
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A nitrogen–sulfur co-doped porous graphene matrix as a sulfur immobilizer for high performance lithium–sulfur batteries†
Jing
Xu‡
a,
Dawei
Su‡
a,
Wenxue
Zhang
ab,
Weizhai
Bao
a and
Guoxiu
Wang
*ac
aCentre for Clean Energy Technology, School of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, NSW 2007, Australia. E-mail: Guoxiu.Wang@uts.edu.au
bSchool of Materials Science and Engineering, Chang’an University, Xi’an 710064, China
cCollege of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing, Jiangsu 210016, China
First published on 5th October 2016
Abstract
Owing to their overwhelming advantage of theoretical energy density, lithium–sulfur (Li–S) batteries are considered as one of the most promising candidates for next-generation electrochemical energy storage systems. The primary challenges in the development of Li–S batteries include the suppression of the polysulfide shuttle effect and maintenance of high sulfur loading. Herein, we report a strategy of strong chemisorption for polysulfides utilizing a chemically activated graphene matrix co-doped with nitrogen and sulfur. The dopant N and S atoms act as electron attracting atoms, leading to the nearby C atoms and causing oxygen functional groups to be polarized and more active for anchoring sulfur and polysulfides. Meanwhile, highly developed defects and edges, as well as the porous structure derived from graphene chemical activation, not only achieve a high sulfur loading in a well dispersed amorphous state, but also serve as polysulfide reservoirs to alleviate the shuttle effect. When applied as cathode hosts for lithium–sulfur batteries, the nitrogen–sulfur co-doped porous graphene architecture exhibited a high specific capacity of 1178 mA h g−1 at 0.2C, 1103 mA h g−1 at 0.5C, and 984 mA h g−1 at 1C rate, and excellent cycling stability for 600 cycles with a retained capacity of 780 mA h g−1 (0.2C).
Introduction
The rapid depletion of fossil fuels and increasing environmental pollution issues are the major stimulating forces for the development of clean energy technologies. Renewable energy sources, such as solar, wind, and tide, are attractive in this regard. However, efficient utilization of these intermittent resources requires efficient and economical energy storage (EES) systems. Advanced rechargeable batteries are the most suitable option for EES. However, confined by their limited theoretical energy density, as well as high cost, state-of-the-art lithium-ion batteries (LIBs) are not capable of fulfilling the specific energy requirements for future electric vehicles (EVs) and large-scale smart grid applications. Therefore, it is of great importance to develop advanced rechargeable batteries to satisfy future needs. Invented in the 1960s, rechargeable lithium–sulfur batteries have now attracted extensive interest, mainly due to their high theoretical energy density (≈2567 W h kg−1), which is estimated to be 5 times higher than that of state-of-the-art lithium-ion batteries. Due to the natural abundance, low cost and environmental friendliness, Li–S batteries are one of the most promising rechargeable battery technologies for a wide range of applications.1–5Despite considerable advantages of lithium–sulfur batteries, their commercialization still suffers from some tough obstacles, such as the insulating nature of sulfur, large volume expansion/contraction (∼80%) during cycling, dissolution of polysulfides (Li2Sx, 4 ≤ x ≤ 8) in organic electrolytes, and the shuttle effect of polysulfides, which inevitably result in the low electrochemical utilization of sulfur and rapid capacity degradation.2,6–10 Many approaches have been devoted to addressing these issues. These include: (1) encapsulating sulfur particles into various carbon matrix materials and/or conducting polymers, such as mesoporous carbon, carbon nanofiber interlayers, and reduced graphene oxide, polyaniline (PAni), polypyrrole (PPy), and their derivatives.3,4,8,11,12 (2) Strengthening sulfur's chemical bonds via chemical interactions or surface-mediated redox reactions with metal oxides (e.g. SiO2, TiO2, metal–organic frameworks) and metal carbides (MXene phases).13,16,18 (3) Adding novel electrolyte additives such as P2S5 or Cs+ to suppress the polysulfide shuttle.17,19 (4) Modifying the surface chemistry of the hosts to prevent the shuttle effect of the soluble polysulfides between the cathode and the anode, enabling better cycling performance.20,22 The chemical properties of sp2 carbon materials can be further modified by introducing heteroatom doping, such as phosphorus, sulfur, boron and nitrogen, which demonstrated significant potential for lithium polysulfide confinement.9 Song et al. reported that nitrogen doping in mesoporous carbon effectively promoted the chemical adsorption of sulfur atoms on oxygen-containing functional groups because surface modification with nitrogen atoms greatly enhances the electrochemical reactivity and electronic conductivity of carbon matrices.14,51 Additionally, sulfur-doped porous carbon also attracted wide attention for battery applications due to the fact that sulfur-doping can change the charge state of the neighbouring carbon atoms. Therefore, sulfur-doped carbon materials exhibit enhanced adsorption ability. Most importantly, when sulfur and nitrogen are simultaneously doped into the carbon matrix, synergistic effects can occur due to the highly active multiple doping elements.15,21,24,26 The co-doping not only strengthens the chemical bonding between the carbon host and sulfur chains in the heat treatment during the sulfur loading process, ensuring the uniform distribution of sulfur, but also generates more active sites and increases their activity, which greatly improve the adsorption of soluble lithium polysulfide intermediates, thus increasing the cycle ability and coulombic efficiency.29 Therefore, the co-doping strategy can achieve superior electrochemical activity beyond that of mono-element doping.
Graphene, a free-standing atom-thick layer of sp2 carbon atoms, has emerged as the most significant carbon material in the last decade due to its unique physical and chemical properties.23,25,41,42,53–55 Nitrogen–sulfur co-doped graphene is a promising nanoscale immobilizer as a cathode host material for lithium–sulfur batteries. However, graphene nanosheets tend to form agglomerates or even restack through van der Waals interactions during the preparation processes and subsequent electrode fabrication,27 resulting in the loss of specific surface area. This effect will also lead to lower polysulfide adsorption due to the decrease of the accessible active sites on the graphene surface. Therefore, it is very important to prevent graphene aggregation by creating a porous structure of nitrogen–sulfur co-doped graphene nanosheets.
Herein, we have developed a highly crumpled nitrogen–sulfur co-doped 3D graphene nanosheet matrix as a sulfur immobilizer for lithium–sulfur batteries. These highly porous nitrogen–sulfur co-doped graphene nanosheets were synthesized through a facile synthesis approach, in which polypyrrole (PPy) serves as a nitrogen source and ammonium persulfate (APS) as a sulfur source. Highly developed defects and edges, as well as hierarchical pore structures derived from graphene chemical activation endow nitrogen–sulfur co-doped graphene nanosheets with large surface area (1012 m2 g−1) and a wide pore size distribution of 2 nm and 25–40 nm. The as-prepared materials have strong chemisorption capabilities for polysulfides and high sulfur loading. When applied as cathode materials in lithium–sulfur batteries, the cathodes exhibit a high initial capacity of 1178 mA h g−1 at 0.2C and excellent long life cycling performance with a retained capacity of 780 mA h g−1 after 600 cycles.
Experimental
Synthesis of NSG
Graphene oxide (GO) was synthesized using natural graphite flakes by a modified Hummers' method. 50 mg of the as-made graphene oxide was well suspended in 50 mL DI water after ultrasonically stirring for 2 h, then 0.73 g cetyltrimethylammonium bromide (CTAB) and 1.37 g ammonium persulfate (APS) were dissolved into the prepared graphene solution magnetic stirring for 1 h. After that, 0.83 mL of 99% pyrrole monomer was added and the mixture was stirred in an ice water bath for about 24 h. When the reaction finished, the resultant products were washed with DI water and dried in a vacuum at 80 °C for 24 h to remove any residual solvents, this final product is denoted as NSG.Synthesis of A-NSG
Typically, 400 mg NSG powder was dispersed in 20 mL 3 M K2CO3 solution and stirred for 24 hours. The extra K2CO3 solution was removed by briefly filtering the mixture through a polycarbonate membrane (Whatman, 0.2 μm); then the mixture was dried in the lab environment at 70 °C for 24 hours. The NSG/K2CO3 mixture was put in a tube furnace under flowing nitrogen at 800 °C for 2 h (heating rate 5 °C min−1). Subsequently, the final product (denoted as A-NSG) was thoroughly washed with 0.1 M HCl and DI water until the pH value reached 7 and finally dried in a vacuum at 80 °C for 20 h.Synthesis of A-NSG@S
A-NSG was homogeneously dispersed in DI water by ultrasonication, then transferred to an appropriate amount of elemental sulfur/CS2/ethanol solution with a mass ratio of mA-NSG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mS = 1:3, the mixed solution was magnetically stirred for 24 h over an ice water bath. During this time period, the CS2 and ethanol were allowed to completely evaporate while stirring, then the A-NSG@S composite was filtered and dried at 50 °C in a vacuum oven for 12 h. Finally, the as-synthesized A-NSG@S was heated to 155 °C at a heating rate of 2 °C min−1 in a tube furnace under flowing argon at 100 sccm before re-heating at 230 °C for another 2 h under the same conditions.
:mS = 1:3, the mixed solution was magnetically stirred for 24 h over an ice water bath. During this time period, the CS2 and ethanol were allowed to completely evaporate while stirring, then the A-NSG@S composite was filtered and dried at 50 °C in a vacuum oven for 12 h. Finally, the as-synthesized A-NSG@S was heated to 155 °C at a heating rate of 2 °C min−1 in a tube furnace under flowing argon at 100 sccm before re-heating at 230 °C for another 2 h under the same conditions.
Characterization of materials
The morphology of the obtained materials was characterized by field emission scanning electron microscopy (FESEM, Zeiss Supra 55VP) and transmission electron microscopy (TEM, Model JEM-2011, JEOL). The crystallographic information for the samples was collected on a Siemens D5000 diffractometer using Cu-K radiation with a scanning step of 0.02° per second. Raman spectra were measured by a Renishaw in Via Raman spectrometer system (Gloucestershire, UK) equipped with a Leica DMLB microscope (Wetzlar, Germany) and a 17 mW 633 nm Renishaw helium neon laser at 50% power. XPS analysis was performed on an ESCALAB MK II X-ray photoelectron spectrometer. FTIR spectra were recorded on a NEXUS 670FTIR spectrometer using KBr disks. Thermogravimetric analysis (TGA) was used for the analysis of weight loss from the precursor to the final product. Nitrogen adsorption–desorption measurements were conducted on a 3 Flex surface characterization analyzer to determine the Brunauer–Emmett–Teller (BET) specific surface area using a Quadrasorb SI analyzer at 77 K. The BET surface area was calculated using experimental points at a relative pressure of P/P0 = 0.05–0.25. The UV-vis spectra were measured in the spectral range of 300–1000 nm using a Cary 60 UV-vis variable wavelength spectrophotometer. All the samples for UV-vis measurement were prepared and sealed in an argon-filled glove box.Electrochemical characterization
Working electrodes were made from 90 wt% of active materials, 2 wt% of conductive agent (carbon black) and 8 wt% of binder (polyvinylidene difluoride). The mass loading of sulfur on the electrode is around 1.5 mg cm−2. CR2032 coin cells were assembled in an argon-filled glove box (Mbraun, Unilab, Germany), in which both the moisture and oxygen contents were controlled to be less than 0.1 ppm. Lithium foil was used as a counter electrode. The electrolyte was 1 M lithium bis(trifluoromethanesulfonyl) imide (LiTFSI) and 1 wt% lithium nitrate (LiNO3) in 1,3-dioxolane and 1,2-dimethoxy-ethane (volume ratio 1:1), for each electrode, around 30 μL electrolyte was added into the coin cell. The electrodes were dried at 60 °C under vacuum for 12 h. Electrochemical measurements were conducted using a LAND-CT2001C battery test system. The cells were discharged and charged galvanostatically in the fixed voltage range of 1.7–2.7 V with a current rate of 0.1C, 0.5C, 1C, 2C, and 5C. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were conducted with a CHI660C electrochemistry workstation in the voltage range of 1.7 to 2.7 V (vs. Li+/Li).
Computational methods
The simulations are based on density functional theory (DFT), which is provided by DMOL3. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof scheme (PBE) is adopted for the exchange–correlation potential to optimize geometrical structures and calculate properties. The All-Electron Relativistic Kohn–Sham wave functions (AER) and double numeric plus polarization (DNP) basis set are adopted in the local atomic orbital basis set with the global orbital cutoff set to 4.4 Å. A single-layer of graphene was modelled using a hexagonal 4 × 4 supercell. The nearest distance between nanosheets in neighboring cells is greater than 15 Å to ensure no interactions between different layers. For geometric optimization, both the cell and the atomic positions are allowed to fully relax. The Brillouin zone is sampled at 6 × 6 × 1 k-points for all structures in the geometric optimization calculations, which brings out the convergence tolerance of energy of 1.0 × 10−5 Ha (1 Ha = 27.2114 eV), maximum force of 0.002 Ha per Å, and maximum displacement of 0.005 Å. The adsorption energy (Ea) is defined as:| Ea = Etotal − Eads − Esubstrate |
Results and discussion
The 3D nitrogen–sulfur co-doped porous graphene matrix was synthesized via chemical activation of polypyrrole (PPy) functionalized graphene sheets using K2CO3 as shown in Scheme 1. PPy was chosen as the nitrogen source due to its high nitrogen content. More importantly, because of the π–π interaction between PPy and graphene oxide (GO), the PPy monomers will polymerize on the surfaces of graphene oxide (GO) during the polymerization process. The doping of sulfur was achieved by ammonium persulfate (APS). After carbonization, nitrogen and sulfur atoms are expected to dope in the graphene nanosheets. Scanning electron microscopy (SEM) images of nitrogen and sulfur co-doped graphene (denoted as NSG) composites are shown in Fig. S1 (ESI†), it is clear that the NSG composites show quite similar morphologies to the rGO, displaying a thin smooth surface. The as-prepared NSG was chemically activated by K2CO3 to obtain final porous nitrogen–sulfur co-doped graphene (denoted as A-NSG). Fig. 1a, 2a and S2 (ESI†) show the scanning electron microscopy (SEM) images and transmission electron microscopy (TEM) images of A-NSG. The corrugated morphology with wrinkles and folded regions is clearly visible, which is markedly different from rGO and NSG. Meanwhile, most of the A-NSG layers are discontinuous, giving rise to a large number of defects such as edges and pores. Fig. 1d shows the typical AFM image of the A-NSG, which further reveals that the thin wall typically consists of only a few graphene layers as the A-NSG nanosheets with an average thickness of about 0.396 nm. The crumpling of the A-NSG can effectively prevent them from agglomerating and restacking, thus increasing the electrolyte accessible surface area. The corresponding energy-dispersive X-ray spectroscopy (EDX) illustrates uniform distribution of carbon, nitrogen and sulfur in A-NSG (Fig. 1b).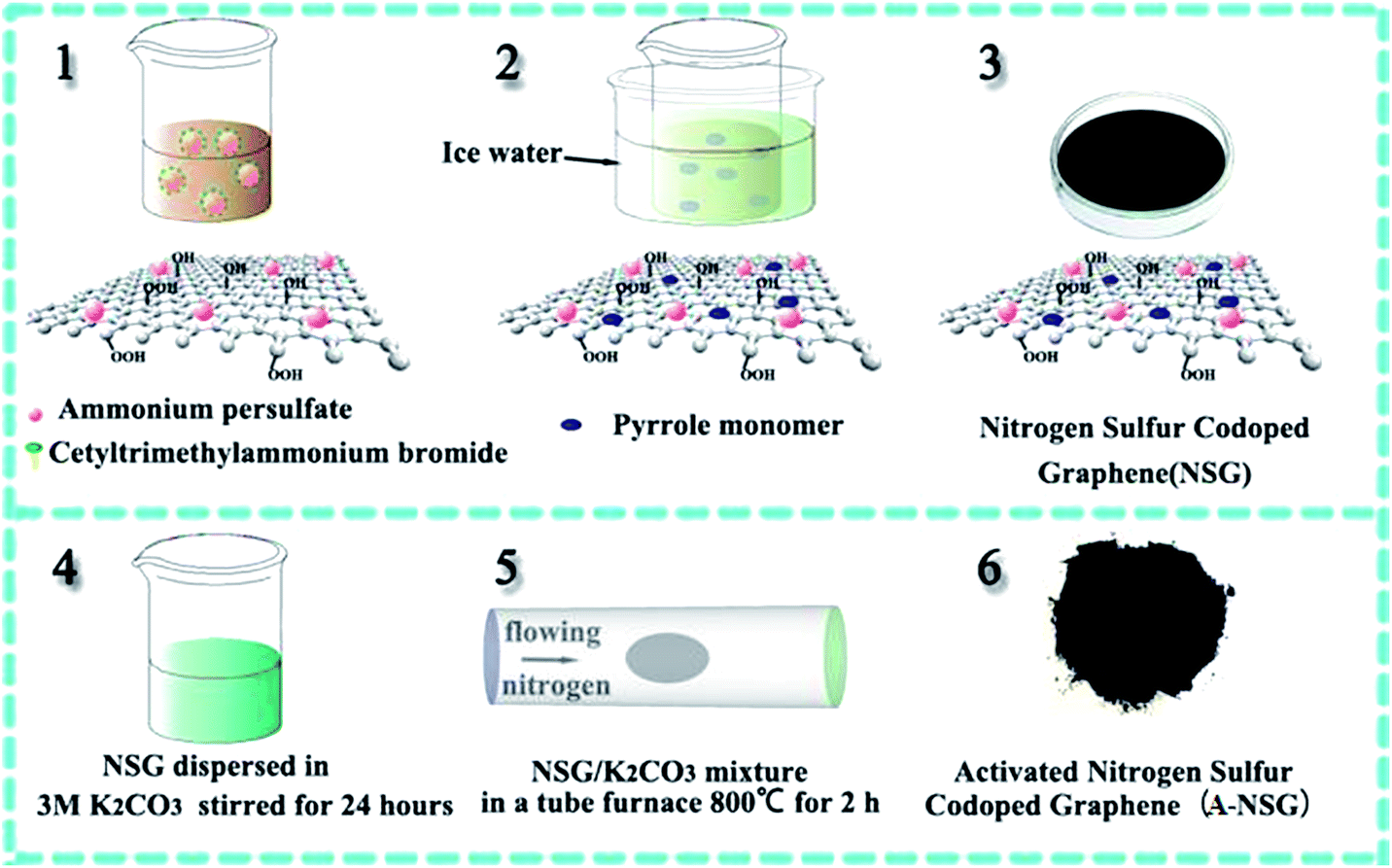 | ||
| Scheme 1 Schematic illustration of the formation of A-NSG. | ||
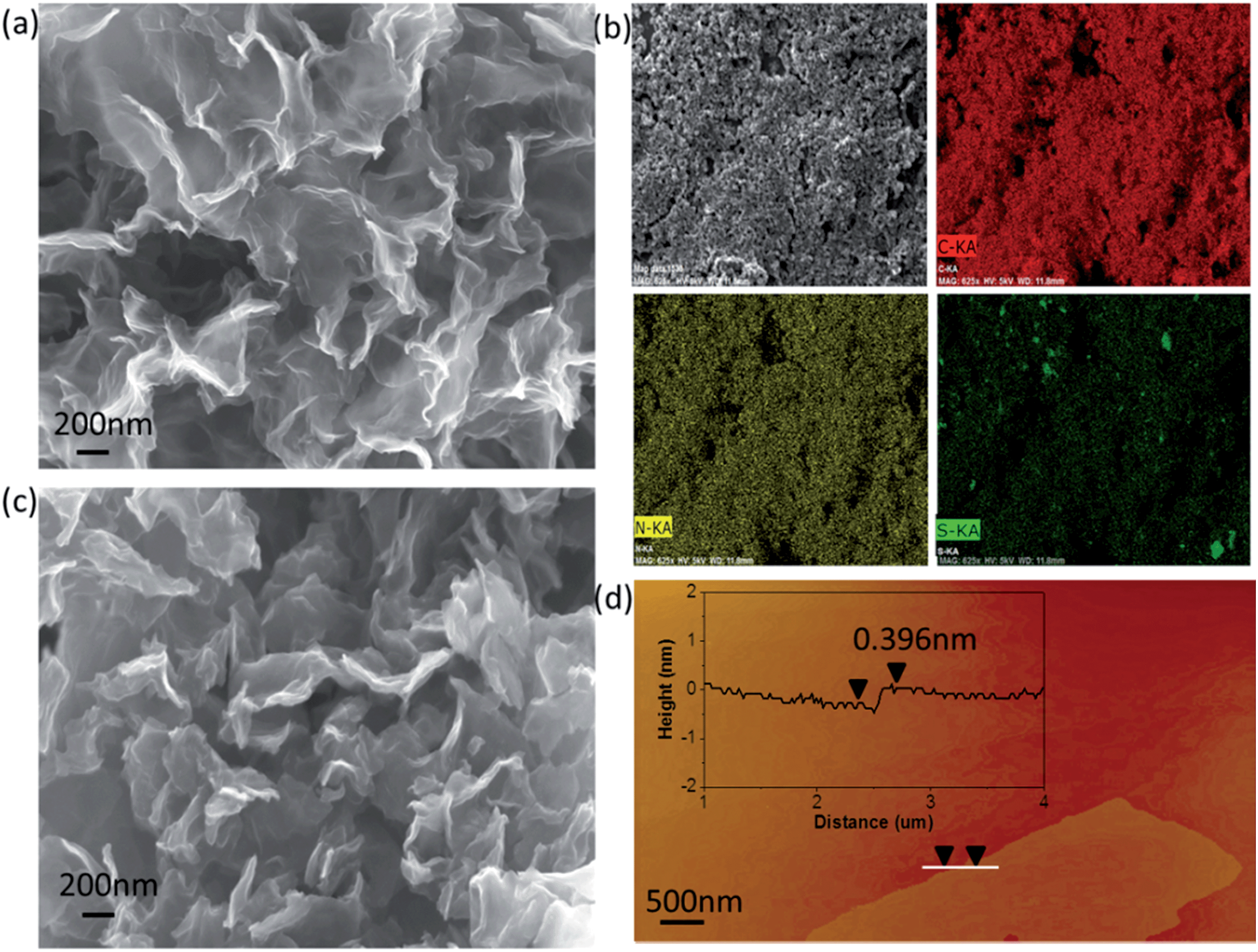 | ||
| Fig. 1 (a) SEM image of A-NSG. (b) EDX elemental mapping for A-NSG. (c) SEM image of the A-NSG@S. (d) AFM image of A-NSG. | ||
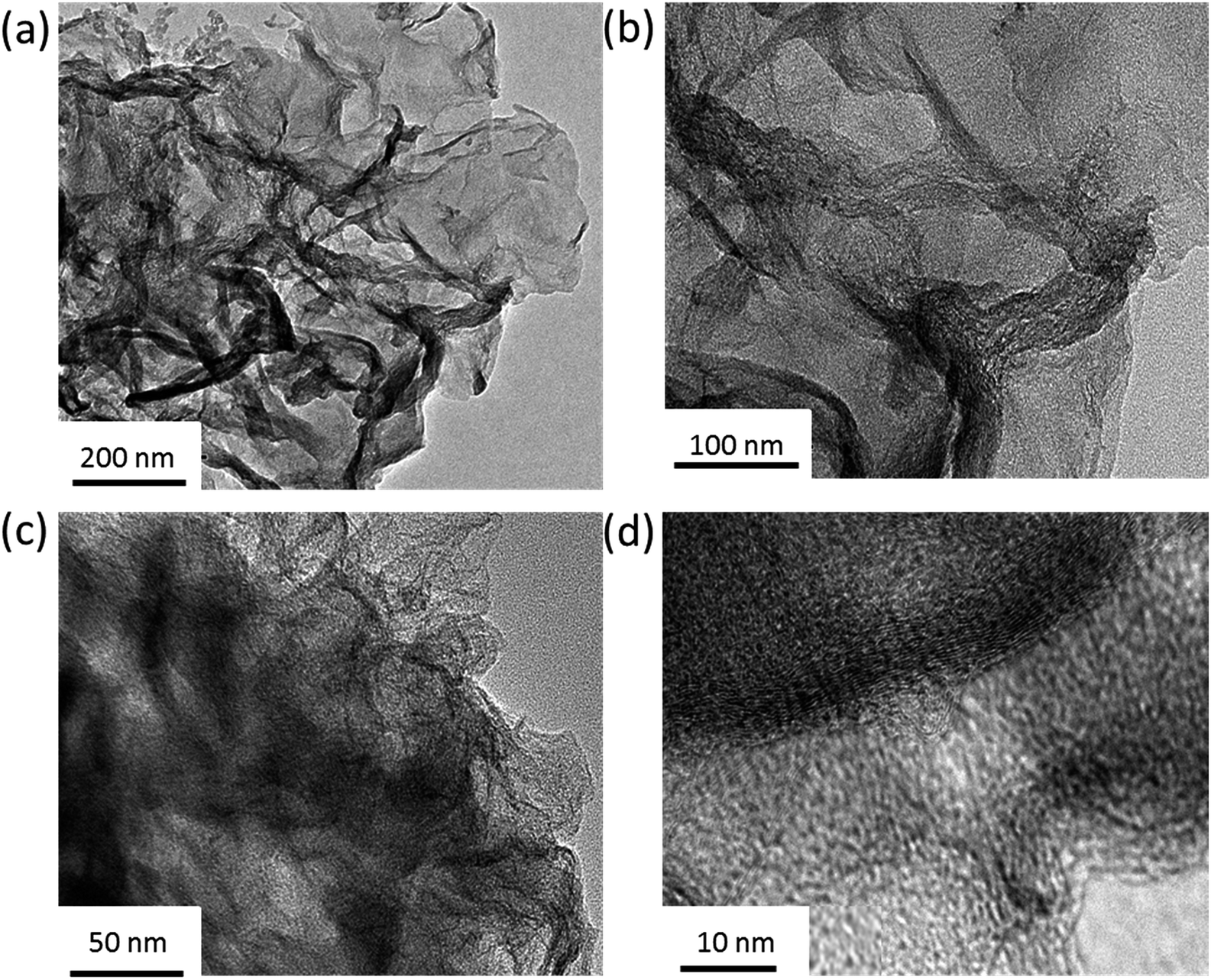 | ||
| Fig. 2 (a and b) TEM images of A-NSG. (c and d) High-magnification TEM images of the A-NSG@S. | ||
Applying the 3D A-NSG hierarchical network structure as the sulfur host, we loaded sulfur into the A-NSG matrix by liquid-phase infiltration and melt vulcanization processes. The initial liquid phase infiltration process ensures the uniform distribution of sulfur in the A-NSG. The melt vulcanization processes include two major steps: (i) a typical step at 155 °C for 10 hours, aiming to encapsulate sulfur into the porous graphene 3D frameworks (denoted as A-NSG@S-155), and (ii) heating at 230 °C, to trigger the ring-opening polymerization of elemental sulfur (S8) into linear polysulfides along porous graphene (denoted as A-NSG@S-230). In the A-NSG@S composites, a uniform layer of sulfur is homogeneously anchored on the surface of A-NSG without any obviously aggregated sulfur particles being observed in the TEM image (Fig. 2c) or FSEM image (Fig. 1c). The sulfur content in the A-NSG@S nanocomposites was determined by thermogravimetric analysis (TGA) under a nitrogen atmosphere with a heating rate of 10 °C min−1, as shown in Fig. S3 (ESI†). The TGA analyses reveal that the sulfur loading in A-NSG@S was 72.4 wt%, thus the mass loading of sulfur in each electrode is around 65%. To verify the structural characteristics of A-NSG@S, X-ray diffraction (XRD) was conducted on pure sulfur, A-NSG@S-155, and A-NSG@S-230 (Fig. 3a). It should be noted that the overlapped broad peak at about 26° appear in both A-NSG@S-155 and A-NSG@S-230, which can be ascribed to the A-NSG from thermal restoration of sp2 carbon.21 Compared with the intact crystalline peaks of pure sulfur and A-NSG@S-155, the absence of sulfur crystal peaks for A-NSG@S-230 is noteworthy (Fig. 3a). This indicated that sulfur now exists in a highly dispersed amorphous state. Meanwhile, as shown in the Raman spectra (Fig. 3b), the pure sulfur powder exhibits characteristic peaks in the range of 100–500 cm−1, associated with the vibration of the S–S bond in S8 species while both the A-NSG@S-155 and A-NSG@S-230 did not show noticeable characteristic sulfur peaks, which is also consistent with the conclusion from XRD patterns.28 Additionally, the SAED pattern (Fig. S4, ESI†) of A-NSG@S reveals a typical sixfold symmetry expected for graphene without any sign of crystallized sulfur, confirming its amorphous state. This implies that S8 molecules were loaded into the A-NSG matrix without long-range ordering, similar to small sulfur molecule dispersion. Other groups have reported that the amorphous state of sulfur could result in the high utilization of sulfur in electrochemical lithiations.23
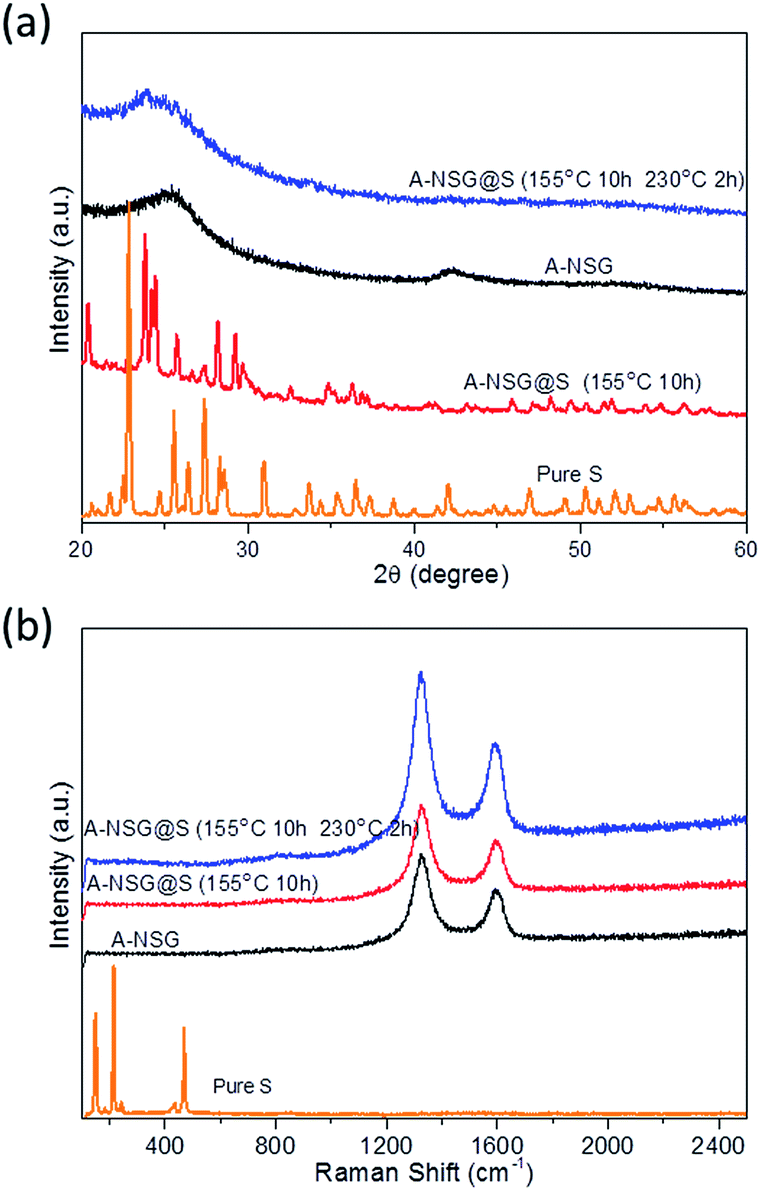 | ||
| Fig. 3 XRD patterns and Raman spectra of the pure sulfur powder, A-NSG, A-NSG@S-155, and A-NSG@S-230. | ||
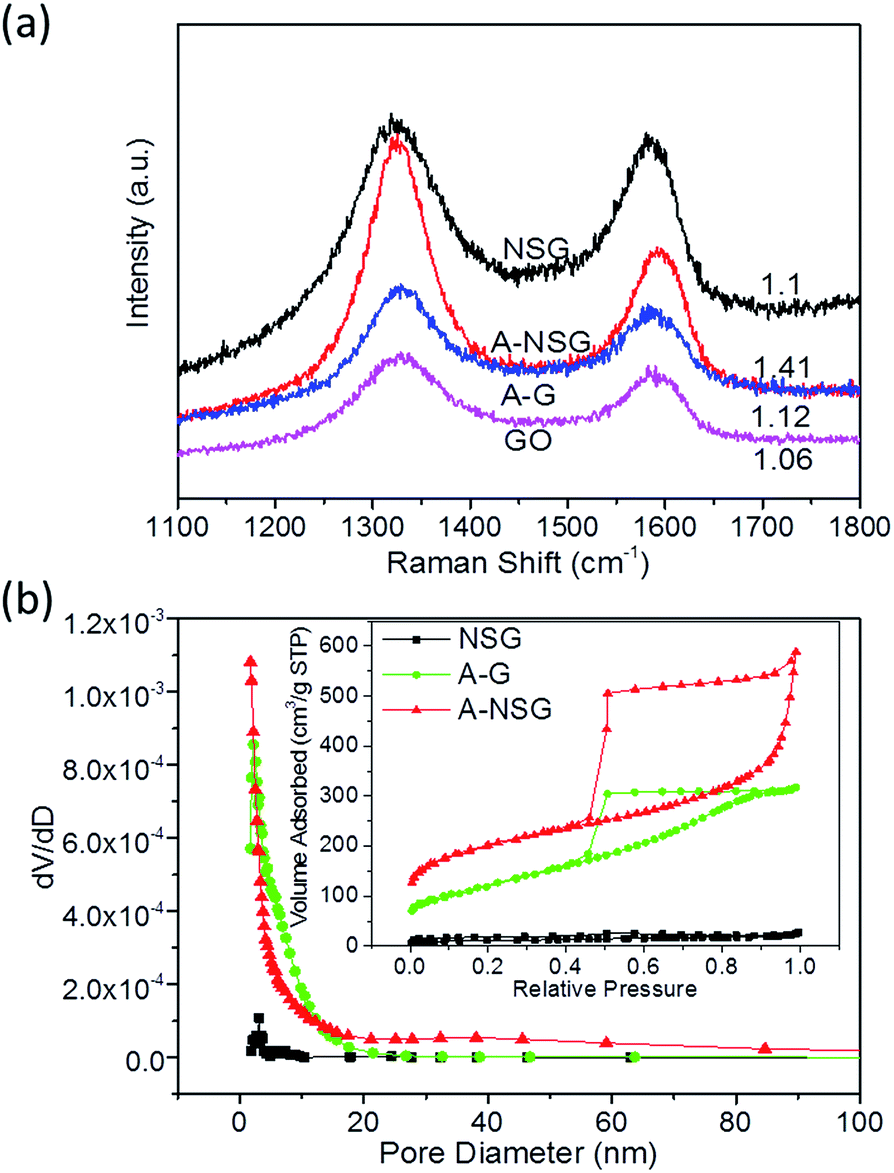
To investigate the elemental composition and functional groups in the A-NSG, X-ray photoelectron spectroscopy (XPS) measurements were performed. As shown in Fig. 4a, the XPS survey spectrum of A-NSG exhibits four characteristic peaks located at ca. 164, 400.23, 285.07 and 532.25 eV, corresponding to S 2p, N 1s, C 1s, and O 1s. The elemental content of S and N in A-NSG is 0.85 at% and 4.18 at%, respectively. This suggests the effective doping of N and S into the graphene sheets. In the N 1s spectrum of A-NSG (Fig. 4b), three components including pyridinic N (21.68 wt%), pyrrolic N (50.16 wt%), and graphitic N (28.16 wt%), can be identified at 398.4, 400.2 and 402.1 eV, respectively.22,30 It is also reported that the dominant pyridinic and pyrrolic N had been proven to contribute to the improvement of the affinity and binding energy of non-polar carbon atoms with polar and rate capability of Li–S batteries.7,22 We also displayed the molecular geometries and the adsorption energies of Li2S and Li2S4 on pyridinic and pyrrolic N sites in Fig. S5a–d (ESI†). In the most stable configuration, terminal Li atoms in Li2S and Li2S4 tended to directly bind to the pyridinic N atom. There was distinctive electron concentration between N and Li atoms, suggesting a strong Li–N electrostatic interaction. The electron migration can be well explained by the Lewis acid–base theory.43,44 The pyridinic N with an extra pair of electrons was considered as an electron-rich donor that naturally acted as a Lewis-base site to interact with the strong Lewis acid of terminal Li atom in lithium (poly)sulfides. Similarly, as indicated by Fig. S5b and d,† pyrrolic N played an analogous role as pyridinic N, which were consistent with Hou et al.'s theoretical results.44 In addition, the dependence of adsorption strength between LiPSs and N doping configuration can be seen from Li–N distances. The Li–N distances of Li2S on pyridinic and pyrrolic N sites were 1.99 and 2.02 Å, which are consistent with the large Ea obtained from Li2S on the pyridinic N site. In the case of Li2S4 on pyridinic and pyrrolic N sites, the Li–N distances were calculated to be about 2.04 and 2.05 Å, which further confirmed the strong anchoring effect between pyridinic, pyrrolic N and acid terminated Li in Li2Sx. On the other hand, in the high resolution C 1s spectra (Fig. 4a), due to the removal of most oxygen groups as well as the partial reconstruction of the graphitic carbon network, peaks corresponding to C–O (286.2 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.2 eV) and O–CO (289.6 eV)21 decrease considerably for A-NSG, compared with that of GO;7 two particular new peaks located at 286.1 and 287.2 eV suggest the bond formation of graphene; the sharp peak in the C 1s spectrum of A-NSG, still attributed to the sp2-hybridized graphitic carbon atoms, shifts to higher binding energy and its full width of half-maximum (fwhm) at 284.8 eV increases with the presence of nitrogen content (Table S1, ESI†).31,32,41 All of these results confirm the formation of C–N bonding configurations in the A-NSG. Moreover, in the X-ray photoelectron spectroscopy (XPS) analysis, the S 2p spectrum for the A-NSG nanocomposites in Fig. 4c has 2p3/2 and 2p1/2 spin–orbit levels with an energy separation of 1.27 eV and intensity ratio of 2:1, confirming the effective doping of sulfur atom into the carbon lattice. Two sulfur species are assigned to carbon-bonding: thiophenic S (163.6 eV) and sulfonic S (167.9 eV); the thiophenic S (sulfur atom bonded with carbon directly by a C–S bond) is believed to make the carbon matrix positively charged, thereby increasing its affinity to absorb polysulfides. At the same time, it is also interesting to find that the C/O ratio of GO is 2.4 and this value increases to 17.52 for A-NSG (Table S2, ESI†), indicating that nitrogen–sulfur co-doping increases the reduction efficiency of GO. This seems to be further supported by the Raman spectroscopy analysis. In Fig. 5a the Raman spectra for A-NSG exhibit two remarkable peaks at around 1350 and 1580 cm−1. The G band located at 1580 cm−1 is related to the E2g vibration mode of sp2 carbon atoms while the D band at 1350 cm−1 is attributed to the defects and disorder hybridized vibrational mode of graphene. Therefore, the higher intensity ratio (ID/IG) of A-NSG than GO (1.41 vs. 1.06) should be ascribed to more defect sites generated by N and S co-doping, resulting in the high disorder of graphene nanosheets. More importantly, these defects can provide considerable active sites for lithium polysulfide adsorption.21,33,40
O (288.2 eV) and O–CO (289.6 eV)21 decrease considerably for A-NSG, compared with that of GO;7 two particular new peaks located at 286.1 and 287.2 eV suggest the bond formation of graphene; the sharp peak in the C 1s spectrum of A-NSG, still attributed to the sp2-hybridized graphitic carbon atoms, shifts to higher binding energy and its full width of half-maximum (fwhm) at 284.8 eV increases with the presence of nitrogen content (Table S1, ESI†).31,32,41 All of these results confirm the formation of C–N bonding configurations in the A-NSG. Moreover, in the X-ray photoelectron spectroscopy (XPS) analysis, the S 2p spectrum for the A-NSG nanocomposites in Fig. 4c has 2p3/2 and 2p1/2 spin–orbit levels with an energy separation of 1.27 eV and intensity ratio of 2:1, confirming the effective doping of sulfur atom into the carbon lattice. Two sulfur species are assigned to carbon-bonding: thiophenic S (163.6 eV) and sulfonic S (167.9 eV); the thiophenic S (sulfur atom bonded with carbon directly by a C–S bond) is believed to make the carbon matrix positively charged, thereby increasing its affinity to absorb polysulfides. At the same time, it is also interesting to find that the C/O ratio of GO is 2.4 and this value increases to 17.52 for A-NSG (Table S2, ESI†), indicating that nitrogen–sulfur co-doping increases the reduction efficiency of GO. This seems to be further supported by the Raman spectroscopy analysis. In Fig. 5a the Raman spectra for A-NSG exhibit two remarkable peaks at around 1350 and 1580 cm−1. The G band located at 1580 cm−1 is related to the E2g vibration mode of sp2 carbon atoms while the D band at 1350 cm−1 is attributed to the defects and disorder hybridized vibrational mode of graphene. Therefore, the higher intensity ratio (ID/IG) of A-NSG than GO (1.41 vs. 1.06) should be ascribed to more defect sites generated by N and S co-doping, resulting in the high disorder of graphene nanosheets. More importantly, these defects can provide considerable active sites for lithium polysulfide adsorption.21,33,40
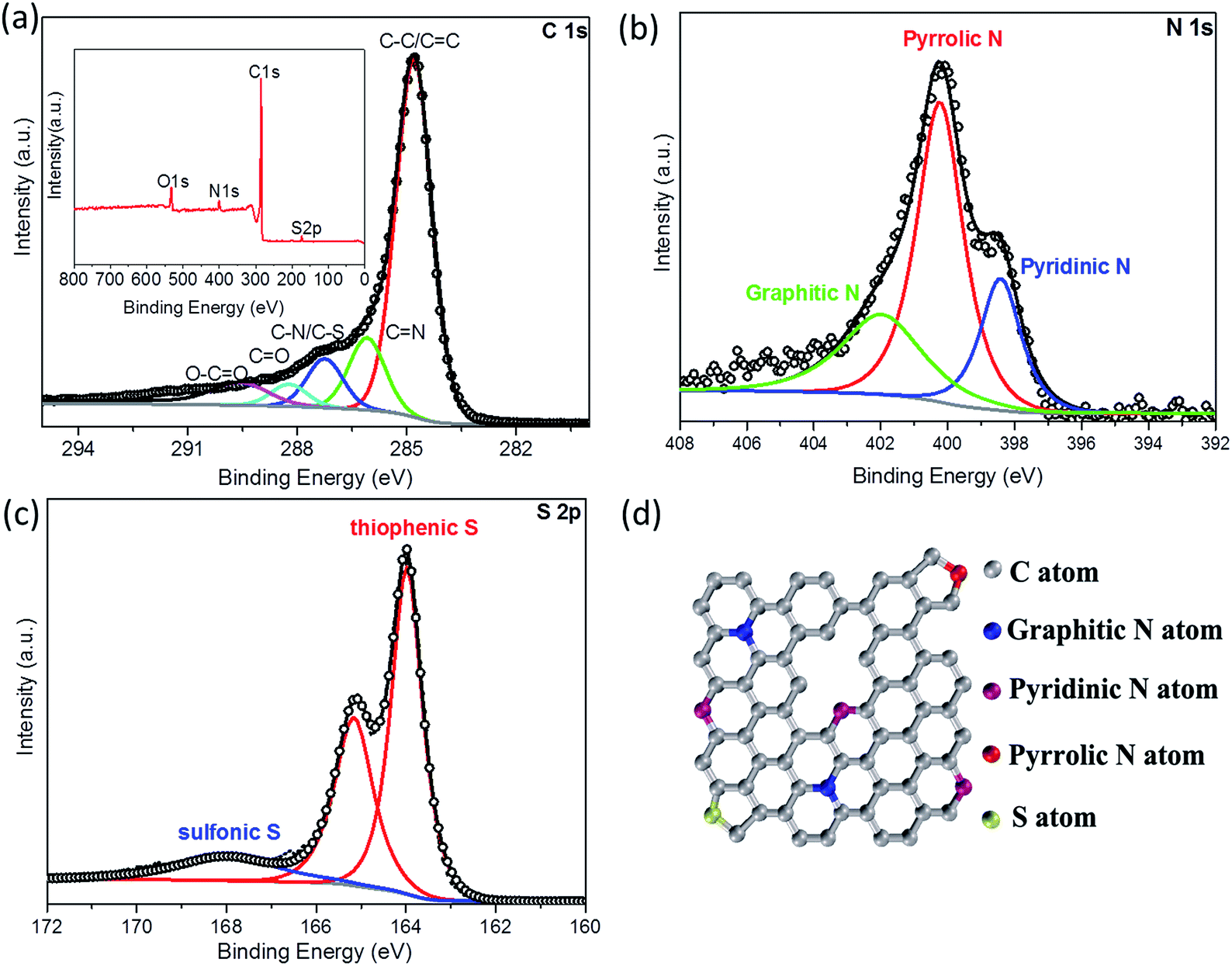 | ||
| Fig. 4 (a–c) High-resolution C 1s, N 1s and S 2p XPS spectrum of A-NSG and corresponding XPS survey spectra of A-NSG. (d) Schematic structure of A-NSG. | ||
 | ||
| Fig. 5 (a) Raman spectra of GO, NSG, A-G and A-NSG. (b) Pore characterization of the NSG, A-G and A-NSG materials. | ||
To further prove the interaction between polysulfides and A-NSG, the Li2S4 solution was prepared by dissolving Li2S and sublimed sulfur (with a stoichiometric ratio of 1:3) in the mixed solvents of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) (1:1 v/v). Then, 15 mg of N–S co-doped graphene was soaked in 15 mL of 5 mM Li2S4 solutions at 30 °C for 24 h, respectively. The upper solution was carefully collected and sealed for UV-vis spectra measurement. Fig. S6a–d (ESI†) display digital photos of pristine Li2S4 solution and the color changes of the Li2S4 solution at various periods after adding N–S co-doped graphene composites. After adding N–S co-doped graphene into the Li2S4 solution, a homogeneous black suspension was initially formed, and it took several minutes for N–S co-doped graphene to precipitate. After aging for 3 and 24 h, the color of Li2S4 turns from the initial yellow to light yellow, indicating that much of the Li2S4 has been adsorbed by the N, S graphene. The result indicates the adsorption capability of N–S co-doped graphene for lithium polysulfides. The corresponding UV-vis spectra in Fig. S7 (ESI†) shows that the reflect index at 533 nm of the Li2S4 solution was increased after soaking the N, S graphene composite for 24 h, confirming that large amount of Li2S4 was absorbed by N–S co-doped graphene.
We also conducted X-ray photoelectron spectroscopy (XPS) analysis to identify the interaction between polysulfides and N, S-graphene. Fig. S8a (ESI†) shows the Li 1s XPS spectrum of the electrode after discharging to 2.1 V. It exhibits a single asymmetric peak at around 56.0 eV. Therefore, an additional peak with a +1.1 eV shift was fitted and attributed to the Li in the polysulfides interacting with doped N (Li–N) because normally, the Li 1s XPS spectrum only shows the symmetric peak around 55.5 eV. This result is consistent with the observation for nitrogen/sulfur-doped cellulose bonded LiPSs,57 suggesting that N, S doped graphene can trap the polysulphides. The S 2p spectrum also confirms the interaction between the lithium polysulfide and N–S co-doped graphene. The new peak at around 164.6 eV could be ascribed to the –CS– bonding, which is derived from the interaction between polysulfide and the N, S doped graphene because this peak was not detected in the S 2p spectrum of N–S co-doped graphene@S (Fig. S9, ESI†).58 This further proved the interaction between polysulfide and the N–S co-doped graphene. We also note that the peaks at 170.64 eV and 169.4 eV in the N, S co-doped graphene electrode discharged to 2.1 V (Fig. S8b, ESI†) could originate from (i) surface oxy-group of graphene and (ii) oxidization due to the exposure to the atmosphere during the transferring to the XPS measurement.
To further confirm and quantitatively characterize the polysulfide adsorption ability of N–S co-doped graphene, N doped graphene and none doped graphene, we carried out quantum mechanical calculations based on density functional theory, the results are shown in Table S3,† and the corresponding models for N–S co-doped graphene and none doped graphene are in Fig. 6, and for N-doped graphene in Fig. S5 (ESI†). The adsorption energies of Li2S and Li2S4 on pristine graphene are −0.94 and −0.49 eV, respectively. The adsorption energies of Li2S and Li2S4 on N doped graphene are around −1.41 and −1.01 eV, respectively, while for Li2S and Li2S4 on N, S co-doped graphene, our DFT calculations predict that thiophene-like sulfur can exist adjacent to pyridinic N, as shown in Fig. 6c and d. The adsorption energies are −1.85 and −1.25 eV, respectively, which exhibits much stronger bonding effects than those on pristine graphene and N doped graphene. Furthermore, there are 0.227 and 0.019 electrons transferred from Li2S and Li2S4 to pristine graphene while 0.555 and 0.317 electrons are transferred from Li2S and Li2S4 to N, S co-doped graphene, suggesting that more charge migrates into co-doped graphene as lithiation proceeds.43,44 Therefore, the co-doping of N and S in graphene can significantly enhance the adsorption of lithium polysulphides as well as lithium sulfides.
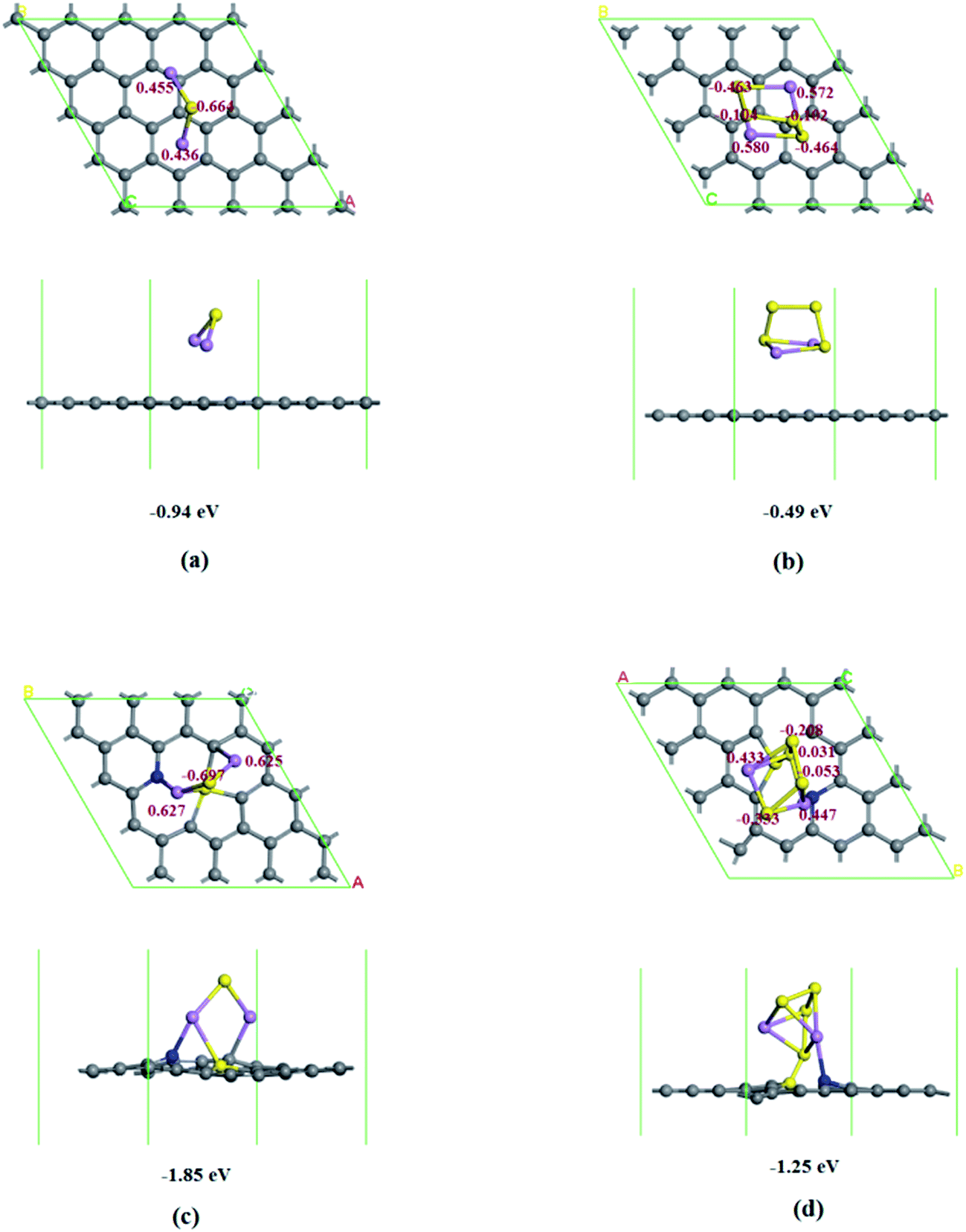 | ||
| Fig. 6 Optimized configurations for the adsorption of Li2S and Li2S4 on pristine graphene (a and b) and N, S codoped graphene (c and d) with corresponding adsorption energies in eV and atom Mulliken charge. Gray, blue, yellow, and purple balls represent C, N, S, and Li atoms, respectively. | ||
The Brunauer–Emmett–Teller (BET) results also elucidate the function of K2CO3 chemical activation. As shown in Fig. 5b, the optimized A-NSG showed a high BET surface area of 1012 m2 g−1 and a large pore volume of 1.8 cm3 g−1. A hierarchical nanoporosity including both micropores (2 nm) and mesopores (25–40 nm) resulted from the gas evolution upon pyrolysis of the polymers and K2CO3 chemical activation. In contrast, the NSG sample, which only experienced a nitrogen–sulfur co-doping process without K2CO3 chemical activation, exhibits a low surface area of 230 m2 g−1 and a pore volume of 0.3 cm3 g−1. Therefore, the K2CO3 chemical activation greatly contributes to the nanoporosity of the material. Moreover, the large pore volume plays a vital role in embedding sulfur efficiently and providing enough void space for volume expansion, while the high surface area is important for interfacial chemisorption of lithium polysulfides. On the other hand, the ID/IG intensity ratio of A-G of Raman spectrum analysis is clearly higher than that of GO or NSG (1.12 vs. 1.06, 1.1), which indicates that K2CO3 activation could also induce GO to generate more defects for effective reactions. In summary, the activation by K2CO3 yields a high BET surface area, and creates porous structures and continuous 3D networks.
The electrochemical performance of A-NSG@S nanocomposite electrodes was tested by cyclic voltammetry (CV) and charge/discharge cycling behaviors of sulfur cathodes. Two cathodic peaks at 2.35 V and 2.05 V (Fig. 7a) are consistent with the voltage plateaus in Fig. 7b. The upper plateau at 2.35 V contributes to the transformation of cyclo-octasulfur to long-chain soluble lithium polysulfides. The lower plateau at 2.05 V is associated with the decomposition of those polysulfides to insoluble short-chain lithium sulfides, which contributes to the major capacity of the A-NSG@S electrode. Notably, there are no shape or position changes of the redox peaks, implying excellent reversibility and stability of the A-NSG@S nanocomposite electrodes.34,35Fig. 7d shows the long-term cycling performance of the A-NSG@S at 0.2C up to 600 cycles. The electrode delivered an initial capacity of 1178 mA h g−1 and retained a high capacity of 780 mA h g−1 after 600 cycles. It should be noted that the capacity gradually increases in the first 10 cycles upon cycling at 0.2C current rate, which has been reported as a common phenomenon for electrodes applied in lithium–sulfur batteries.45,46 As shown in Fig. 7c and d, the total capacity increased is mainly derived from the capacity accumulated in the low voltage plateau. There is almost no polysulfide loss in the high voltage plateau during the first few cycles. Normally, for sulfur batteries, the capacity is gradually decreased due to the dissolution of polysulfides at the low voltage plateau. However, due to the strong polysulfide absorption effect of N–S co-doping A-NSG, the polysulfides could be effectively trapped by the N–S co-doping graphene. Additionally, we suppose that not all sulfur takes part in the reaction with lithium ions due to the poor conductivity in the initial several cycles, and because of the “sulfur activation” in the first initial cycles,47 more and more sulfur participated in the discharge reaction step by step, thus the capacity generated in the low voltage plateau gradually improved, resulting in the increase of total capacity. Furthermore, the capacity experienced only a slight decrease during the 100th to 600th cycles, which further demonstrates long term cycling stability. When increasing the current rates to 1C (Fig. S10, ESI†), the A-NSG@S electrodes achieved a capacity of 994 mA h g−1 in the initial cycle, and showed superior cycling performance with the capacity degradation rate of 0.81% per cycle up to 300 cycles. The cycling performance is much better than that of the previously reported data with high-loaded sulfur cathodes.
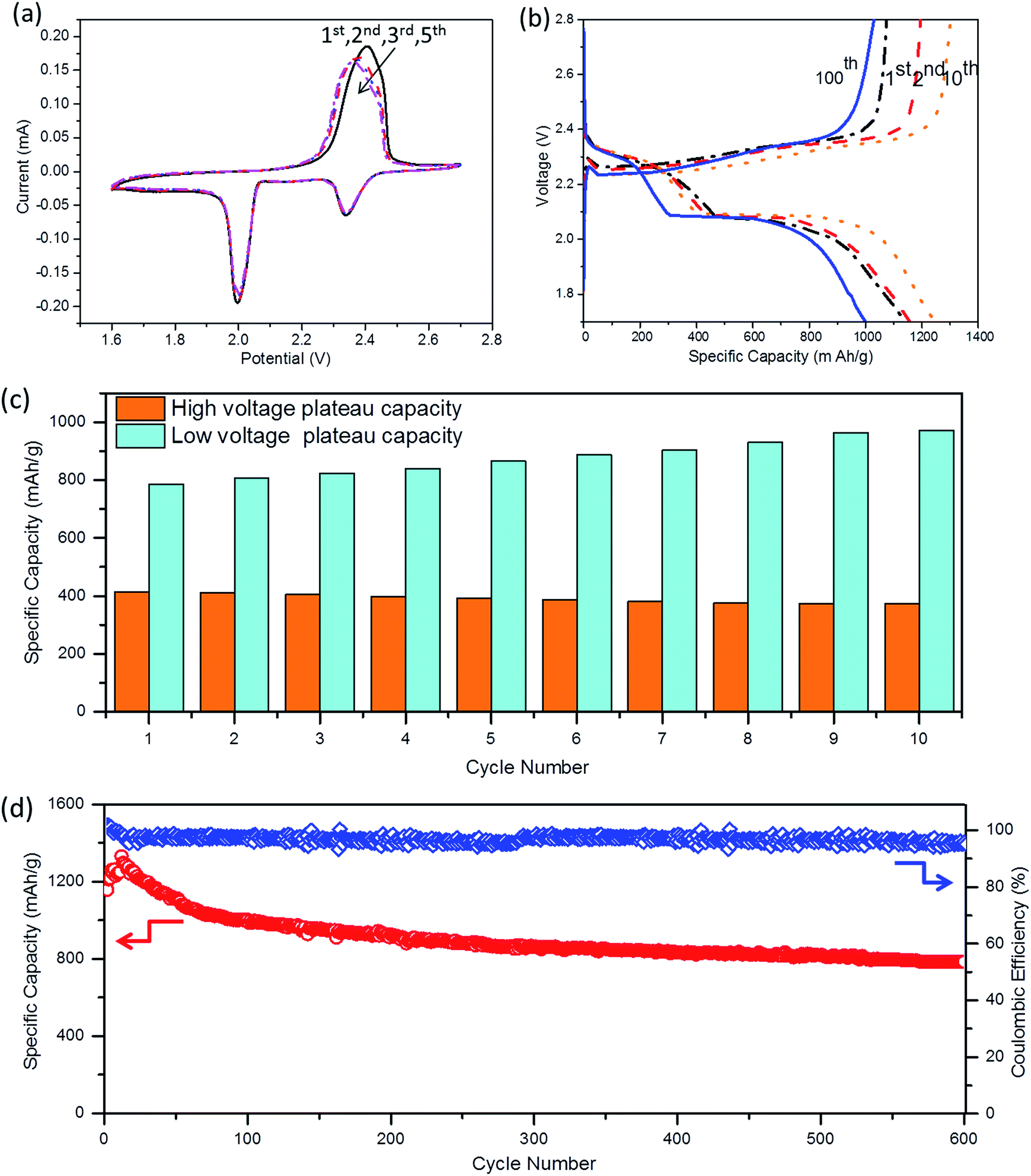 | ||
| Fig. 7 Electrochemical characterization of the A-NSG material as the cathode of the Li–S battery. (a) Cyclic voltammetry (CV) measured between 1.7 and 2.7 V at a sweep rate of 0.1 mV s−1 for the first, second, third, and fifth cycles. (b) Galvanostatic charge and discharge profiles for different cycles at 0.2C. (c) The discharge capacities of high and low voltage plateaus. The onset voltage of the low plateau was defined around 2.0 V. (d) Long term cycling performance test of the A-NSG@S electrode at 0.2C discharge rate and the corresponding coulombic efficiency. | ||
The high conductivity and porous structure enables the A-NSG@S composites to be kinetically sustainable for a wide range of current rates. Generally, the rate performance of a battery can be reflected from two characteristics i.e. the capacity maintenance at stepped rates and polarization during high-current charge and discharge processes. To explore the rate performance of A-NSG@S cathodes, we tested batteries at different current rates. The voltage gap between charge and discharge plateaus was slightly enlarged when the current rate was increased tenfold, and was stable for 200 cycles (Fig. 8b). When increasing current rates, reversible capacities of 1203, 1120, 987, and 855 mA h g−1 were obtained at current rates of 0.1C, 0.5C, 1C, and 2C (Fig. 8c), respectively. Even at a high current rate of 5C, a reversible capacity of 651 mA h g−1 was still retained. When the current rate was gradually returned back to 0.1C, a reversible capacity of 1021 mA h g−1 was recovered, showing excellent high rate capability.
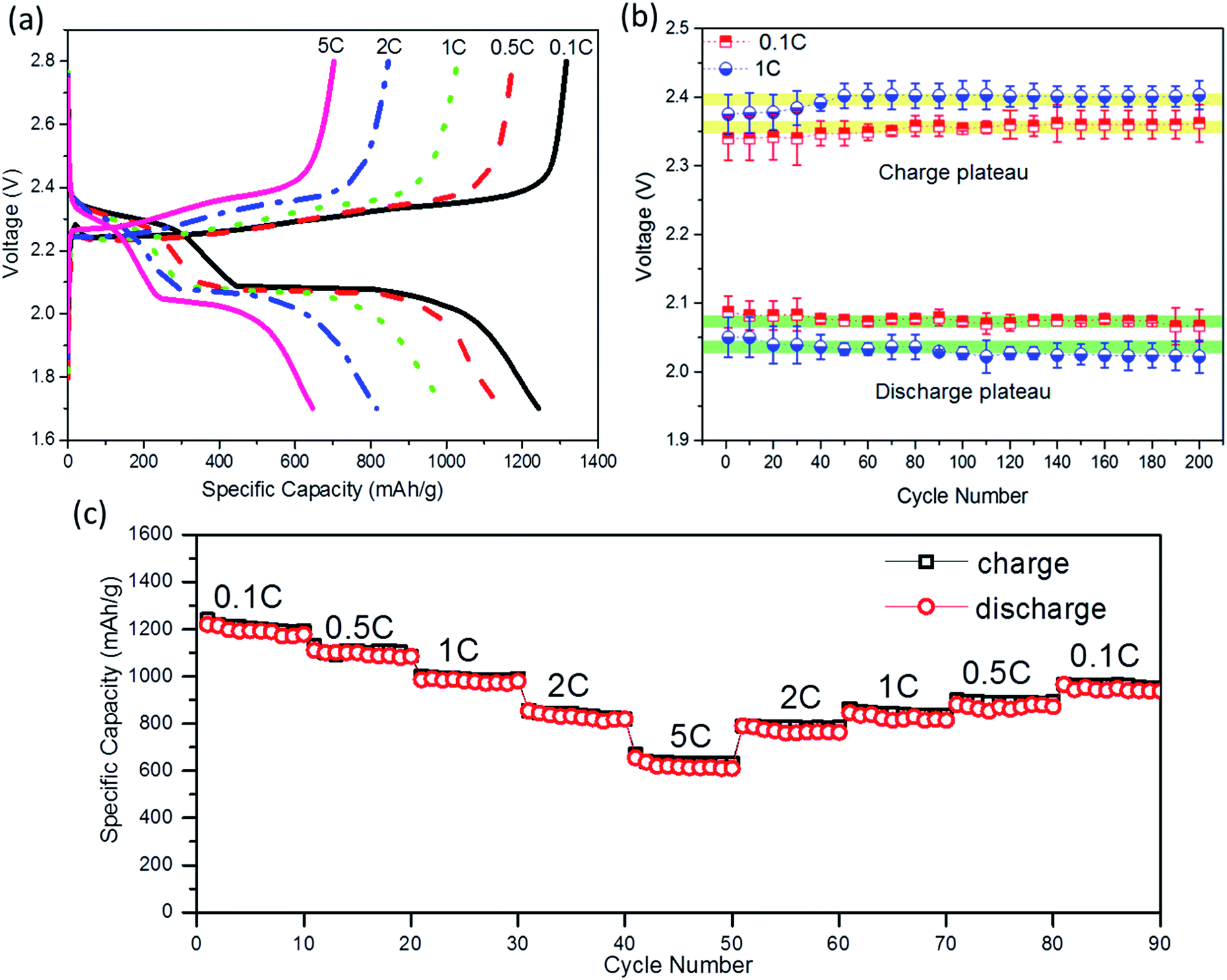 | ||
| Fig. 8 Electrochemical characterization of the A-NSG material as the cathode of the Li–S battery. (a) Galvanostatic charge and discharge profiles at 0.1C, 0.5C, 1C, 2C, and 5C. (b) Voltage plateaus for charge and discharge processes over 200 cycles at 0.1 and 1C. (c) Discharge/charge capacity cycled at various rates from 0.1C, 0.5C, 1C, 2C, and 5C. | ||
The Incremental Capacity Analysis (ICA) allows us to probe any gradual changes in the electrochemical behavior of rechargeable batteries during a cycle-life test with greater sensitivity than those based on the conventional charge and discharge curves. The advantage of ICA is achieved by transforming either the voltage or inflection points on V vs. Q curves into clearly identifiable ΔQ/ΔV peaks on the ICA curves, depicting the solid solution or phase transformation characteristics.36,37 By monitoring the evolution of these ΔQ/ΔV peaks upon cycling, we can access key information on the cell chemistry behavior. Fig. 9a shows the incremental capacity peaks derived from the discharge curves (0.2C) for different cycles. The ICA peak located at 2.05 V is slightly broadened upon cycling, suggesting that the characteristics of charge transfer might have been altered, which may be associated with an increased diffusion path for lithium polysulfides due to the volume expansion during cycling. However, this voltage vibration caused by volume expansion is almost negligible compared with other reports,23,27 because our 3D porous graphene networks and graphene layers can buffer the volume expansion/contraction of activated materials upon cycling. Meanwhile, both positions of the two peaks remain almost unchanged, implying that polarization is almost unchanged for 600 cycles. This result is consistent with the outcomes shown in Fig. 8b and 9b. Moreover, although a slight intensity reduction appears in both peaks due to the inevitable active material loss, the position as well as the sharpness of both peaks is maintained unchanged for 600 cycles, which further confirms that electrode materials preserve excellent electrochemical stability. In particular, the chemically activated A-NSG@S functions as a 3D immobilizer to accommodate a large amount of S active materials, and the internal graphene layer greatly increases the polysulfide utilization due to the improved overall electrical conductivity of the structure.
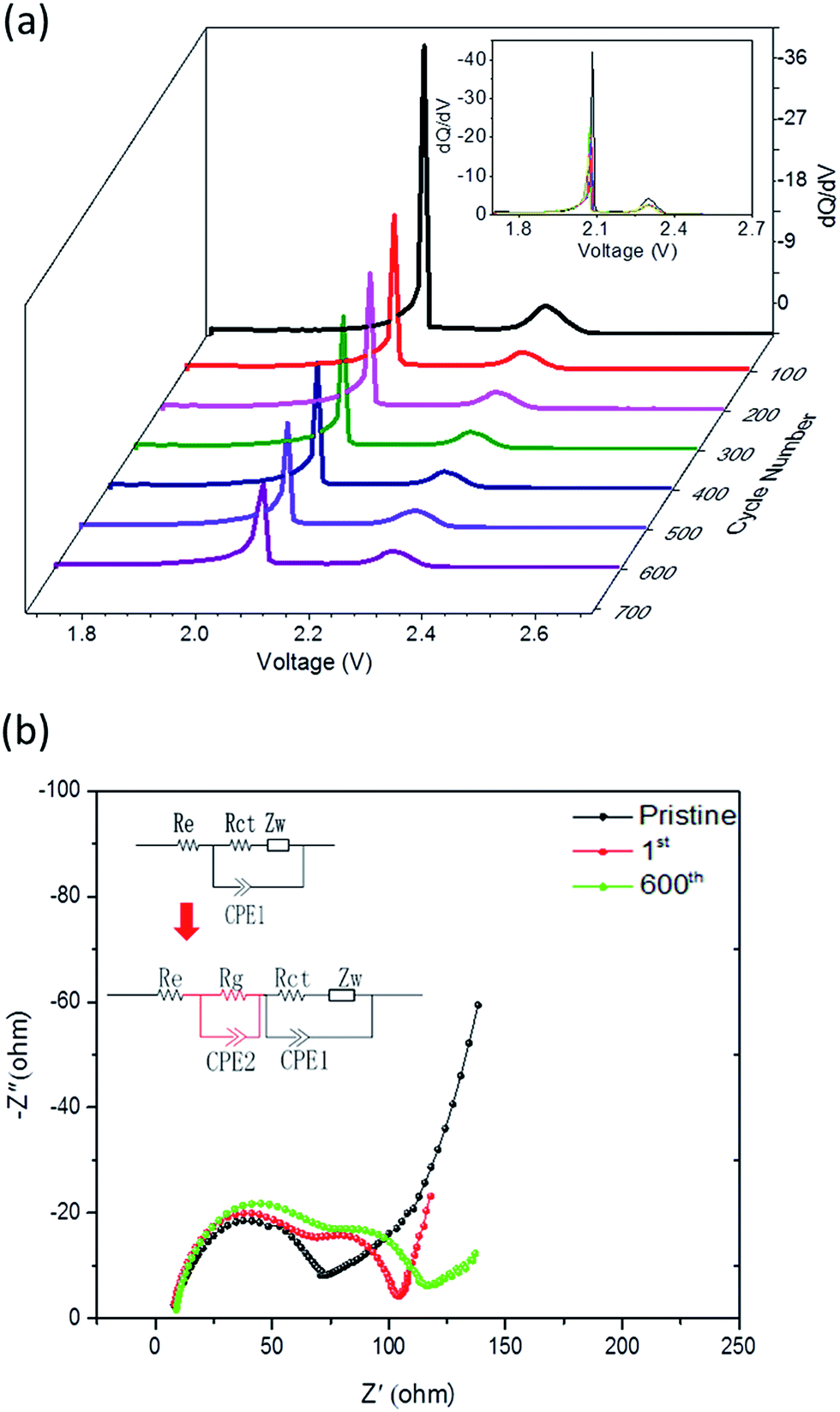 | ||
| Fig. 9 (a) dQ/dV plots of the A-NSG@S electrode. (b) Electrochemical impedance spectra of the A-NSG@S lithium–sulfur battery at different cycles. | ||
To further understand the improved electrochemical performance of A-NSG@S composites, electrochemical impedance spectroscopy (EIS) was employed to characterize the cathode before cycling, after the first cycle and 600 cycles. Nyquist plots are shown in Fig. 9b. For the cathode before cycling, the impedance curve is composed of one depressed semicircle at high frequency and a short inclined line in the low frequency regions. The semicircle corresponds to the internal resistance of the cathode including bulk impedance and interfacial impedance. In addition, the inclined line at low frequency reflects the lithium ion diffusion into the active mass.38,39 In contrast, the cathode after cycling exhibited two depressed semicircles followed by a long sloping line. The depressed semicircle in the high frequency region represents the charge transfer process at the carbon matrix interface, which also dominates the reduction reaction along the upper voltage plateau, while the semicircle at middle frequencies could be ascribed to the formation of insoluble Li2S, Li2S2, which controls the lower voltage plateau.13,40 Based on this analysis, the two features of impedance spectra in Fig. 9b can be used to deduce the equivalent circuits 1 and 2, respectively (shown as the inset). In the equivalent circuits, Re represents the resistance of the electrolyte, Rct is the charge transfer resistance at the conductive agent interface, and CPE is a constant phase element, which is associated with the roughness of the particle surface. CPE2 describes the space charge capacitance of the Li2S (or Li2S2) film and Rg is the resistance in the Li2S (or Li2S2) film. ZW is the Warburg impedance due to the diffusion of the polysulfides within the cathode. The fitting results are shown in Fig. 9b. Parameter identification results are summarized in Table S4 (ESI†), according to the equivalent circuit. It can be seen that Re experienced a slight increase during cycling due to the inevitable dissolution of polysulfides. In contrast, Rg increased remarkably during the first cycle, compared with that of a pristine cell, while it shows a modest rise in the following cycles. We can draw a conclusion that the solid Li2S (or Li2S2) appeared at the very beginning of the second voltage plateau, but due to the dominant pyridinic and pyrrolic N in the carbon lattice of A-NSG@S, the electron distribution is modified and the affinity for insoluble Li2S is improved, and the reversible conversion of Li2S/polysulfide/S is correspondingly promoted. Similarly, Rct exhibits the same trend as Rg during cycling. As for the equivalent component CPE2, which contains two parameters Y and n, the dimension for Y is Ω−1 cm−2 s−n or S cm−2 s−n, and the parameter n is a dimensionless index. The admittance response expression for CPE is:
| Y = Y0ωncos(nπ/2) + jY0ωnsin(nπ/2) | (1) |
However, after 600 cycles, both Y and n mostly remained stable as shown in Table S4 (ESI†), showing that the electrochemical stability of CPE2 is excellent after 600 cycles. Most importantly, this electrochemical stability also contributes greatly for the improvement of the electrochemical performance of lithium–sulfur batteries.
To demonstrate whether the co-doped heteroatoms of N and S and the graphene chemically activated by K2CO3 have positive functions on the electrochemical performance of Li–S batteries, several other comparison materials were prepared and tested. These include sulfur composites with about 70 wt% of sulfur prepared via a two step melt-diffusion method to produce nitrogen–sulfur co-doped graphene (NSG), K2CO3 activated graphene (A-G), nitrogen–sulfur co-doped and K2CO3 activated graphene (A-NSG), and graphene (rGO), denoted as NSG@S, A-G@S, A-NSG@S, and rGO@S, respectively. For comparison, the cycle performances of these electrodes were tested at the same current density, 0.2C rate, within a voltage window of 1.7–2.7 V, as shown in Fig. S11 and S12 (ESI†). The capacity retention for rGO@S, NSG@S and A-G@S electrodes up to 100 cycles is 38%, 55.2%, and 56.8%, respectively, which is not comparable with that of A-NSG@S. It also further confirmed the effective function of co-doping as well as K2CO3 activation. Furthermore, we compared the stability of A-NSG@S with other co-doped hosts as shown in Table S5.†48–52 Apparently, the sulfur electrode made of A-NSG exhibits a decay rate of 0.056% per cycle up to 600 cycles, which it superior to that of other co-doped hosts. Additionally, we can see that the cycle performance of the N, S doped carbon framework is better than that of N, O doped. This is because that N and S co-doped carbon exhibits asymmetric charge density distribution, namely, N bears a negative charge and S bears a positive charge, which benefits charge transfer.52,56 Therefore, the excellent electrochemical performance of the A-NSG@S electrodes is clearly associated with their unique structure: (i) the 3D porous graphene network and graphene layers facilitate electron transfer, and remarkably buffer the volume expansion/contraction of active materials upon cycling. (ii) The inner defects, edges and porous structure not only permit a high sulfur loading in a homogeneously dispersed amorphous state, but also are beneficial for electrolyte access to the active sulfur component, leading to efficient reactions with Li+. (iii) Combined physical adsorption of lithium polysulfides onto porous graphene and the chemical binding of polysulfides to N and S sites in the A-NSG promote reversible Li2S/polysulfide/S conversion, realizing high performance Li–S batteries with long cycle life and high-energy density.
Conclusions
In summary, we designed an efficient strategy for preparing crumpled N–S co-doped porous graphene (A-NSG) via chemical activation of polypyrrole (PPy) functionalized graphene sheets with K2CO3. The A-NSG@S electrodes with a high sulfur loading of 72.4 wt% exhibit low polarization, stable cycling performance, and excellent rate capability, compared with rGO@S, NSG@S and A-G@S electrodes. The excellent performance could be attributed to synergic effects, including the high surface area and high conductivity of the porous matrix, as well as the unique lithium polysulfide binding capability of the N, S functional groups in the A-NSG sheets. Furthermore, pyridinic nitrogen and thiophenic sulfur in the carbon lattice of A-NSG@S can modify the electron distribution and improve the affinity for Li2S as well as Li2S4, leading to the improvement in reversibility of Li2S/polysulfide/S conversion. This work not only presents a simple method to synthesize N–S co-doped hierarchical porous graphene, but also demonstrates the excellent performance of A-NSG cathode materials for lithium–sulfur batteries.Acknowledgements
This project is financially supported by the Australian Renewable Energy Agency (ARENA) through an ARENA project (ARENA 2014/RND106). The author Jing Xu gratefully acknowledges the support from the Chinese Scholarship Council (CSC, No. 201307090011).Notes and references
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- X. Fang, W. Weng, J. Ren and H. Peng, Adv. Mater., 2016, 28, 491–496 CrossRef CAS PubMed.
- L. Qie, C. Zu and A. Manthiram, Adv. Energy Mater., 2016, 7, 23–25 Search PubMed.
- H. Kim, J. Lee, H. Ahn, O. Kim and M. J. Park, Nat. Commun., 2015, 6, 7278 CrossRef CAS PubMed.
- H. Chen, Q. Zou, Z. Liang, H. Liu, Q. Li and Y. C. Lu, Nat. Commun., 2015, 6, 5877 CrossRef CAS PubMed.
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS PubMed.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- H. Chen, C. Wang, C. Hu, J. Zhang, S. Gao, W. Lu and L. Chen, J. Mater. Chem. A, 2015, 3, 1392–1395 CAS.
- Z. Ma, S. Dou, A. Shen, L. Tao, L. Dai and S. Wang, Angew. Chem., 2015, 54, 1888–1892 CrossRef CAS PubMed.
- M. Wang, H. Zhang, Q. Wang, C. Qu, X. Li and H. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 3590–3599 CAS.
- Q. Sun, X. Fang, W. Weng, J. Deng, P. Chen, J. Ren, G. Guan, M. Wang and H. Peng, Angew. Chem., 2015, 54, 10539–10544 CrossRef CAS PubMed.
- X. Zhao, H. J. Ahn, K. W. Kim, K. K. Cho and J. H. Ahn, J. Phys. Chem. C, 2015, 119, 7996–8003 CAS.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., 2015, 54, 4325–4329 CrossRef CAS PubMed.
- S. Xiao, S. Liu, J. Zhang and Y. Wang, J. Power Sources, 2015, 293, 119–126 CrossRef CAS.
- P. Strubel, S. Thieme, T. Biemelt, A. Helmer, M. Oschatz, J. Brückner, H. Althues and S. Kaskel, Adv. Funct. Mater., 2015, 25, 287–297 CrossRef CAS.
- X. G. Sun, Y. Fang, X. Jiang, K. Yoshii, T. Tsuda and S. Dai, Chem. Commun., 2016, 52, 292–295 RSC.
- M. Rao, X. Geng, X. Li, S. Hu and W. Li, J. Power Sources, 2012, 212, 179–185 CrossRef CAS.
- M. Liu, D. Zhou, Y. B. He, Y. Fu, X. Qin, C. Miao, H. Du, B. Li, Q. H. Yang, Z. Lin, T. S. Zhao and F. Kang, Nano Energy, 2016, 22, 278–289 CrossRef CAS.
- Y. Y. L. Ding, P. Kopold, K. Hahn, P. A. van Aken, J. Maier and Y. Yu, Adv. Funct. Mater., 2015, 34, 578–584 Search PubMed.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed.
- Y. Qiu, W. Li, W. Zhao, G. Li, Y. Hou, M. Liu, L. Zhou, F. Ye, H. Li, Z. Wei, S. Yang, W. Duan, Y. Ye, J. Guo and Y. Zhang, Nano Lett., 2014, 14, 4821–4827 CrossRef CAS PubMed.
- L. Fei, X. Li, W. Bi, Z. Zhuo, W. Wei, L. Sun, W. Lu, X. Wu, K. Xie, C. Wu, H. L. Chan and Y. Wang, Adv. Mater., 2015, 27, 5936–5942 CrossRef CAS PubMed.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- J. P. Rong, M. Y. Ge, X. Fang and C. W. Zhou, Nano Lett., 2014, 14, 473–479 CrossRef CAS PubMed.
- M. Agostini, B. Scrosati and J. Hassoun, Adv. Energy Mater., 2015, 78, 785–793 Search PubMed.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai and P. J. Ferreira, Science, 2011, 332(6037), 1537–1541 CrossRef CAS PubMed.
- Y. Xi, Z. Long, Z. Fan and H. Yi, ACS Nano, 2014, 8(5), 5208–5215 CrossRef PubMed.
- C. Zu and A. Manthiram, Adv. Energy Mater., 2014, 4(18), 6784–6789 Search PubMed.
- Z. Li and L. Yin, ACS Appl. Mater. Interfaces, 2015, 7, 4029–4038 CAS.
- S. Zhen, S. Lin, C. Jing, B. Wen and W. Feng, ACS Nano, 2011, 5(6), 4350–4358 CrossRef PubMed.
- Y. Shao, S. Zhang, M. H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I. A. Aksay and Y. Lin, J. Mater. Chem., 2010, 20, 7491 RSC.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef CAS PubMed.
- S. Chen, X. Huang, H. Liu, B. Sun, W. Yeoh, K. Li, J. Zhang and G. Wang, Adv. Energy Mater., 2014, 4, 159–165 Search PubMed.
- C. Wang, K. Su, W. Wan, H. Guo, H. H. Zhou, J. T. Chen, X. X. Zhang and Y. H. Huang, J. Mater. Chem. A, 2014, 2, 5018–5023 CAS.
- J. Ryu, D. Hong, S. Choi and S. Park, ACS Nano, 2016, 10, 2843–2851 CrossRef CAS PubMed.
- J. Nelson, S. Misra, Y. Yang, A. Jackson, Y. J. Liu, H. L. Wang, H. J. Dai, J. C. Andrews, Y. Cui and M. F. Toney, J. Am. Chem. Soc., 2012, 134, 6337–6343 CrossRef CAS PubMed.
- C. Barchasz, J. C. Leprêtre, F. Alloin and S. Patoux, J. Power Sources, 2012, 199, 322–330 CrossRef CAS.
- Z. Li, J. T. Zhang, Y. M. Chen, J. Li and X. W. Lou, Nat. Commun., 2015, 6, 8850 CrossRef CAS PubMed.
- L. Yuan, X. Qiu, L. Chen and W. Zhu, J. Power Sources, 2009, 189, 127–132 CrossRef CAS.
- J. Zhang, Z. Dong, X. Wang, X. Zhao, J. Tu, Q. Su and G. Du, J. Power Sources, 2014, 270, 1–8 CrossRef CAS.
- C. Xu, Y. Wu, X. Zhao, X. Wang, G. Du, J. Zhang and J. Tu, J. Power Sources, 2015, 275, 22–25 CrossRef CAS.
- T. Z. Hou, X. Chen, H. J. Peng, J. Q. Huang, B. Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283–3291 CrossRef CAS PubMed.
- T.-Z. Hou, H.-J. Peng, J.-Q. Huang, Q. Zhang and B. Li, 2D Mater., 2015, 2, 014011 CrossRef.
- L. Miao, W. Wang, K. Yuan, Y. Yang and A. Wang, Chem. Commun., 2014, 50, 13231–13234 RSC.
- G. Zhou, L. Li, C. Ma, S. Wang, Y. Shi, N. Koratkar, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2015, 11, 356–365 CrossRef CAS.
- L.-C. Yin, J. Liang, G.-M. Zhou, F. Li, R. Saito and H.-M. Cheng, Nano Energy, 2016, 25, 203–210 CrossRef.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. B. Wu, C. Hao, S. Liu, J. Qiu and X. W. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- H.-J. Peng, T.-Z. Hou, Q. Zhang, J.-Q. Huang, X.-B. Cheng, M.-Q. Guo, Z. Yuan, L.-Y. He and F. Wei, Adv. Mater. Interfaces, 2014, 1, 1400227 CrossRef.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., 2015, 54, 4325–4329 CrossRef CAS PubMed.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed.
- J. Guo, J. Zhang, F. Jiang, S. Zhao, Q. Su and G. Du, Electrochim. Acta, 2015, 176, 853–860 CrossRef CAS.
- Y. Liu, J. Guo, J. Zhang, Q. Su and G. Du, Appl. Surf. Sci., 2015, 324, 399–404 CrossRef CAS.
- Y. Wu, C. Xu, J. Guo, Q. Su, G. Du and J. Zhang, Mater. Lett., 2014, 137, 277–280 CrossRef CAS.
- M. Hagen, P. Schiffels, M. Hammer, S. Dorfler, J. Tubke, M. J. Hoffmann, H. Althues and S. Kaskel, J. Electrochem. Soc., 2013, 160, A1205–A1214 CrossRef CAS.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., 2015, 54, 2131–2136 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta05878g |
| ‡ Jing Xu and Dawei Su contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |