
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of intermediate sites on bulk diffusion barriers: Mg intercalation in Mg2Mo3O8†
Gopalakrishnan Sai
Gautam‡
 abc,
Xiaoqi
Sun‡
d,
Victor
Duffort
d,
Linda F.
Nazar
d and
Gerbrand
Ceder
*bc
abc,
Xiaoqi
Sun‡
d,
Victor
Duffort
d,
Linda F.
Nazar
d and
Gerbrand
Ceder
*bc
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
bMaterials Science Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: gceder@berkeley.edu; gceder@lbl.gov
cDepartment of Materials Science and Engineering, University of California Berkeley, CA 94720, USA
dDepartment of Chemistry, Waterloo Institute of Nanotechnology, University of Waterloo, ON N2L3G1, Canada. E-mail: lfnazar@uwaterloo.ca
First published on 18th October 2016
Abstract
The ongoing search for high voltage positive electrode materials for Mg batteries has been primarily hampered by poor Mg mobility in bulk oxide frameworks. Motivated by the presence of Mo3 clusters that can facilitate charge redistribution and the presence of Mg in a non-preferred (tetrahedral) coordination environment, we have investigated the Mg (de)intercalation behavior in layered-Mg2Mo3O8, a potential positive electrode. While no electrochemical activity is observed, chemical demagnesiation of Mg2Mo3O8 is successful but leads to amorphization. Subsequent first-principles calculations predict a strong thermodynamic driving force for structure decomposition at low Mg concentrations and high activation barriers for bulk Mg diffusion, in agreement with experimental observations. Further analysis of the Mg diffusion pathway reveals an O–Mg–O dumbbell intermediate site that creates a high Mg2+ migration barrier, indicating the influence of transition states on setting the magnitude of migration barriers.
Rechargeable Mg batteries have received interest as an energy storage system that potentially offers high energy density. The major advantage relies on the benefits of Mg metal as the negative electrode, which, in addition to being inexpensive, abundant and safe in handling and storage, also provides high volumetric capacity (3833 mA h cm−3) and can be free of dendrite growth when operating in an electrochemical cell.1–3 However, the development of corresponding positive electrode materials has been slow.2 Since the discovery of the first seminal functional Mg insertion positive electrode – the Chevrel phase (CP, Mo6S8),4 only recently have two other structures been shown to be suitable for Mg (de)intercalation in a full cell arrangement with a Mg anode, namely the spinel and layered titanium sulfide.5,6 The above materials take advantage of a “soft” anionic framework that interacts weakly with the Mg2+ and assists its mobility. In contrast, sluggish multivalent ion mobility is generally observed in oxide lattices. Nevertheless, oxides are still of great interest due to their potentially higher operating voltage.7–15
Levi et al. have speculated that the presence of Mo6 clusters in the CP structure is one of the key factors for facile Mg2+ mobility by the promotion of charge redistribution.8 The possibility that a similar principle may apply to oxides guided us to Mg2Mo3O8 (Fig. 1a), which has Mo3 clusters in the Mo3O8 layers (Fig. 1b).16,17 In this structure, Mg occupies both octahedral and tetrahedral sites between the layers. While octahedral Mg shares both edges and corners with MoO6 octahedra (Fig. 1a), tetrahedral Mg shares corners with MoO6 and MgO6 octahedra. Since Mg is also present in a “non-preferred” tetrahedral coordination (Fig. 1a),18 a Mg diffusion pathway with lower migration barriers is expected than when Mg is exclusively found in its preferred octahedral coordination,9 such as in conventional layered oxides. In the case of layered oxides, the Mg diffusion pathways contain an intermediate tetrahedral site presumably with high energy relative to the stable octahedral site, leading to poor Mg mobility.9
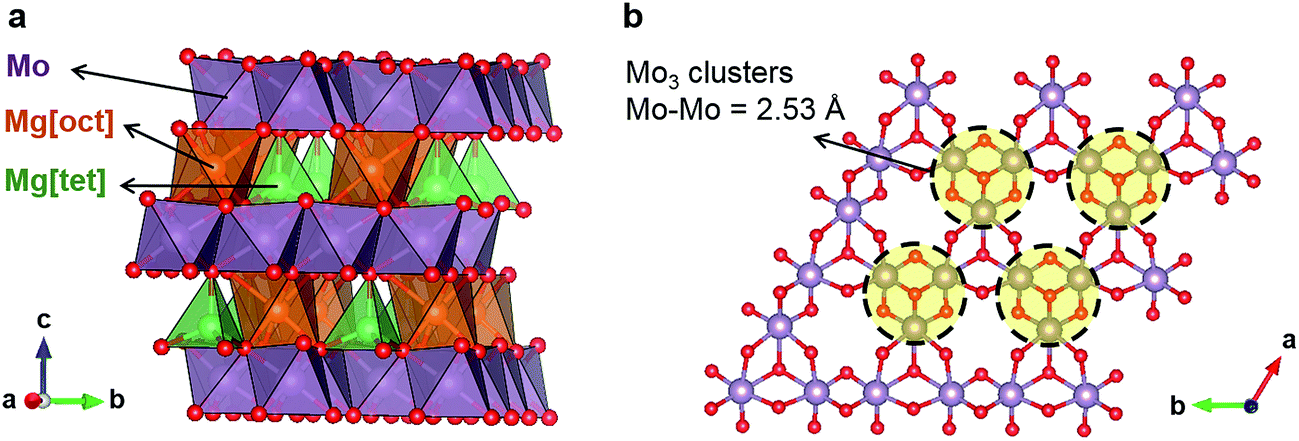 | ||
| Fig. 1 (a) Crystal structure of Mg2Mo3O8. (b) MoO6 octahedra layer showing Mo3 clusters. | ||
We note that the Li analogue (Li4Mo3O8) has previously been examined in a Li cell, offering an initial specific capacity of 218 mA h g−1.19 Other materials with similar structures containing Mo3 clusters, such as LiMoO2 and Li2MoO3, also function well as Li-ion positive electrodes.19–21 On the other hand, only limited work has been done on Mg intercalation in Mo-oxides,12,22,23 motivating us to examine the Mg2+ diffusion properties in Mg2Mo3O8 and its potential to be a positive electrode material for Mg batteries.
Mg2Mo3O8 was obtained by solid-state synthesis (see the ESI†), which provided particles of a few micrometers in size (Fig. 2a). Its X-ray diffraction (XRD) pattern was indexed to the P63mc space group characteristic of this material (Fig. 2b). In order to study the possibility of Mg removal from such a structure, chemical demagnesiation was carried out using NO2BF4, a commonly used oxidizing agent for chemical delithiation.24 Mg2Mo3O8 and NO2BF4 were reacted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio, which would allow complete Mg de-intercalation if each NO2BF4 sustained a one electron reduction as anticipated. Energy dispersive X-ray spectroscopy (EDX) reveals that the majority of the Mg was removed from the structure (Table 1). The particles become smaller after demagnesiation (Fig. 2c), suggesting some changes in the material. Despite these differences, the XRD results indicate no shift of the peaks (Fig. 2d). The atomic positions obtained by Rietveld refinement25 are almost the same as those of the pristine (Table S1 in the ESI†), suggesting that a two-phase reaction takes place, with the demagnesiated phase being amorphous. During this process, Mg is presumably first removed from the outer shell, leading to the destabilization of the parent lattice and eventual amorphization. The amount of the amorphous phase in the demagnesiated product is estimated to be around 87 wt% using Si as an external standard (Fig. 2d, see ESI† for details), giving an overall composition of Mg0.24Mo3O8, which is similar to the cationic ratio determined by EDX (Table 1) and indicates complete demagnesiation of the amorphous component. The ∼13 wt% unreacted Mg2Mo3O8 results from the reduced oxidizing strength of NO2BF4 exhibited near the end of the reaction due to low oxidizer concentration, or other side reactions. Partial demagnesiation of Mg2Mo3O8 was not achieved when the ratio of the oxidizing agent was reduced (Mg2Mo3O8:NO2BF4 = 1:2), as indicated by the preservation of the initial phase obtained by XRD refinement (Fig. S1a and Table S1c†). Together with the decrease of the overall Mg concentration (Mg/Mo ratio of ∼0.53(4)/3 by EDX) and the co-existence of different morphologies (Fig. S1b†), the XRD data suggest that part of Mg2Mo3O8 undergoes complete demagnesiation and becomes amorphous with some fraction of material not participating in the reaction.
:4 ratio, which would allow complete Mg de-intercalation if each NO2BF4 sustained a one electron reduction as anticipated. Energy dispersive X-ray spectroscopy (EDX) reveals that the majority of the Mg was removed from the structure (Table 1). The particles become smaller after demagnesiation (Fig. 2c), suggesting some changes in the material. Despite these differences, the XRD results indicate no shift of the peaks (Fig. 2d). The atomic positions obtained by Rietveld refinement25 are almost the same as those of the pristine (Table S1 in the ESI†), suggesting that a two-phase reaction takes place, with the demagnesiated phase being amorphous. During this process, Mg is presumably first removed from the outer shell, leading to the destabilization of the parent lattice and eventual amorphization. The amount of the amorphous phase in the demagnesiated product is estimated to be around 87 wt% using Si as an external standard (Fig. 2d, see ESI† for details), giving an overall composition of Mg0.24Mo3O8, which is similar to the cationic ratio determined by EDX (Table 1) and indicates complete demagnesiation of the amorphous component. The ∼13 wt% unreacted Mg2Mo3O8 results from the reduced oxidizing strength of NO2BF4 exhibited near the end of the reaction due to low oxidizer concentration, or other side reactions. Partial demagnesiation of Mg2Mo3O8 was not achieved when the ratio of the oxidizing agent was reduced (Mg2Mo3O8:NO2BF4 = 1:2), as indicated by the preservation of the initial phase obtained by XRD refinement (Fig. S1a and Table S1c†). Together with the decrease of the overall Mg concentration (Mg/Mo ratio of ∼0.53(4)/3 by EDX) and the co-existence of different morphologies (Fig. S1b†), the XRD data suggest that part of Mg2Mo3O8 undergoes complete demagnesiation and becomes amorphous with some fraction of material not participating in the reaction.
 | ||
Fig. 2 (a) SEM image and (b) the Rietveld refinement fit of pristine Mg2Mo3O8 (Bragg-Brentano geometry). (c) SEM image of the demagnesiated sample. (d) Rietveld refinement of the demagnesiated sample with the external silicon standard added to evaluate the percentage of the amorphous phase. The mixture was sealed in an X-ray capillary under Ar and was measured in a Debye–Scherrer geometry. Black crosses – experimental data, red lines – fitted data, blue line – difference map between observed and calculated data, green ticks – the P63mc phase of Mg2Mo3O8, pink ticks – the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m phase of Si. m phase of Si. | ||
| Sample | Pristine | Demagnesiated |
|---|---|---|
| Mg/Mo | 1.59(4)/3 | 0.13(6)/3 |
Since the degree of chemical oxidation was hard to control, we attempted to evaluate stepwise the demagnesiation behavior by an electrochemical method. As it has been suggested that the Mg desolvation process depends on the solvent,26,27 and this is critical for the electrochemical mechanism at the positive electrode,28,29 Mg2Mo3O8 was examined in both non-aqueous (all phenyl complex – APC30) and aqueous (Mg(ClO4)2 in water) systems. A demagnesiation voltage similar to the delithiation voltage of Li4Mo3O8 (average of ∼2.4 V vs. Mg),19 or at ∼2.6 V as predicted by first principles calculations (Fig. S4, see the ESI† for details) could be expected. Both electrolytes offer a stable voltage window for this range; however, no electrochemical activity was observed in either system (Fig. S2†). Such results potentially indicate the existence of a high Mg2+ diffusion barrier in the structure, hence kinetics being the main limitation. Chemical oxidation, on the other hand, might involve a mechanism other than simple cation diffusion, such as a partial dissolution/re-precipitation process. This helps in lowering the kinetic barrier and establishes successful Mg removal.
In order to understand the amorphization upon chemical demagnesiation and rationalize the lack of electrochemical activity in Mg2Mo3O8, we carried out first principles calculations to determine the energy above hull (Ehull) indicating the stability of the structure, and the activation barriers for Mg diffusion within the structure (methodological details of the calculations are provided in the ESI†).
The energy above the convex ground state hull (Ehull) of the MgXMo3O8 structure, calculated with respect to the stable compounds in the Mg–Mo–O ternary phase diagram, can be used to evaluate the thermodynamic stability of the structure on demagnesiation.13,15 Typically, a thermodynamically stable structure will have an Ehull of 0 meV per atom, while more positive Ehull values indicate a greater driving force to form other phases, which may be reflected as a difficulty in synthesizing a compound, or as decomposition during (de)intercalation. Also, Ehull values are evaluated at 0 K and entropic contributions can stabilize a structure at higher temperatures. The values listed in Table 2 have been determined from the available compounds in the Materials Project database.31 The trends in Table 2 indicate an increasing Ehull with increasing Mg removal from the Mg2Mo3O8 structure, corresponding to an increase in the thermodynamic driving force for decomposition. The Ehull values at lower Mg concentrations are very high – consistent with the experimentally observed amorphization during chemical Mg extraction from Mg2Mo3O8 (Fig. 2) and the naturally amorphous occurrence of Mo3O8.32,33
| Composition | E hull | Decomposition products | Comments |
|---|---|---|---|
| Mg2Mo3O8 | 51 | MoO2 + MgO | Chemically synthesizable |
| MgMo3O8 | 180 | MoO2 + MgMoO4 | — |
| Mo3O8 | 330 | MoO2 + Mo8O23 | Naturally amorphous32,33 |
To evaluate Mg mobility in the Mg2Mo3O8 structure, the possible Mg diffusion hops within the structure were determined. Being a layered structure, Mg2Mo3O8 can be visualized on a 2D-plane, as shown in Fig. 3, with octahedral Mo, tetrahedral Mg and octahedral Mg indicated by purple, green and orange triangles, respectively. The four possible Mg→Mg hops that can occur in the structure are illustrated by the black circle and arrows in Fig. 3. Three hops (black arrows) occur in the same Mg-plane and the fourth hop (black circle) moves Mg across a Mo-plane. The shortest hops (type 1 and 2) span ∼3.38 Å and ∼4.33 Å, respectively, and involve Mg migration from a tetrahedral site to an octahedral site (or vice versa), while hops 3 and 4 are ∼5.76 Å in distance and involve Mg jumps between similarly coordinated sites (oct → oct or tet → tet). Although hops 3 and 4 are direct between octahedral or tetrahedral Mg sites, they are likely to be constituted by two consecutive hops of type 1 (i.e. an oct → tet hop followed by a tet → oct hop and vice versa). Alternate routes for hops 3 and 4 are not possible due to intermediate Mg tetrahedral sites, which will face-share with MoO6 octahedra and experience strong electrostatic repulsions as a result. Hence, hops 1 and 2 are the relevant Mg migration pathways that need to be considered in calculations.
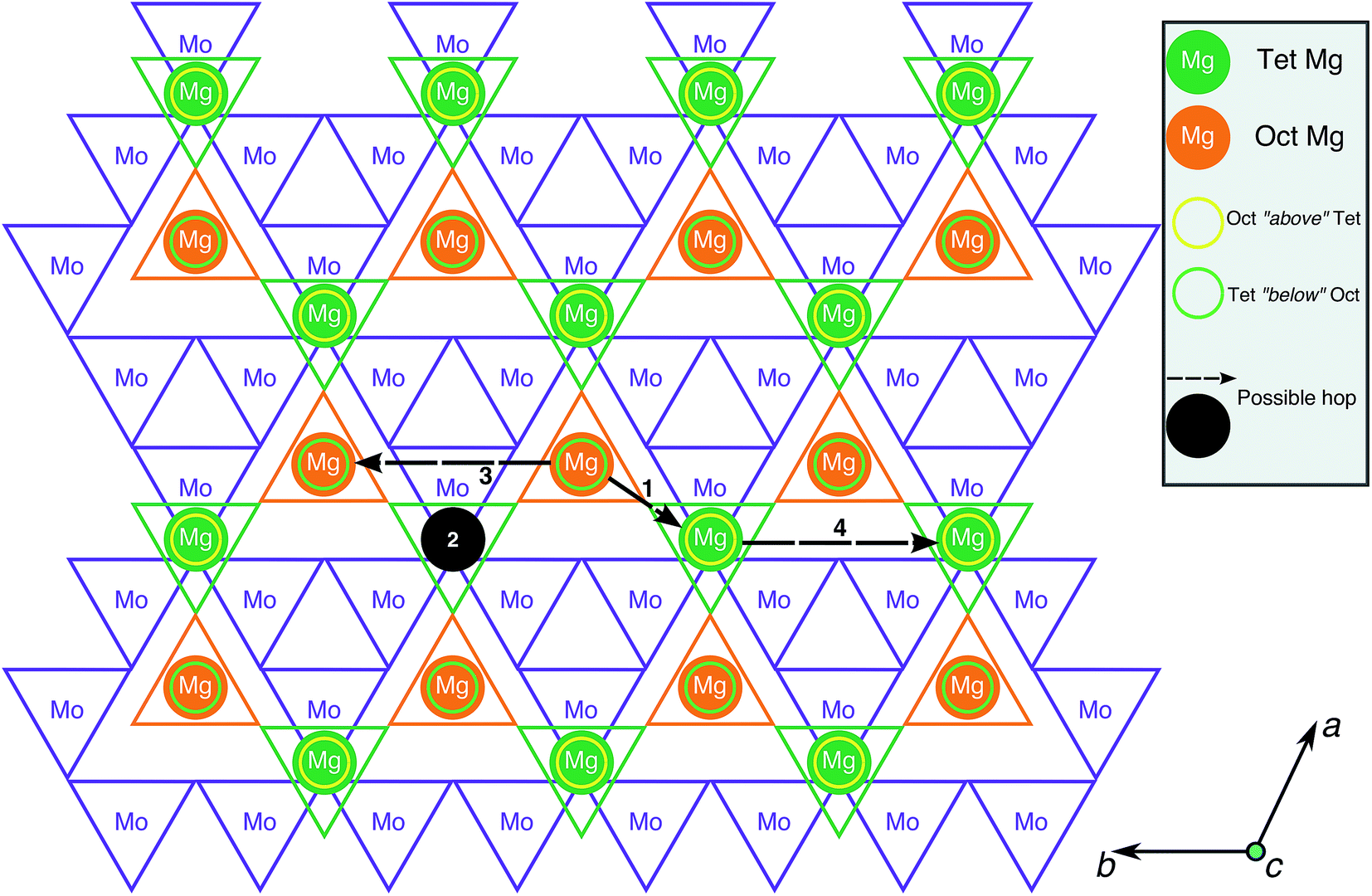 | ||
| Fig. 3 A 2D-view of the Mg2Mo3O8 structure perpendicular to the layer spacing direction (c-axis) is shown. Purple, green and orange triangles indicate MoO6 octahedra, Mg tetrahedra and Mg octahedra, respectively. The yellow and green circles correspond to octahedral and tetrahedral Mg atoms across a Mo-plane. The black circles and arrows indicate possible Mg→Mg hops within the structure. | ||
Fig. 4a displays the calculated Mg migration barriers (at xMg ∼ 2 with a dilute vacancy limit) along the hop 1 (black) and 2 (red) pathways, with the respective hop distances normalized on the x-axis. Both hops begin at a tetrahedral Mg and terminate at an octahedral Mg, explaining the difference in energy between the end points (∼250 meV). Notably, Mg mobility along both hops 1 and 2 is expected to be poor, given the large migration barriers (∼1200 meV and ∼2000 meV for hops 1 and 2, respectively), compared to the 525–650 meV required for bulk Mg mobility at reasonable rates.9 The high migration barriers also explain the lack of electrochemical activity observed. Nevertheless, if any Mg migration is observed in the structure, the Mg2+ ions are likely to diffuse along the in-plane hop 1 pathway.
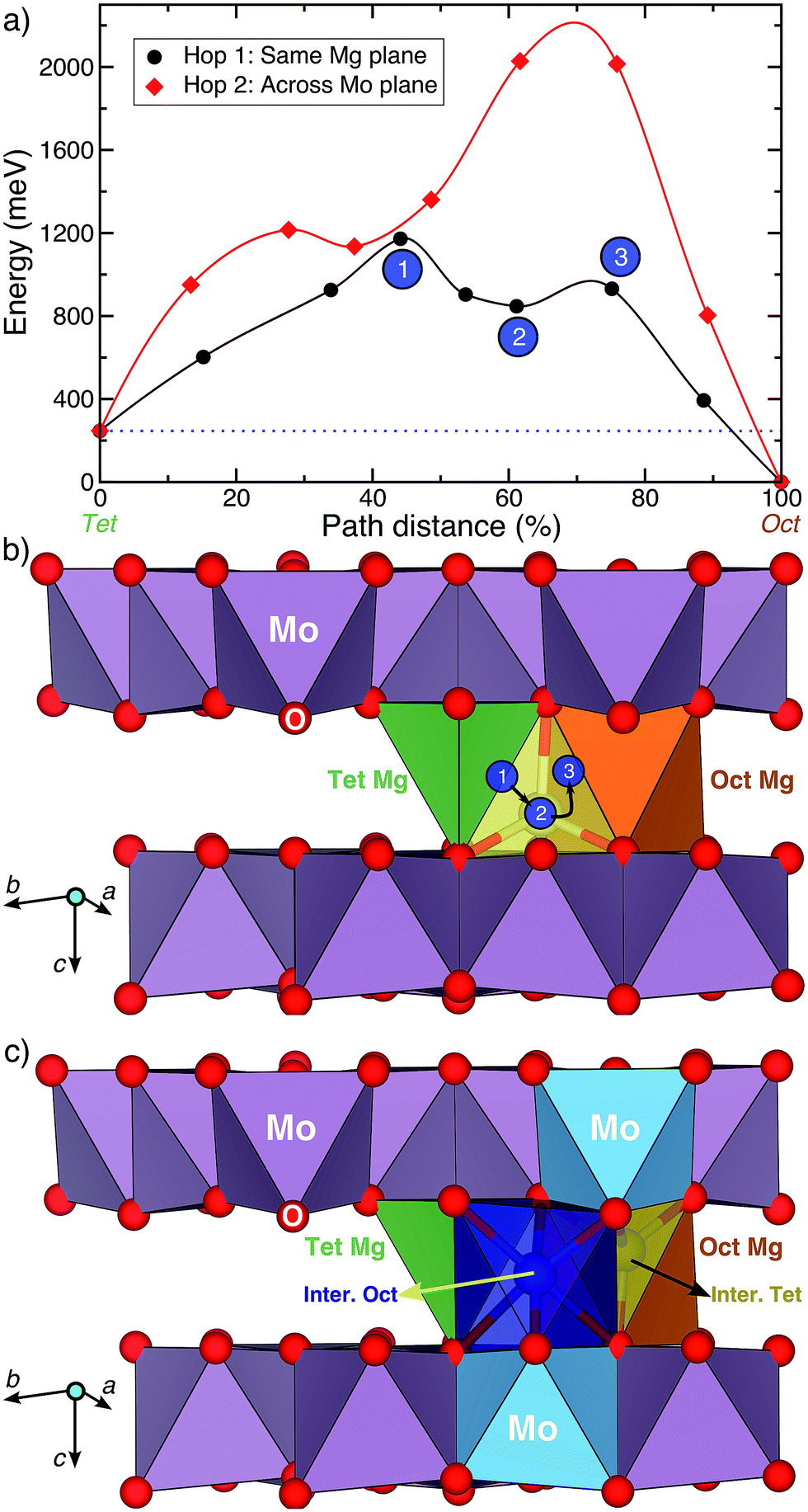 | ||
| Fig. 4 (a) The activation barrier for Mg diffusion along hops 1 and 2 in the Mg2Mo3O8 structure, with the normalized path distance on the x-axis. (b) A closer view of hop 1, where the numbered circles correspond to various intermediate sites along the hop as labeled in (a). The intermediate tetrahedral site, which is edge-sharing with the stable tetrahedral site (green), is indicated in yellow. (c) An alternate pathway for hop 1 that involves intermediate octahedral (dark blue) and tetrahedral (yellow) sites, which are face-sharing with the stable tetrahedral (green) and octahedral (orange) sites, respectively. The intermediate sites in (c) also share a face with the MoO6 octahedra (blue). | ||
While the high barrier for hop 2 is due to the strong electrostatic repulsion Mg experiences from Mo atoms as it passes through a triangular face of oxygen atoms across the Mo3O8 layer, a closer look into hop 1 is required to understand the large barriers. Visualization of the Mg migration along hop 1 is given in Fig. 4b, with intermediate sites and their respective energies (in Fig. 4a) indicated by the numbered circles. Sites 1, 2 and 3 respectively correspond to the O–Mg–O “dumbbell” configuration,9 the intermediate metastable tetrahedral site (yellow) and the triangular face between the intermediate tetrahedral and stable octahedral sites. While site 3 (triangular face, Fig. 4b) has an energy of ∼685 meV with respect to the tetrahedral site (similar to ∼600–800 meV observed in oxide spinels15), the magnitude of the barrier is determined by site 1, where Mg is situated along an O–O bond (edge of the stable tetrahedron), in a dumbbell configuration. Previous evaluations of Mg migration through an O–O dumbbell hop for layered NiO2 have reported high barriers (∼1400 meV),9 similar to the value reported in this work.
Although the O–Mg–O dumbbell hops are precluded from occurring in usual cathode materials9,10 due to the presence of alternate low-energy pathways, no such possibility exists for Mg migration in the Mg2Mo3O8 structure. For example, an alternate pathway for hop 1 that avoids the O–Mg–O dumbbell is shown in Fig. 4c. The intermediate octahedral (dark blue) and tetrahedral (yellow) sites in Fig. 4c share a triangular face with the stable tetrahedral (green) and octahedral (orange) sites, respectively. Additionally, each intermediate site also shares a triangular face with a MoO6 octahedron (blue polyhedron, Fig. 4c). While an intermediate Mg octahedron that face-shares with a higher valent transition metal octahedron need not preclude Mg migration, the intermediate tetrahedral site (yellow site, Fig. 4c) will experience much stronger electrostatic repulsion from the face-sharing MoO6 octahedron, subsequently increasing its energy and preventing any potential Mg migration. Indeed, Mg migration calculations initializing hop 1 as displayed in Fig. 4c relax to a path similar to the O–Mg–O hop (Fig. 4b) with a similar barrier (∼1150 meV, Fig. S5†). Notably, scenarios involving a Mg2+ ion diffusing through an intermediate (tetrahedral) site that face-shares with a transition metal polyhedron lead to high migration barriers in oxides (e.g., high Mg barriers in layered NiO2 (ref. 9)), while analogous trends have been observed for Li-diffusion in disordered rock-salt structures.34 Thus, the high Mg migration barrier in Mg2Mo3O8 can be attributed to the intermediate O–Mg–O dumbbell configuration, which occurs in the absence of alternate low energy pathways. This indicates the importance of intermediate sites along a diffusion path, determined by the specific topology of cation sites in an anion lattice, in addition to the occurrence of the mobile cation with a non-preferred coordination and a preferentially coordinated metastable site.9
One of the challenges towards the development of high energy density secondary Mg batteries is the design of an ideal positive electrode, which can reversibly intercalate Mg at a high voltage with high capacities at reasonable rates. The Mg2Mo3O8 structure used in this study was primarily motivated by the presence of Mo3 clusters (similar to the Mo6 clusters in the Chevrel-positive electrodes) and the occurrence of Mg in a non-preferred tetrahedral coordination (satisfying one of the design rules known in the literature9). While Mg could be chemically extracted from the structure, albeit with significant amorphization, no electrochemical activity was observed. Further analysis using first-principles calculations revealed high Ehull values (structural instability) at a low Mg content and high Mg migration barriers (poor bulk Mg mobility in the structure), explaining the aforementioned experimental observations. The high activation barrier for Mg diffusion in Mg2Mo3O8 arises from the O–Mg–O dumbbell hop, reflecting the impact of intermediate sites along a diffusion pathway besides cation coordination preferences. Thus, in the search for high Mg-mobility oxide positive electrodes, a careful analysis of the diffusion pathway and the topology of cation sites is advantageous – such as identifying low-energy intermediate sites – in addition to the requirement of Mg being found in a non-preferred coordination environment.9 Such understanding of Mg diffusion pathways will help to find suitable positive electrodes for multivalent batteries.
Acknowledgements
This work was fully supported by the Joint Center for Energy Storage Research (JCESR), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science and Basic Energy Sciences. The authors would like to thank the National Energy Research Scientific Computing Center (NERSC) for providing computing resources. Natural Sciences and Engineering Research Council of Canada (NSERC) is acknowledged by LFN for a Canada Research Chair.References
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 CAS.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, Energy Environ. Sci., 2016, 9, 2273–2277 CAS.
- X. Sun, P. Bonnick and L. F. Nazar, ACS Energy Lett., 2016, 1, 297–301 CrossRef CAS.
- G. Amatucci, F. Badway, A. Singhal, B. Beaudoin, G. Skandan, T. Bowmer, I. Plitz, N. Pereira, T. Chapman and R. Jaworski, J. Electrochem. Soc., 2001, 148, A940–A950 CrossRef CAS.
- E. Levi, M. Levi, O. Chasid and D. Aurbach, J. Electroceram., 2009, 22, 13–19 CrossRef CAS.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- G. S. Gautam, P. Canepa, A. Abdellahi, A. Urban, R. Malik and G. Ceder, Chem. Mater., 2015, 27, 3733–3742 CrossRef.
- S.-H. Bo, C. P. Grey and P. G. Khalifah, Chem. Mater., 2015, 27, 4630–4639 CrossRef CAS.
- G. Gershinsky, H. D. Yoo, Y. Gofer and D. Aurbach, Langmuir, 2013, 29, 10964–10972 CrossRef CAS PubMed.
- G. S. Gautam, P. Canepa, R. Malik, M. Liu, K. Persson and G. Ceder, Chem. Commun., 2015, 51, 13619–13622 RSC.
- G. S. Gautam, P. Canepa, W. D. Richards, R. Malik and G. Ceder, Nano Lett., 2016, 16, 2426–2431 CrossRef PubMed.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974 CAS.
- W. H. McCarroll, L. Katz and R. Ward, J. Am. Chem. Soc., 1957, 79, 5410–5414 CrossRef CAS.
- F. A. Cotton, Inorg. Chem., 1964, 3, 1217–1220 CrossRef CAS.
- I. Brown, Acta Crystallogr., Sect. B: Struct. Sci., 1988, 44, 545–553 CrossRef.
- J. Barker, M. Y. Saidi and J. L. Swoyer, Lithiated molybdenum oxide active materials, US Pat. 6,908,710, 2005.
- K. Ben-Kamel, N. Amdouni, H. Groult, A. Mauger, K. Zaghib and C. Julien, J. Power Sources, 2012, 202, 314–321 CrossRef CAS.
- J. Ma, Y.-N. Zhou, Y. Gao, X. Yu, Q. Kong, L. Gu, Z. Wang, X.-Q. Yang and L. Chen, Chem. Mater., 2014, 26, 3256–3262 CrossRef CAS.
- J. T. Incorvati, L. F. Wan, B. Key, D. Zhou, C. Liao, L. Fuoco, M. Holland, H. Wang, D. Prendergast and K. R. Poeppelmeier, et al. , Chem. Mater., 2015, 28, 17–20 CrossRef.
- W. Kaveevivitchai and A. J. Jacobson, Chem. Mater., 2016, 28, 4593–4601 CrossRef CAS.
- A. R. Wizansky, P. E. Rauch and F. J. Disalvo, Powerful Oxidizing Agents for the Oxidative Deintercalation of Lithium from Transition Metal Oxides, Dtic document technical report, 1989 Search PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- P. Canepa, S. Jayaraman, L. Cheng, N. N. Rajput, W. D. Richards, G. S. Gautam, L. A. Curtiss, K. A. Persson and G. Ceder, Energy Environ. Sci., 2015, 8, 3718–3730 CAS.
- P. Canepa, G. S. Gautam, R. Malik, S. Jayaraman, Z. Rong, K. R. Zavadil, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 3317–3325 CrossRef CAS.
- L. F. Wan, B. R. Perdue, C. A. Apblett and D. Prendergast, Chem. Mater., 2015, 27, 5932–5940 CrossRef CAS.
- X. Sun, V. Duffort, B. L. Mehdi, N. D. Browning and L. F. Nazar, Chem. Mater., 2016, 28, 534–542 CrossRef CAS.
- O. Mizrahi, N. Amir, E. Pollak, O. Chusid, V. Marks, H. Gottlieb, L. Larush, E. Zinigrad and D. Aurbach, J. Electrochem. Soc., 2008, 155, A103–A109 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, et al. , APL Mater., 2013, 1, 011002 CrossRef.
- P. Vlek and W. Lindsay, Soil Sci. Soc. Am. J., 1977, 41, 42–46 CrossRef CAS.
- L. W. Staples, Am. Mineral., 1951, 36, 609–614 CAS.
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4, 1400478 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the setup used for the synthesis, electrochemistry, characterization and first principles calculations are included. X-ray refinement data, electrochemical data and calculated voltage data are also included. See DOI: 10.1039/c6ta07804d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |