR (travel grant fellowship) for its financial support. We also thank Tomasz Jaroszewicz, Ewa Kijeńska and Emilia Choińska for their help with DSC, FTIR and TGA devices respectively.References
- M. Navarro, A. Michiardi, O. Castaño and J. A. Planell, J. R. Soc., Interface, 2008, 5, 1137–1158 CrossRef CAS PubMed.
- L. L. Hench and I. Thompson, J. R. Soc., Interface, 2010, 7, S379–S391 CrossRef CAS PubMed.
- L. L. Hench and J. M. Polak, Science, 2002, 295, 1014–1017 CrossRef CAS PubMed.
- I. D. Xynos, A. J. Edgar, L. D. Buttery, L. L. Hench and J. M. Polak, J. Biomed. Mater. Res., 2001, 55, 151–157 CrossRef CAS PubMed.
- S.-H. Rhee, J.-Y. Choi and H.-M. Kim, Biomaterials, 2002, 23, 4915–4921 CrossRef CAS PubMed.
- J. M. Kanczler and R. O. C. Oreffo, Eur. Cells Mater., 2008, 15, 100–114 CAS.
- M. Charles-Harris, M. Navarro, E. Engel, C. Aparicio, M. P. Ginebra and J. A. Planell, J. Mater. Sci.: Mater. Med., 2005, 16, 1125–1130 CrossRef CAS PubMed.
- M. Charles-Harris, M. A. Koch, M. Navarro, D. Lacroix, E. Engel and J. A. Planell, J. Mater. Sci.: Mater. Med., 2008, 19, 1503–1513 CrossRef CAS PubMed.
- M. Navarro, E. Engel, J. A. Planell, I. Amaral, M. Barbosa and M. P. Ginebra, J. Biomed. Mater. Res., Part A, 2008, 85, 477–486 CrossRef CAS PubMed.
- A. Aguirre, A. González, M. Navarro, O. Castaño, J. A. Planell and E. Engel, Eur. Cells Mater., 2012, 24, 90–106 CAS.
- M. Charles-Harris, S. del Valle, E. Hentges, P. Bleuet, D. Lacroix and J. A. Planell, Biomaterials, 2007, 28, 4429–4438 CrossRef CAS PubMed.
- T. Serra, J. A. Planell and M. Navarro, Acta Biomater., 2013, 9, 5521–5530 CrossRef CAS PubMed.
- J.-H. Jang, O. Castano and H.-W. Kim, Adv. Drug Delivery Rev., 2009, 61, 1065–1083 CrossRef CAS PubMed.
- N. Sachot, O. Castaño, M. A. Mateos-timoneda, E. Engel and J. A. Planell, J. R. Soc., Interface, 2013, 10, 1–5 CrossRef PubMed.
- O. Castaño, N. Sachot, E. Xuriguera, E. Engel, J. A. Planell, J.-H. Park, G.-Z. Jin, T.-H. Kim, J.-H. Kim and H.-W. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 7512–7522 Search PubMed.
- N. Sachot, O. Castano, J. A. Planell and E. Engel, J. Biomed. Mater. Res., Part B, 2015, 103, 1287–1293 CrossRef CAS PubMed.
- N. Sachot, M. A. Mateos-Timoneda, J. A. Planell, A. H. Velders, M. Lewandowska, E. Engel and O. Castano, Nanoscale, 2015, 15349–15361 RSC.
- E. S. Sanzana, M. Navarro, M.-P. Ginebra, J. A. Planell, A. C. Ojeda and H. A. Montecinos, J. Biomed. Mater. Res., Part A, 2013, 1–7 Search PubMed.
- M. M. Stevens, Mater. Today, 2008, 11, 18–25 CrossRef CAS.
- H. Oliveira, S. Catros, C. Boiziau, R. Siadous, J. Marti-Munoz, R. Bareille, S. Rey, O. Castano, J. Planell, J. Amédé and E. Engel, Acta Biomater., 2016, 29, 435–445 CrossRef CAS PubMed.
- M. Navarro, M. Ginebra, J. Clément, S. Martinez, G. Avila and J. A. Planell, J. Am. Ceram. Soc., 2003, 86, 1342–1352 CrossRef.
- Q. P. Pham, U. Sharma and A. G. Mikos, Tissue Eng., 2006, 12, 1197–1211 CrossRef CAS PubMed.
- J. Geltmeyer, L. Van der Schueren, F. Goethals, K. De Buysser and K. De Clerck, J. Sol-Gel Sci. Technol., 2013, 67, 188–195 CrossRef CAS.
- T. Ogoshi and Y. Chujo, Compos. Interfaces, 2005, 11, 539–566 CrossRef CAS.
- S. Iannace, L. Sorrentino and E. Di Maio, Biomedical Foams for Tissue Engineering Applications, Elsevier, 2014 Search PubMed.
- J. Wen and J. E. Mark, J. Appl. Polym. Sci., 1995, 58, 1135–1145 CrossRef CAS.
- E. Petinakis, X. Liu, L. Yu, C. Way, P. Sangwan, K. Dean, S. Bateman and G. Edward, Polym. Degrad. Stab., 2010, 95, 1704–1707 CrossRef CAS.
- H. Pan and Z. Qiu, Macromolecules, 2010, 43, 1499–1506 CrossRef CAS.
- D. Carta, D. M. Pickup, R. J. Newport, J. C. Knowles, M. E. Smith and K. O. Drake, Phys. Chem. Glasses, 2005, 46, 365–371 CAS.
- H. Liang and W. Shi, Polym. Degrad. Stab., 2004, 84, 525–532 CrossRef CAS.
- L. H. Perng, C. J. Tsai, Y. C. Ling, S. D. Wang and C. Y. Hsu, J. Appl. Polym. Sci., 2002, 85, 821–830 CrossRef CAS.
- V. Thomas, M. V. Jose, S. Chowdhury, J. F. Sullivan, D. R. Dean and Y. K. Vohra, J. Biomater. Sci., Polym. Ed., 2006, 17, 969–984 CrossRef CAS PubMed.
- N. L. Nerurkar, D. M. Elliott and R. L. Mauck, J. Orthop. Res., 2007, 25, 1018–1028 CrossRef CAS PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- B. Athoff and J. Hilborn, J. Biomed. Mater. Res., Part B, 2007, 80, 121–130 CrossRef PubMed.
- N. Fogh-Andersen, Clin. Chem., 1977, 23, 2122–2126 CAS.
- X. Zhu, H. Fan, D. Li, Y. Xiao and X. Zhang, J. Biomed. Mater. Res., Part B, 2007, 82, 65–73 CrossRef PubMed.
- A. Krajewski, R. Malavolti and A. Piancastelli, Biomaterials, 1996, 17, 53–60 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, J. Mater. Chem., 2007, 17, 4079–4087 RSC.
- C. Liu, Z. Xia and J. T. Czernuszka, Chem. Eng. Res. Des., 2007, 85, 1051–1064 CrossRef CAS.
- M. J. Dalby, M. O. Riehle, H. Johnstone, S. Affrossman and A. S. G. Curtis, Biomaterials, 2002, 23, 2945–2954 CrossRef CAS PubMed.
- G. Le Saux, A. Magenau, T. Böcking, K. Gaus and J. J. Gooding, PLoS One, 2011, 6, e21869 CAS.
- A. González-Vázquez, J. A. Planell and E. Engel, Acta Biomater., 2014, 10, 2824–2833 CrossRef PubMed.
- A. Aguirre, A. González, J. A. Planell and E. Engel, Biochem. Biophys. Res. Commun., 2010, 393, 156–161 CrossRef CAS PubMed.
- R. G. Flemming, C. J. Murphy, G. A. Abrams, S. L. Goodman and P. F. Nealey, Biomaterials, 1999, 20, 573–588 CrossRef CAS PubMed.
- H. Chang and Y. Wang, in Regenerative Medicine and Tissue Engineering – Cells and Biomaterials, ed. D. Eberli, Intech, 2011 Search PubMed.
- L. Ghasemi-Mobarakeh, M. Morshed, K. Karbalaie, M. Fesharaki, M. H. Nasr-Esfahani and H. Baharvand, Yakhteh Med. J., 2008, 10, 179–184 CAS.
- A. K. Ekaputra, G. D. Prestwich, S. M. Cool and D. W. Hutmacher, Biomacromolecules, 2008, 9, 2097–2103 CrossRef CAS PubMed.
- S. Agarwal, J. H. Wendorff and A. Greiner, Adv. Mater., 2009, 21, 3343–3351 CrossRef CAS PubMed.
- E. D. Boland, T. A. Telemeco, D. G. Simpson, G. E. Wnek and G. L. Bowlin, J. Biomed. Mater. Res., Part B, 2004, 71, 144–152 CrossRef PubMed.
- Z. Álvarez, O. Castaño, A. A. Castells, M. A. Mateos-Timoneda, J. A. Planell, E. Engel and S. Alcántara, Biomaterials, 2014, 35, 4769–4781 CrossRef PubMed.
- J. X. Lu, B. Flautre, K. Anselme, P. Hardouin, A. Gallur, M. Descamps and B. Thierry, J. Mater. Sci.: Mater. Med., 1999, 10, 111–120 CrossRef CAS PubMed.
- C. A. Staton, M. W. R. Reed and N. J. Brown, Int. J. Exp. Pathol., 2009, 90, 195–221 CrossRef CAS PubMed.
- E. A. Abou Neel, D. M. Pickup, S. P. Valappil, R. J. Newport and J. C. Knowles, J. Mater. Chem., 2009, 19, 690–701 RSC.
- E. A. Abou Neel and J. C. Knowles, J. Mater. Sci.: Mater. Med., 2008, 19, 377–386 CrossRef CAS PubMed.
- W. Chrzanowski, E. A. Abou Neel, K.-Y. Lee, A. Bismarck, A. M. Young, A. D. Hart, M. J. Dalby and J. C. Knowles, Adv. Eng. Mater., 2010, 12, B298–B308 CrossRef.
- V. Rajendran, A. V. Gayathri Devi, M. Azooz and F. H. El-Batal, J. Non-Cryst. Solids, 2007, 353, 77–84 CrossRef CAS.
- D. M. Pickup, R. J. Speight, J. C. Knowles, M. E. Smith and R. J. Newport, Mater. Res. Bull., 2008, 43, 333–342 CrossRef CAS.
- N. J. Lakhar, J.-H. Park, N. J. Mordan, V. Salih, I. B. Wall, H.-W. Kim, S. P. King, J. V. Hanna, R. A. Martin, O. Addison, F. Mosslemans and J. C. Knowles, Acta Biomater., 2012, 8, 4181–4190 CrossRef PubMed.
- E. A. Abou Neel, T. Mizoguchi, M. Ito, M. Bitar, V. Salih and J. C. Knowles, Biomaterials, 2007, 28, 2967–2977 CrossRef CAS PubMed.
- M. Navarro, M. P. Ginebra, J. A. Planell, C. C. Barrias and M. A. Barbosa, Acta Biomater., 2005, 1, 411–419 CrossRef CAS PubMed.
- C. Colnot, X. Zhang and M. L. Knothe Tate, J. Orthop. Res., 2012, 30, 1869–1878 CrossRef CAS PubMed.
- Z. Lin, A. Fateh, D. M. Salem and G. Intini, J. Dent. Res., 2013, 93, 109–116 CrossRef PubMed.
- X. Zhang, H. A. Awad, R. J. O'Keefe, R. E. Guldberg and E. M. Schwarz, Clin. Orthop. Relat. Res., 2008, 466, 1777–1787 CrossRef PubMed.
- I. Rajzer, E. Menaszek, R. Kwiatkowski, J. A. Planell and O. Castano, Mater. Sci. Eng., C, 2014, 44, 183–190 CrossRef CAS PubMed.
- W. S. Rasband, US Natl. Inst. Heal. Bethesda, USA, 1997, 2014, http://http//imagej.nih.gov/ij/.
- A. Oyane, H.-M. Kim, T. Furuya, T. Kokubo, T. Miyazaki and T. Nakamura, J. Biomed. Mater. Res., Part A, 2003, 65, 188–195 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb02162j |
| This journal is © The Royal Society of Chemistry 2016 |