
Tracking translocation of glucan microparticles targeting M cells: implications for oral drug delivery†
Yunchang
Xie
a,
Xiongwei
Hu
a,
Haisheng
He
a,
Fei
Xia
a,
Yuhua
Ma
a,
Jianping
Qi
a,
Xiaochun
Dong
a,
Weili
Zhao
ab,
Yi
Lu
*a and
Wei
Wu
*a
aSchool of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery of MOE and PLA, Shanghai 201203, China. E-mail: wuwei@shmu.edu.cn; fd_luyi@fudan.edu.cn
bKey Laboratory for Special Functional Materials of the Ministry of Education, Henan University, Kaifeng 475001, China
First published on 4th March 2016
Abstract
Taking advantage of its ability to deal with exogenous pathogens, the M cell passage has proven to be the most reliable pathway for entry of particulates, thus creating opportunities for oral immunization and delivery of biomacromolecules. Albeit a well-known story, the underlying mechanisms of this pathway are not yet well understood, especially concerning direct evidence of translocation of particulates. Herein, model glucan microparticles (GMs) targeting M cells are employed to track translocation through M cell pathways as well as to various organs via the systemic circulation. GMs were first labeled with a novel kind of near-infrared fluorescent water-quenching probe through encapsulation and locking by stearin. In vivo live imaging indicates prolonged residence of GMs in the gastrointestinal tract for as long as 12 h. GMs are found to be gradually absorbed from the ligated ileum segment but little from the jejunum. Histological examination using confocal laser scanning microscopy (CLSM) confirms distribution of GMs to the basolateral side of the ileum through Peyer's patches. However, no detectable fluorescence can be observed in any other organs or tissues until 12 h after administration. After 12 h, GMs can be found in the liver, spleen and lung. At 24 h, GMs accumulate in these organs with approximately 2.3% of the total amount. Repeated administration for three consecutive days augments total accumulation to as high as 4.5%. By tracking GM-bound fluorescence, the particles can be accurately located in these organs. GMs can be transported across Caco-2/Raji and Caco-2/Raji/J774A.1 co-culture monolayers, but not Caco-2 monolayers, in a time-dependent manner. As observed by CLSM, GMs can be voraciously engulfed with as many as 10–15 particles per cell. Evidence of translocation of GMs indicates that GMs can be absorbed through the M cell pathway located at Peyer's patches, especially in the ileum, and translocated to reticulo-endothelial organs.
Introduction
The human body has evolved to absorb small molecules and ions such as nutrients from the gastrointestinal (GI) tract, but block entry of exogenous particulates, so as to keep the body safely nourished. Seemingly only small molecular drugs can take advantage of the oral route to reach the systemic circulation. In fact, the GI tract lining is not so tightly defended, leaving gates to food-bound particulate pathogens, e.g. bacteria, viral particles and various microorganisms, which are normally recognized and internalized by specialized microvilli or microfold cells, commonly known as M cells, through specific or non-specific mechanisms.1–3 The M cells are specialized epithelial cells located in the follicle-associated epithelia (FAE) of intestinal Peyer's patches (PPs),1 whose morphology is typical of poorly organized brush borders and deeply invaginated basolateral membranes, organizing a hosting pocket for immune cells such as antigen presenting cells (APCs) and lymphocytes. The uptake and transportation of foreign particulates by M cells from the lumen to basolateral lymphoid tissues have been extensively documented throughout the entire intestinal segments.2–5 Taking advantage of the residence of APCs in PPs, the oral route has long been tested for possible oral mucosal immunization.6–8 Alternatively, this route has also been attempted for the delivery of very challengeable drugs like peptides and proteins encapsulated into particles.9–11 However, the population of M cells in the human GI tract is relatively small, about 5% of human FAE and less than 1% of the total intestinal surfaces.12 The M cell pathway is believed to be the most reliable way for entrance of particulates to date,13 although several studies propose trans-enterocytic absorption through ligand–receptor interactions.14,15 Despite the ever fervent interest in the M cell pathway, the underlying absorption mechanisms have not been well interpreted yet. The reasons, to a large part, lie in the lack of functional tools to monitor real-time translocation of the particles that are without exception too small to be detectable. Both fluorescent- and radio-labeling often provide ambiguous information because signals of dissociated probes may be misread as the particles. Therefore, in this study we hypothesize to track the translocation of particles through the M cell pathway by using particles targeting M cells that are large enough to be easily identifiable with the help of fluorescent imaging.Within the arsenals, natural glucan microparticles (GMs), also called yeast cell wall particles, prepared from Saccharomyces cerevisiae (Baker's yeast), appear to be more promising because of their distinctive features of a relatively large size of 2–5 μm and hollow cores that can host a large fraction of the payload. The principal component of GMs is β-1,3-D-glucan, whose receptors are CR3 and dectin-1 at the molecular level.16 CR3 and dectin-1 are highly expressed on phagocytes, including macrophages, dendritic cells, and neutrophils.17 Dectin-1 has been proven to play a crucial role in regulating the transcytotic function of M cells.18 Recently, GMs are gaining attention as carriers for oral delivery of biomacromolecules such as siRNAs,19,20 DNAs21 and vaccines.22,23 The hollow and porous structure imparts a capacity to load versatile entities, even nanoparticles24 and self-assemblies, formed inside GMs.25,26 To date, the potential of GMs as drug carriers hasn't been well explored.
Albeit relatively large, there are still difficulties recovering or tracing GMs by discriminating them from biological tissues. Therefore, they should be labeled first-hand for better tracking. Molecular fluorescent probes presently are one of the most efficient tools to monitor the in vivo fate of nanocarriers, either by physical incorporation or covalent linking to either drug molecules or carrier skeletons.27 However, concerns over misreading due to dissociation of probes from particulates greatly compromise the accuracy and precision of imaging. “Smart” fluorescent probes that can sensitively detect and report structural integrity are in high demand. In our previous study,28 water-quenching near-infrared (NIR) fluorescent probes were developed to visualize intra-GI digestion of lipid nanoparticles. The probes emit fluorescence in a well dispersed state when embedded in lipid matrices, but quench immediately upon release from the vehicles based on the aggregation-caused quenching (ACQ) effect. Herein, we employ the same probes to label GMs and track their in vivo fate via oral delivery. Since ACQ probes are highly hydrophobic, they are firstly solubilized in melted stearin, and then embedded into the hollow cores of GMs and solidified to fix by cooling. Tracking begins with live imaging of the intra-GI fate of GMs, after which translocation of GMs to intestinal epithelia and various organs is imaged. To confirm M cell-associated uptake of GMs by macrophages, trans-monolayer permeation is also studied.
Materials and methods
Fluorescent labeling of GMs
GMs were prepared from Baker's yeast (Fleishmann's Instant Yeast, ACH Food Companies, Inc., Canada) by successive alkaline and acidic treatments as previously reported.21 The particles obtained were dried at room temperature. With the intention of live imaging in animals and ex vivo tissue samples, ACQ probes, either P2 or P4 (prepared and validated in our lab),28 were encapsulated in GMs. P2, with maximum absorption/emission wavelengths (λabs/λem) of 708/732 nm, is suitable for live imaging, whereas P4, with λabs/λem of 651/662 nm, can be used for confocal laser scanning microscopy (CLSM) observation. Briefly, an appropriate amount of P2 or P4 acetonitrile solution was added into melted stearin (Suppocire®, Gattefosse, France), and mixed homogeneously at 50 °C. Then GMs were added under gentle stirring and incubated for 1 h at the same temperature. The soaked GMs were harvested by centrifugation at 1000 rpm for 5 min, washed with hot water three times, and dried at room temperature to obtain NIR fluorescently labeled GMs. The total level of GM-bound fluorescence was adjusted to about 8 × 10−3% for P2 and 4 × 10−3% for P4, respectively. To highlight the importance of interior solidification on the locking of the probes, non-solidified samples were prepared by physical adsorption of the probes, but without the use of stearin, for comparison.Live imaging of the in vivo fate of GMs
The in vivo fate of P2-labeled GMs was investigated by the IVIS Spectrum Live Imaging System (PerkinElmer, USA) after oral administration. Male SD rats (180–200 g) were raised in rooms controlled at 23 ± 1 °C and 55 ± 5% relative humidity with 12 h light/12 h dark time cycles. Standard laboratory chow diet and tap water were provided during acclimatization. Six rats were divided randomly into two groups, the interior solidification group and the non-solidification control group, and fasted for 12 h before oral administration by gavage of 0.8 mL of the test samples. Images were taken after 0.5, 1, 2, 3, 4, 8, 12 and 24 h in the live imaging system by monitoring P2 signals (Ex/Em = 710/760 nm). During the image-capturing process, animals were narcotized by an on-line gas anesthesia system using isoflurane (Shandong Keyuan Pharmaceutical Co., Ltd, China).To investigate the retention of GMs in different intestinal segments and the translocation of GMs to other tissues and organs, the animals were sacrificed at time intervals and dissected to visualize the GI tract and various organs of the reticulo-endothelial system (RES) such as liver, lung, spleen and kidney. In total, 21 rats, fasted for 12 h before the experiment, were included and given the same two test samples; three rats were sacrificed at each time point of 0.5, 1, 2, 4, 8, 12 and 24 h. The whole GI tract and vital organs were collected immediately and imaged ex vivo using the live imaging system. Another group of three rats were dosed with GMs for three consecutive days to study accumulative distribution. All procedures involving animals were approved by the Institutional Animal Care and Use Committee at School of Pharmacy, Fudan University.
Absorption through the intestinal loop model
In situ absorption of GMs was carried out to identify the main absorption site in the GI tract. The jejunum and ileum of the same rat were exposed to well-maintained mesenteric and capillary function at 37 °C. About 3 cm of each intestinal segment was ligated, and injected with 1.5 mL of the P2/P4-GM suspension. Images of both intestinal segments were taken immediately, and at 0.5 h and 1 h. Fluorescence intensity was calculated to evaluate absorption in the jejunum and ileum.Identification of GMs in ex vivo tissues
Tissue samples (intestinal segments, organs) that showed significant accumulation of GMs were collected and frozen in OCT compound (Surgipath®, Leica, USA) at −20 °C for 20 min. Then the frozen tissues were cut into slices of 10 μm in thickness using a Leica Microm CM3050S cryostat (Leica Inc., Germany) and stained with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc., USA). The images were captured with a Zeiss LSM 510 confocal laser scanning microscope (Carl Zeiss Inc., Germany) equipped with a laser operating at 405 nm for DAPI to visualize the nucleus, and 633 nm for P4 to track integral GMs, respectively. Images were superimposed using LAS-AF-Lite software.In vitro phagocytosis of GMs by macrophages
Three cell lines, human colorectal adenocarcinoma Caco-2 cells, human Burkitt's lymphoma Raji cells and mouse reticulum cell sarcoma J774A.1 macrophages were obtained from the American Type Culture Collection and cultured at 37 °C in an atmosphere of 90% relative humidity and 5% CO2. Caco-2 cells were cultured in DMEM (Gibco, USA), whereas Raji and J774A.1 cells were cultured in RPMI-1640 medium (Gibco, USA). Each cell culture medium was supplemented with 10% fetal bovine serum, 100 units per mL penicillin, 100 μg mL−1 streptomycin and other essential nutritive elements.Results
In vivo fate of GMs in the GI tract
Fig. 1A shows the real-time images after oral administration of interiorly solidified P2-GMs, with non-solidified counterparts as a control. As expected, the non-solidified control group shows a more limited residence time than their interiorly solidified counterparts, due to quick release of the dyes. The P2 signals vanish within hours, confirming the water quenching features of P2. Remarkable findings are obtained from the results of interiorly solidified P2-GMs. Due to interior solidification by stearin, the fluorescence remains in relatively high intensity for as long as 8 h and does not disappear until 12 h (Fig. 1A). Quantification of total fluorescence intensity reveals a sharp contrast between physical adsorption and interior solidification (Fig. 1B), highlighting the sensitivity of P2 to water-quenching in the GI tract, as well as the efficiency of interior solidification by stearin to protect P2 from leakage. Water-quenched dyes give negligible signals in images of live animals, the GI tract and various organs (Fig. S3–S5, ESI†). This sets the foundation for further live imaging to track the translocation of integral GMs to various organs and tissues. Another interesting finding is that GMs reside much longer in the GI tract than other formulations that we studied previously like liposomes31 and solid lipid nanoparticles (SLNs).28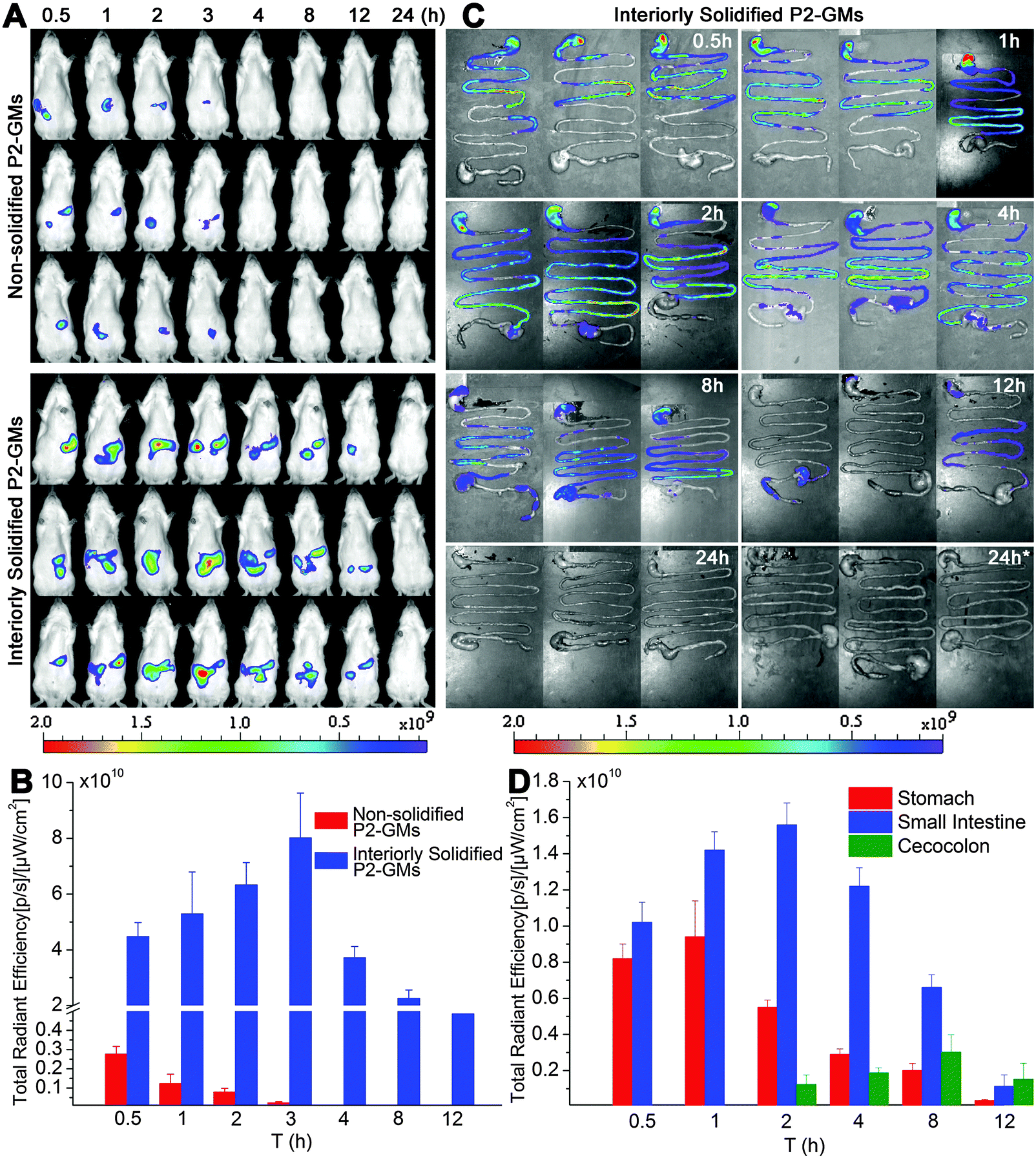 | ||
| Fig. 1 In vivo and ex vivo live imaging of SD rats after gavage administration of P2-labeled GMs under a fasted state. (A) In vivo live imaging shows faint P2 signals for the non-solidified group, confirming quick release of the dye and high sensitivity to water-quenching, whereas the interiorly solidified group shows prolonged resident time of integral GMs in the GI tract; (B) total fluorescence vs. time profile; (C) live imaging of the whole isolated GI segments following oral administration of P2-GMs; (D) quantification of total fluorescence of ex vivo GI segments indicates a gradual transit of GMs from the stomach to the small intestine, with an intestinal peak time at 2 h. “24 h*” indicates sampling 24 h after multiple dosing once a day for 3 consecutive days. | ||
The transit of GMs through the GI tract is further imaged with whole GI segments isolated after sacrificing the rats (Fig. 1C). The overall resident time of GMs is in line with the results of live imaging. Fluorescence can be detected for at least 8 h. In comparison with our previous findings with SLNs (<4 h),32 GMs maintain sufficient robustness to withstand damage by the harsh GI environment. Obviously, GMs reside in the upper GI tract (stomach and jejunum) within 1 h, and then transit gradually to the posterior intestine (ileum) till at least 8 h. It is very interesting to find out that a large fraction of GMs are retained in the ileum, where more PPs and FAE are located. Fig. 1D indicates the real-time transit of GMs in the GI segments by fractionized quantification of fluorescence. Within 1 h, there is a certain amount of GMs retained in the stomach, which however transit to the small intestine afterwards. There is a relatively high amount of GMs residing in the small intestine for 8 h. Taken together, the resident time of GMs in the GI tract is long enough to provide a sufficient chance for intimate contact between GMs and the GI epithelia.
To identify the virtual absorption site in the GI tract, the intestinal loop model was used. Fig. 2A shows live imaging of the fate of GMs in the ligated jejunum and ileum segments. A decreasing tendency in ileum-bound fluorescence can be barely discerned in the live images, but quantification of the average radiant efficiency (ARE) further confirms a near linear decrease of about 32% in fluorescent intensity within the investigated time duration of 60 min (Fig. 2B). However, there is only a slight decrease of less than 5% in jejunum-bound fluorescence. This result implies that the ileum may be the right site for absorption of GMs. Coincidentally, the observation of prolonged residence of GMs in the ileum favors absorption.
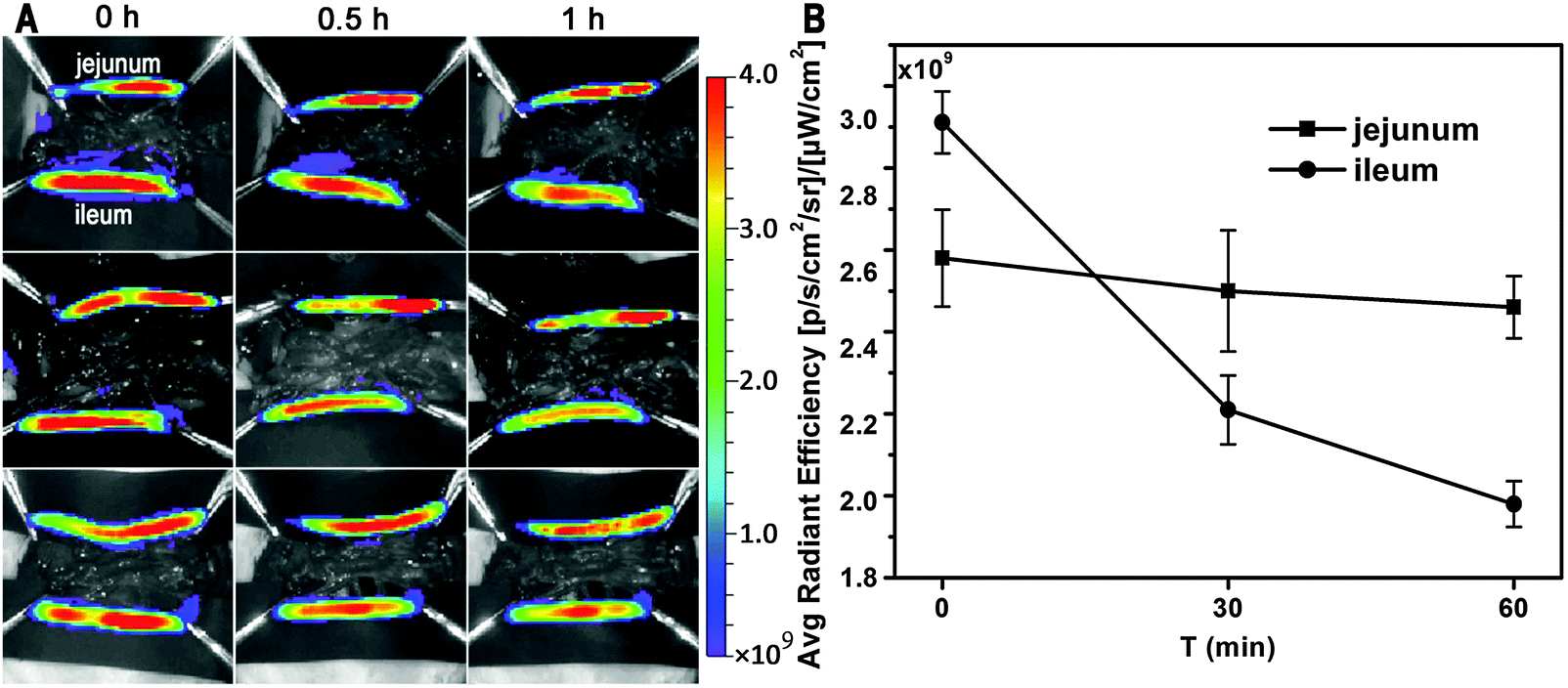 | ||
| Fig. 2 Ex vivo imaging of uptake of interiorly solidified P2-GMs by the duodenum, jejunum and ileum (A), and ARE vs. time profiles (B). | ||
Translocation of GMs to tissues and organs
Besides kinetic investigation on the in vivo fate of GMs, we carried out observations on frozen sections of ex vivo samples dissected from small intestinal segments with the aim to identify GMs, as represented by fluorescent dots (P4 signals). One of the advantages of employing GMs as the subject of investigation is their relatively large size, which helps to identify the particles themselves easily under the microscope. In Fig. 3, GMs can be found in either the duodenum or jejunum or ileum sections. However, GMs are found mostly in the lumen for both duodenum and jejunum, adhering to the villi surfaces, in sharp contrast with the ileum, where GMs can penetrate deep into the tissues. By careful comparison with previous findings, the GM-enriched area in the ileum, the dome, can be identified as a PP.22,33 It is very interesting to find out that GMs locate to an arc belt in the sub-FAE area. This serves as solid evidence of phagocytosis and residence of GMs in sub-FAE lymphoid tissues. After browsing through enough numbers of slices, it can be summarized that GMs can be absorbed through the ileum, specifically through FAE.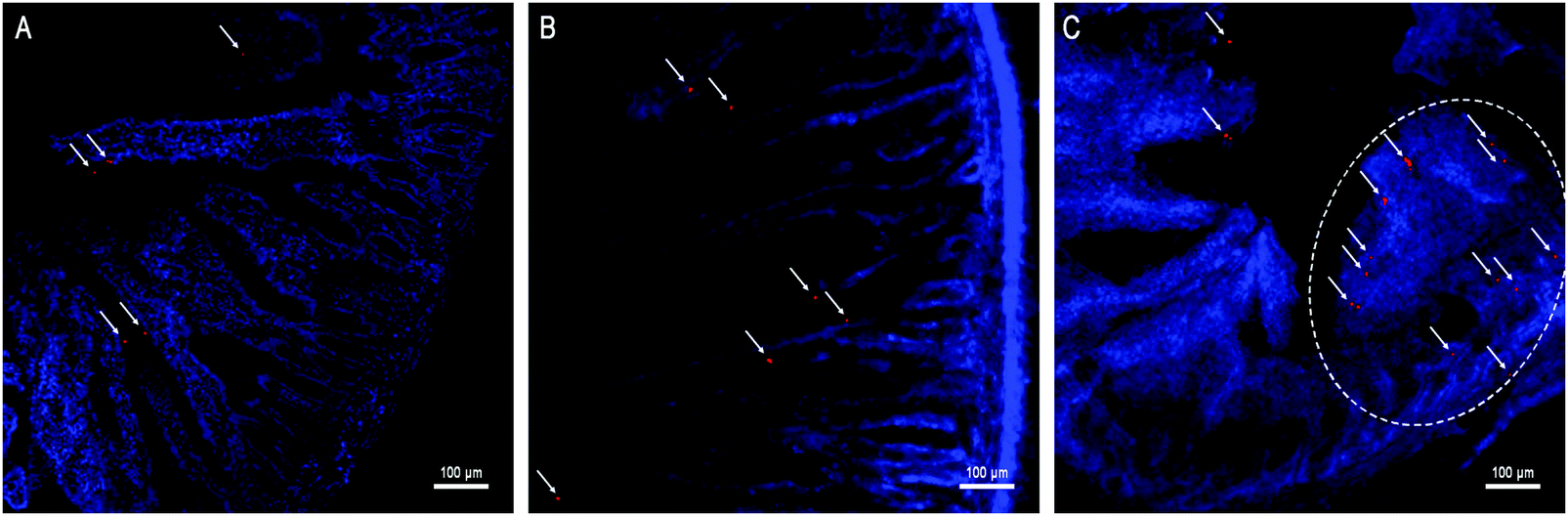 | ||
| Fig. 3 Frozen section of the duodenum (A), jejunum (B) and ileum (C) isolated from rats after oral administration of interiorly solidified P4-GMs for 2 h. The arrows point to fluorescent dots (red) of P4 signals that represent integral GMs. The circled domain in (C) represents a whole Peyer's patch, where GMs can be observed distributing to the basolateral side. | ||
In order to track the translocation of GMs to other organs or tissues, ex vivo samples of RES organs that are rich in mononuclear phagocytes, e.g. liver, lung, spleen and kidney, are observed following an in vivo fate investigation as outlined in Fig. 1C. Fluorescent signals begin to appear in the liver, lung, spleen and kidney 12 h after administration, and the signals intensify at 24 h in all of the observed organs (Fig. 4). After multiple administrations, the organ-associated fluorescence is further intensified, indicating obvious accumulation. This finding highlights the translocation of GMs through the M cell pathway to lymphoid tissues, and finally through the systemic circulation to RES organs. To find evidence of translocation of GMs, frozen tissue slices were browsed to identify integral GMs. The fluorescence signals of P4 can be identified in tissues of the liver, lung and spleen, but not the kidney due to a lower distribution of GMs, by CLSM, confirming translocation of integral GMs (Fig. 5). With the help of the positioning by fluorescent signals, GMs can also be identified visibly in the bright field.
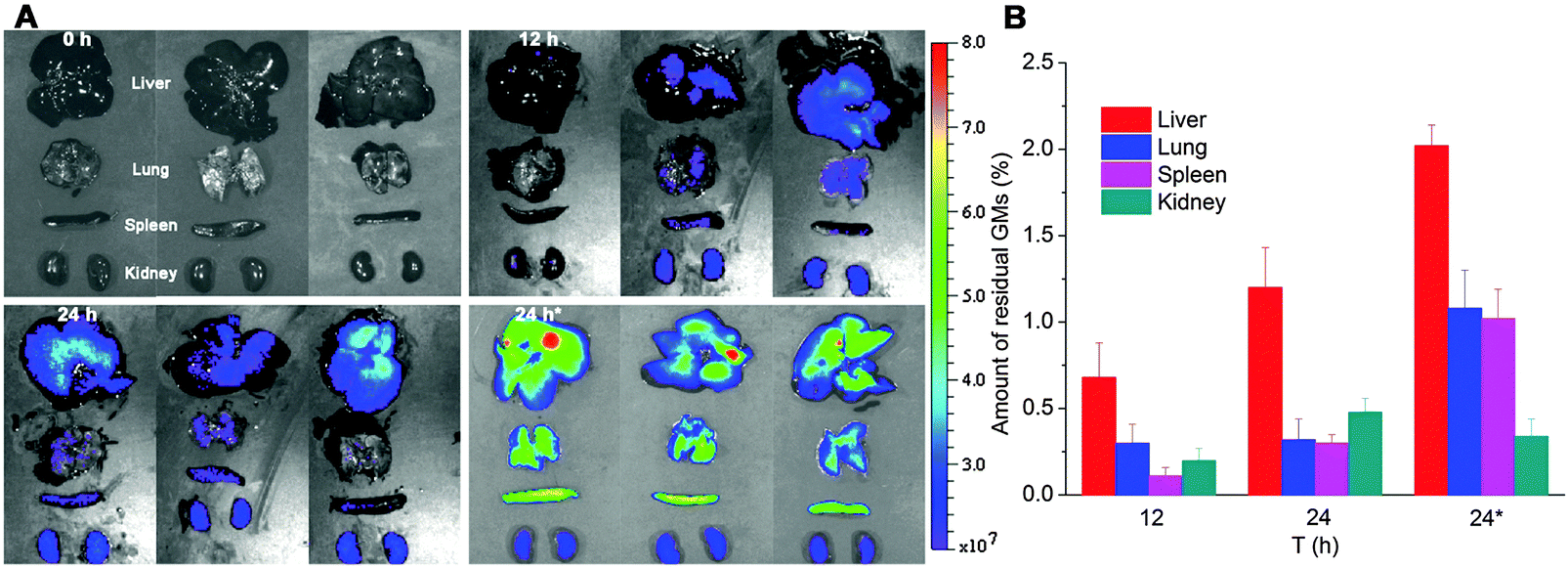 | ||
| Fig. 4 In vivo imaging (A) of RES organs isolated from rats following oral administration of interiorly solidified P2-GMs at 12 and 24 h. Faint fluorescence can be detected at 12 h, and an obvious tendency of accumulation is observed at 24 h, especially after multiple dosing. “24 h*” indicates sampling 24 h after multiple dosing once a day for 3 consecutive days. Quantification of total fluorescence (B) in each organ in reference to the total fluorescence administered, as calculated from GI-bound total fluorescence in Fig. 1C, confirms a very small amount of GMs translocated to organs, with a total accumulation of 1.29 ± 0.43%, 2.3 ± 0.48% and 4.46 ± 0.61% at 12, 24 and 24 h*, respectively. | ||
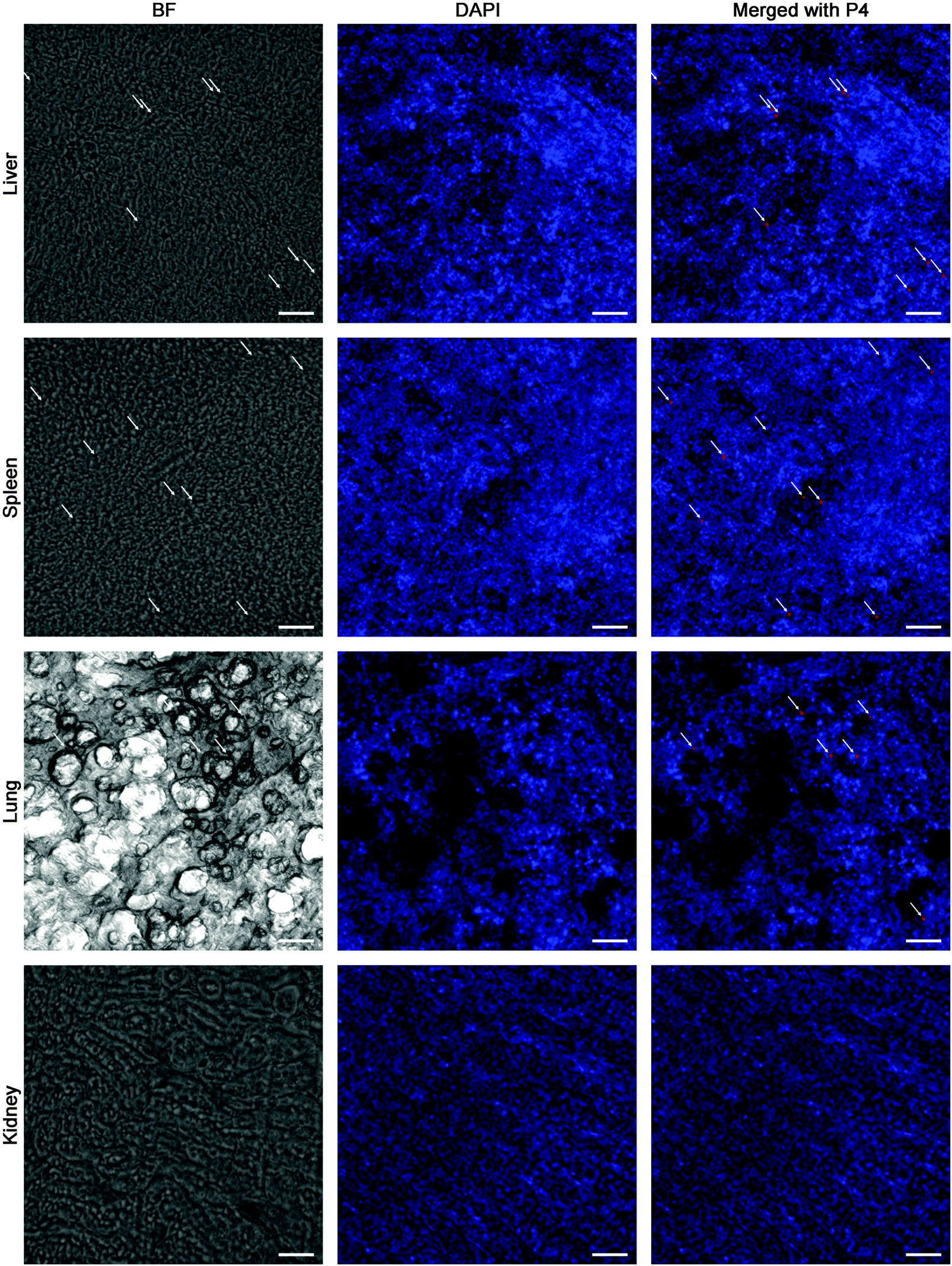 | ||
| Fig. 5 Frozen section of RES-rich organs. The rat was dosed with interiorly solidified P4-GMs every day for 3 days, then 24 h after the final administration, the organs were isolated and observed. Scale bar = 100 μm. | ||
Transport study using the in vitro FAE model
The in vitro FAE model consisting of Caco-2 and Raji cells has been widely used to investigate the absorption mechanism by M cells.34 Due to the limitation of the in vivo study in which it is very difficult to capture the interaction of GMs with bio-tissues, we use an in vitro model. Herein, a modified in vitro FAE model was used to simulate the physiological structure of PPs by adding macrophages to the BL side of Caco-2/Raji cell co-culture monolayers. From the AP to BL side of this model, there are closely packed Caco-2 monolayers and sporadically dispersed Raji cells (stained green) to the AP side (Fig. 6A), whereas to the BL side J774A.1 cells can be observed adhering to Caco-2/Raji cell monolayers (Fig. 6B), representing the existence of phagocytes in the BL pocket of FAE. After instillation with P4-labeled GMs to the AP side and incubation for 4 h, GMs are found phagocytosed by J774A.1 cells (Fig. 6C). GMs can be clearly identified in the phagocytes, rather than in Raji cells, which indicates that the transportation by Raji cells may be very quick. The transport rate of GMs in various cell monolayers is compared (Fig. 6D). Transport across Caco-2 monolayers is negligible, confirming that the trans-enterocyte pathway is not favorable for particles as large as GMs (2–5 μm). When Caco-2/Raji monolayers are used, the overall transport of GMs is significantly enhanced, highlighting the contribution of Raji cells. In comparison with the results in Caco-2 monolayers, a mechanism of receptor-mediated transport can be proposed for GMs. Although the addition of macrophages to the BL side accelerates the initial stage of trans-monolayer transport, the overall transport profile is similar to that without macrophages. Besides, the particle transportation is temperature-dependent, confirming energy-consuming active transportat mechanisms.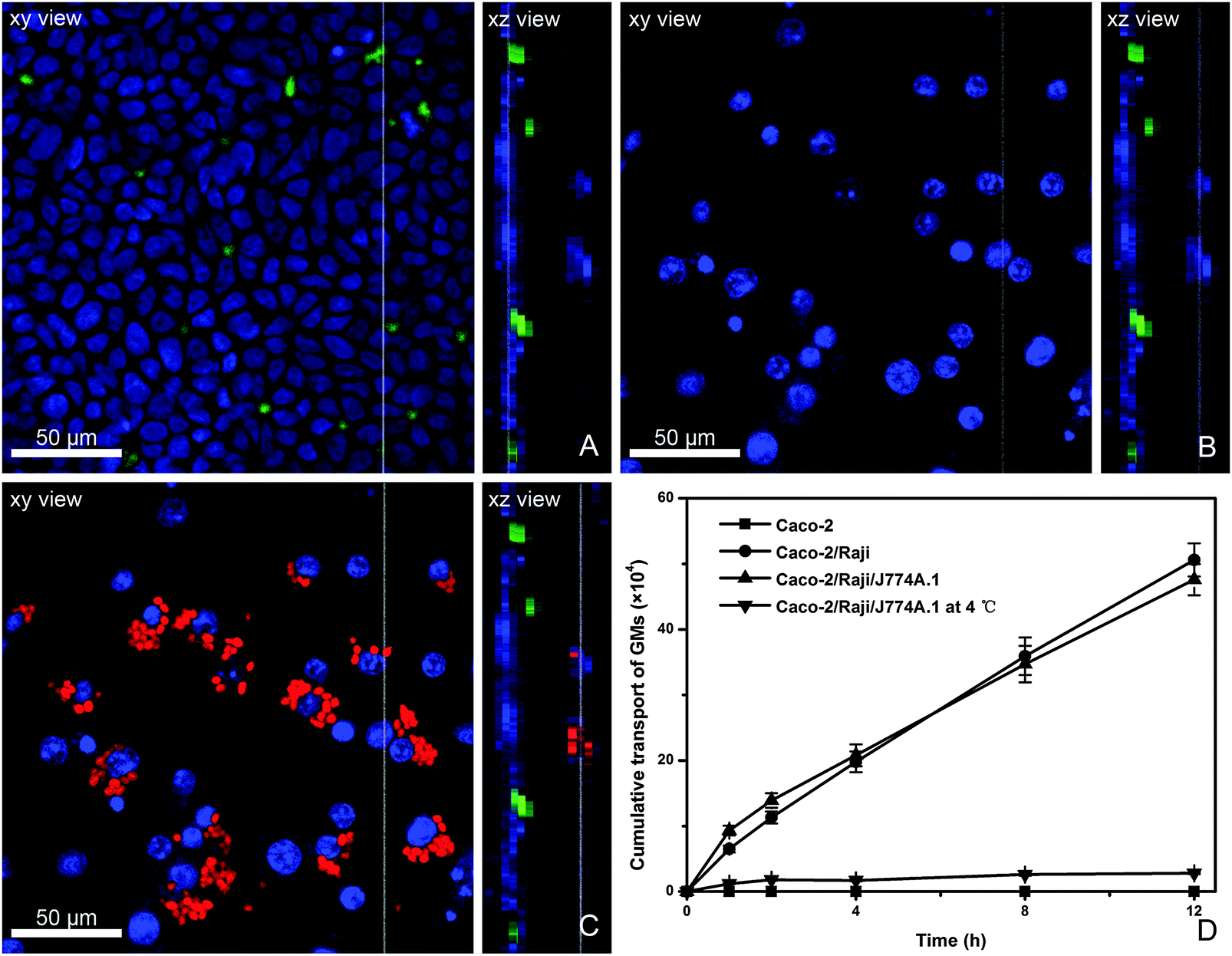 | ||
| Fig. 6 Display of the Caco-2/Raji/J774A.1 co-culture cell model. The tight Caco-2 monolayer was planted in the apical side (blue nuclei) with Raji cells (green color) inserted (A). The scattered J774A.1 cells were in the basolateral side (blue nuclei) below the other cells (B). The red channel was added, revealing the transport and phagocytosis of P4-labeled GMs (C). The transport profile of P4-GMs labeled GMs across different cell monolayers (D). | ||
Discussion
Although the M cell pathway for delivery of particulates has a long history,35 it remains to be the research focus not only because it is to date the most reliable way for entry of particulates, but also because the underlying mechanisms have not been fully understood yet. Earlier work managed to identify transportation of PLGA microspheres smaller than 5 μm by efferent lymphatics and retention of those with a diameter of 5 μm.36 With the help of fluorescent tools and histological examination, the transcytosis of β-glucan microparticles via M cells were captured.22 An in-depth investigation revealed the in vivo fate of glucan microparticles and PLGA nanoparticles in gut-associated lymphoid tissues, which accumulated and remained in dendritic cells.33 The most significant contribution of previous works is the presentation of evidence of uptake and transportation by M cells. However, the post-PP fate of orally absorbed particles has been seldom explored, which however plays crucial roles in deciphering the mechanisms of both pharmacological and toxicological actions. The results of this study provides a sketch of the in vivo fate of GMs from the GI tract to terminal organs with emphasis on tracking the translocation of the particles per se.In contrary to protocols of particle labeling using conventional probes,22,33 we use novel NIR fluorescent ACQ probes, which quench upon release from the particles and thus are able to represent the integral particles. Fig. S6 (ESI†) is a CLSM image of P4-labelled GMs. The key point is that what we observed as fluorescent dots can be regarded as the GMs themselves. Of course, GM-bound fluorescence should be kept stable during in vivo translocation. This has been well justified by the fluorescence stability study in simulated media, e.g. buffers and bio-relevant media (Fig. S1 and S2, ESI†). Based on the above assumption, the scenario of translocation of GMs can be roughly delineated.
The story begins when GMs are orally administered into the stomach by gavage. It is obvious that GMs can remain in the GI tract for at least 8 h. Semi-quantitative analysis indicates a gradual decrease of fluorescence after 4 h, which is an indicator of excretion, maybe through feces, and more importantly, absorption. It is strange to see a gradual increase of total fluorescence within the first 2 h, which nevertheless can be explained by the assumption that GM beads in a concentrated state may undercount total fluorescence as within 2 h an obvious retention of GMs in the stomach is seen. It is well accepted that PPs are commonly distributed in the ileum,37 a site that coincidentally finds a greater residence of GMs in this study (Fig. 1C). This provides more opportunities for absorption of particulates through the M cell route. The result of the intestine loop experiment that the ileum-, but not jejunum-, associated fluorescent ARE decrease confirms the above assumption, and is further strengthened by the facts of the uptake of GMs by M cells and translocation to PPs as obtained by identifying GMs in tissue slices. The finding that GMs accumulate in sub-FAE domains of PPs is in line with results by Jesus et al.,33 while GMs found in the lamina propria are prone to translocating to the systemic circulation.
A rough scanning by live imaging excludes any sign of distribution of GMs to tissues other than the abdominal region up to 12 h, which means that peripheral distribution, albeit may exist, must be at levels below the detection limit. Dissection to expose the bowel helps to increase the sensitivity of detection, and fluorescence can be observed in RES organs like the liver, lung, spleen and kidney 12 h after a single dose. Histological examination identifies GMs in the liver, lung and spleen, strongly supporting translocation of GMs to these organs. Together with the 24 h accumulation tendency, it is summarized that: (1) the translocation of GMs to RES organs must be via lymphatics because it complies with the slow flow rate of the lymph; (2) the redistribution of GMs aims at tissues rich in macrophages; (3) GMs have exceptional robustness to withstand damage by the physiological environment as well as intracellular lysosomal enzymes. Although at the current stage of this study we cannot outline the whole picture of trans-lymphatic transportation yet, the translocation pathway via M cells to lymphatics and then to the circulation is verified with direct and solid evidence.
Another hard yet very important issue concerning the M cell pathway is how to evaluate its overall contribution to oral bioavailability. Conventional pharmacokinetics give highly variable bioavailability ranging from less than 1% to as high as 7%.35 However, owing to limitations of the assaying tools used, this issue needs to be verified repeatedly whenever innovative strategies are available. The use of ACQ probes provides a nice tool to estimate the overall bioavailability of the particles administered simply by monitoring fluorescence. To compensate for the undercounting of fluorescence from biological samples, the initial (0.5 h) total fluorescence of ex vivo samples of the whole GI tract is taken as absolute fluorescence. On this basis, the estimated accumulation of GMs in the four organs (liver, lung, spleen and kidney) accounts to approximately 1.3% and 2.3% at 12 h and 24 h, respectively. Repeated administration for three consecutive days augments the total accumulation to as high as 4.5%. However, it should be noted that this figure is quite conservative because there is a possibility that GMs residing in tissues other than RES organs hasn't been taken into account.
Though the uptake of particles by M cells and transportation to sub-FAE lymphoid tissues is very quick, GMs accumulate and reside there for a sustained time.37,38 It implies that there might be saturable mechanisms governing the overall translocation extent and rate. Since it is difficult to observe this phenomenon in vivo, we study it in simulated in vitro cell lines by mimicking the architecture of FAE. Negligible transport across Caco-2 monolayers explains that GMs neither target Caco-2 cell lines nor are taken up through non-specific endocytosis mechanisms. The comparable total transport across both Caco-2/Raji and Caco-2/Raji/J774A.1 monolayers implies phagocytosis by macrophages is not the rate-limiting process. It seems that GMs have been quickly taken up by macrophages, and this possibly contributes to the slightly higher transportation rate at the initial stages. Meanwhile, the phagocytosis phenomenon clearly confirms that there is a hand-over from M cells (Raji) to macrophages (J774A.1). However, it should be noted that the results do not necessarily prove the co-existence of a process of exocytosis that makes up the total transportation. The virtual process may involve a quick saturation of macrophages and subsequent retention for a prolonged time. Actually, the macrophages contribute little to late stage trans-monolayer transport. It should be kept in mind the drawbacks of the simulated FAE model when being used to explain in vivo results. Exactly underneath the FAE of PPs, lymphocytes may exist in relative compactness and do not allow free para-cellular passage of GMs; the uptake and retention of particles in sub-FAE domains (Fig. 3C) possibly work as a deposit to amass particles. The substantially delayed translocation of the particles to the circulation surely creates a barrier to systemic drug delivery, which however is beneficial for oral mucosal immunization.
In order to achieve therapeutic levels of absorption of particulates, several barriers within the GI tract should be circumvented. First of all, there is the lumen-to-FAE barrier, comprising of the mucous layer to the lumen side and the FAE. Due to the small population of M cells,37–39 enhancement of mucus penetration and pre-ileum residence time sounds plausible, but should be controlled under the saturation level. Beyond uptake and hand-over by M cells, particulates accumulate within sub-FAE domains, forming the second barrier. It is hypothesized that designing escape strategies may work out. If the particles have been successfully transported through the lymphatics to the circulation, there happens to be the third barrier of redistribution to organs and tissues. It can be easily deduced that most of the particles might be delivered to macrophage-enriched organs such as those in RES. Triggering release or surface modification to divert the particles to targets should be taken into consideration.
In general, there are still limited weapons within the arsenal to fight barriers for oral absorption of particulates. However, detailed understanding of the translocation mechanisms of particulates in vivo will facilitate development in this area. Difficult as it is, it also creates challenges and opportunities for future study.
Conclusions
GMs can be transported to the posterior intestine in 2 h, and reside throughout the whole GI tract for an extended time as long as over 8 h. The ileum, rather than the jejunum, is the main absorption site for GMs due to the abundance of M cells in this intestinal segment. Histological examination by identification of GMs as red fluorescent dots confirms uptake and hand-over of GMs to sub-FAE lymphocytes, as well as transportation to lamina propria. Translocation of GMs to various RES organs appears at 12 h and augments at 24 h with about 1.3% and 2.3% distribution of the particles to the four RES organs, liver, lung, spleen and kidney, in total. Histological examination also reveals the existence of GMs in either the liver or lung or spleen. Voracious uptake of GMs by macrophages can be observed. Trans-monolayer transport in a modified tri-culture Caco-2/Raji/J774A.1 cell model indicated the comparable trans-monolayer transport extent and rate to Caco-2/Raji cell monolayers, but only negligible transport for Caco-2 cell monolayers. It is summarized that GMs can be transported by the M cell pathway and translocated to RES organs.Acknowledgements
This study was financially supported in part, by Shanghai Commission of Science and Technology (14JC1490300), National Natural Science Foundation of China (81573363, 21372063), and National Basic Research Program of China (2015CB931800).References
- P. J. Sansonetti and A. Phalipon, Semin. Immunol., 1999, 11, 193–203 CrossRef CAS PubMed.
- M. R. Neutra, E. Pringault and J. P. Kraehenbuhl, Annu. Rev. Immunol., 1996, 14, 275–300 CrossRef CAS PubMed.
- M. R. Neutra, A. Frey and J. P. Kraehenbuhl, Cell, 1996, 86, 345–348 CrossRef CAS PubMed.
- P. N. Gupta, K. Khatri, A. K. Goyal, N. Mishra and S. P. Vyas, J. Drug Targeting, 2007, 15, 701–713 CrossRef CAS PubMed.
- T. E. Rajapaksa, M. Stover-Hamer, X. Fernandez, H. A. Eckelhoefer and D. D. Lo, J. Controlled Release, 2010, 142, 196–205 CrossRef CAS PubMed.
- H. Długońska and M. Grzybowski, Ann. Parasitol., 2012, 58, 1–8 Search PubMed.
- S. H. Kim, K. Y. Lee and Y. S. Jang, Immune Netw., 2012, 12, 165–175 CrossRef PubMed.
- S. H. Kim and Y. S. Jang, Exp. Mol. Med., 2014, 46, e85 CrossRef CAS PubMed.
- C. Damge, M. Aprahamian, W. Humbert and M. Pinget, J. Pharm. Pharmacol., 2000, 52, 1049–1056 CrossRef CAS PubMed.
- F. Cui, K. Shi, L. Zhang, A. Tao and Y. Kawashima, J. Controlled Release, 2006, 114, 242–250 CrossRef CAS PubMed.
- M. A. Clark, M. A. Jepson and B. H. Hirst, Adv. Drug Delivery Rev., 2001, 50, 81–106 CrossRef CAS PubMed.
- P. J. Giannasca, K. T. Giannasca, A. M. Leichtner and M. R. Neutra, Infect. Immun., 1999, 67, 946–953 CAS.
- M. K. Yoo, S. K. Kang, J. H. Choi, I. K. Park, H. S. Na, H. C. Lee, E. B. Kim, N. K. Lee, J. W. Nah, Y. J. Choi and C. S. Cho, Biomaterials, 2010, 31, 7738–7747 CrossRef CAS PubMed.
- X. Zhang, J. Qi, Y. Lu, W. He, X. Li and W. Wu, Nanomedicine, 2014, 10, 167–176 CAS.
- E. M. Pridgen, F. Alexis, T. T. Kuo, E. Levy-Nissenbaum, R. Karnik, R. S. Blumberg, R. Langer and O. C. Farokhzad, Sci. Transl. Med., 2013, 5, 213ra167 CrossRef PubMed.
- H. B. Huang, G. R. Ostroff, C. K. Lee, S. Agarwal, S. Ram, P. A. Rice, C. A. Specht and S. M. Levitz, J. Immunol., 2012, 189, 312–317 CrossRef CAS PubMed.
- G. D. Brown and S. Gordon, Nature, 2001, 413, 36–37 CrossRef CAS PubMed.
- N. Rochereau, D. Drocourt, E. Perouzel, V. Pavot, P. Redelinghuys, G. D. Brown, G. Tiraby, X. Roblin, B. Verrier, C. Genin, B. Corthésy and S. Paul, PLoS Biol., 2013, 11, e1001658 CAS.
- M. Aouadi, G. J. Tesz, S. M. Nicoloro, M. Wang, M. Chouinard, E. Soto, G. R. Ostroff and M. P. Czech, Nature, 2009, 458, 1180–1184 CrossRef CAS PubMed.
- G. J. Tesz, M. Aouadi, M. Prot, S. M. Nicoloro, E. Boutet, S. U. Amano, A. Goller, M. Wang, C. A. Guo, W. E. Salomon, J. V. Virbasius, R. A. Baum, M. J. J. O'Connor, E. Soto, G. R. Ostroff and M. P. Czech, Biochem. J., 2011, 436, 351–362 CrossRef CAS PubMed.
- E. R. Soto and G. R. Ostroff, Bioconjugate Chem., 2008, 19, 840–848 CrossRef CAS PubMed.
- R. De Smet, T. Demoor, S. Verschuere, M. Dullaers, G. R. Ostroff, G. Leclercq, L. Allais, C. Pilette, M. Dierendonck, B. G. De Geest and C. A. Cuvelier, J. Controlled Release, 2013, 172, 671–678 CrossRef CAS PubMed.
- H. Huang, G. R. Ostroff, C. K. Lee, C. A. Specht and S. M. Levitz, Clin. Vaccine Immunol., 2013, 20, 1585–1591 CrossRef CAS PubMed.
- E. R. Soto, A. C. Caras, L. C. Kut, M. K. Castle and G. R. Ostroff, J. Drug Delivery, 2012, 143524 Search PubMed.
- F. Garello, R. Stefania, S. Aime, E. Terreno and D. D. Castelli, Mol. Pharmaceutics, 2014, 11, 3760–3765 CrossRef CAS PubMed.
- X. Zhang, Y. Y. Zhao, Y. Xu, Y. Pan, F. Chen, A. Kumar, G. Zou and X. J. Liang, J. Mater. Chem. B, 2014, 2, 5882–5890 RSC.
- C. Casteleyn, W. Van den Broeck, A. Gebert, B. R. Tambuyzer, S. Van Cruchten and C. Van Ginneken, Comp. Immunol. Microbiol. Infect. Dis., 2013, 36, 353–364 CrossRef PubMed.
- X. Hu, J. Zhang, Z. Yu, Y. Xie, H. He, J. Qi, X. Dong, Y. Lu, W. Zhao and W. Wu, Nanomedicine, 2015, 11, 1939–1948 CAS.
- A. des Rieux, V. Fievez, I. Theate, J. Mast, V. Preat and Y. J. Schneider, Eur. J. Pharm. Sci., 2007, 30, 380–391 CrossRef CAS PubMed.
- K. Miyazawa, T. Kanaya, I. Takakura, S. Tanaka, T. Hondo, H. Watanabe, M. T. Rose, H. Kitazawa, T. Yamaguchi, S. Katamine, N. Nishida and H. Aso, J. Virol., 2010, 84, 12285–12291 CrossRef CAS PubMed.
- M. Niu, Y. Tan, P. Guan, L. Hovgaard, Y. Lu, J. Qi, R. Lian, X. Li and W. Wu, Int. J. Pharm., 2014, 460, 119–130 CrossRef CAS PubMed.
- X. Hu, W. Fan, Z. Yu, Y. Lu, J. Qi, J. Zhang, X. Dong, W. Zhao and W. Wu, Nanoscale, 2016 10.1039/C5NR07474F.
- M. De Jesus, G. R. Ostroff, S. M. Levitz, T. R. Bartling and N. J. Mantis, PLoS One, 2014, 9, e91002 Search PubMed.
- E. Gullberg, M. Leonard, J. Karlsson, A. M. Hopkins, D. Brayden, A. W. Baird and P. Artursson, Biochem. Biophys. Res. Commun., 2000, 279, 808–813 CrossRef CAS PubMed.
- A. T. Florence, Pharm. Res., 1997, 14, 259–266 CrossRef CAS.
- J. H. Eldridge, C. J. Hammond, J. A. Meulbroek, J. K. Staas, R. M. Gilley and T. R. Tice, J. Controlled Release, 1990, 11, 205–214 CrossRef CAS.
- D. J. Brayden, M. A. Jepson and A. W. Baird, Drug Discovery Today, 2005, 10, 1145–1157 CrossRef CAS PubMed.
- A. des Rieux, V. Fievez, M. Garinot, Y. J. Schneider and V. Préat, J. Controlled Release, 2006, 116, 1–27 CrossRef CAS PubMed.
- H. Miller, J. Zhang, R. KuoLee, G. B. Patel and W. Chen, World J. Gastroenterol., 2007, 13, 1477–1486 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb02706c |
| This journal is © The Royal Society of Chemistry 2016 |