
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The nature and effects of rhodium and antimony dopants on the electronic structure of TiO2: towards design of Z-scheme photocatalysts†
E. N. K.
Glover
 ,
S. G.
Ellington
,
G.
Sankar
and
R. G.
Palgrave
*
,
S. G.
Ellington
,
G.
Sankar
and
R. G.
Palgrave
*
University College London, Chemistry Department, Christopher Ingold Laboratories, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: r.palgrave@ucl.ac.uk
First published on 1st February 2016
Abstract
The nature and effects of rhodium and antimony doping in TiO2 have been investigated using X-ray diffraction (XRD), X-ray Photoelectron Spectroscopy (XPS), Extended X-ray Absorption Fine Structure (EXAFS) analysis, X-ray Absorption Near Edge Structure (XANES) analysis and diffuse reflectance spectroscopy. Together these techniques build up a comprehensive picture of the dopant chemistry in this system. A range of Sb/Rh ratios have been analysed, and it is shown that the Fermi level can be tuned within the band gap through control of dopant stoichiometry, offering the possibility of designing Z-scheme components with specific band offsets. Spontaneous spatial segregation of dopants is measured using TiO2 (110) single crystals. This is shown to have a direct impact on the electronic structure in the region of the semiconductor surface, creating band bending that is expected to be critical for photocatalytic activity of these materials.
Introduction
Photocatalysis has applications in many different important processes such as water splitting, water cleaning, and microbial deactivation.1–3 TiO2 is an abundant, naturally-occurring mineral that has been widely studied since its photocatalytic properties were discovered;4 as a result a large amount of research has been devoted specifically to understanding and improving the photocatalytic processes that can occur at its surface.5–8Photocatalysis can only take place for a given reaction when the conduction band edge of the catalyst material lies at a more negative potential than the reduction potential of the reaction and the valence band edge at a more positive potential than the oxidation potential of the reaction.9 TiO2 fulfils this criterion for water photolysis and can therefore successfully split water into hydrogen and oxygen. However, since the two common forms of TiO2, rutile and anatase, have bands gaps of 3.00 eV and 3.20 eV respectively,10 they are active under Ultra Violet (UV) radiation only. Since UV light accounts for just 4–5% of solar radiation that reaches the Earth's surface,11 much research has focused on modifying TiO2 based photocatalysts to enable their activation under visible light and improve the overall efficiency of these catalysts. Since the conduction band edge is positioned appropriately to enable H+ reduction, modification is usually focussed on changes to the valence band. Addition of dopants can act by mixing of the dopant and basis material valence band orbitals (as in N- and S-doping12–14) or by introduction of discrete energy levels above the basis material valence band (as in C- and B-doping).15–19 Noble metal doping in titanate semiconductor materials has been an area of interest for a number of years, with much research indicating that these materials produce a visible light response.20–25
However, despite intensive research for decades, to date there is no stable single phase material, based on TiO2 or otherwise, capable of efficiently (i.e. >10% quantum yield) carrying out overall (i.e. non-sacrificial) water splitting using sunlight. It may be that more complex doping regimes are necessary in order to achieve efficient visible light photocatalysis, and recently TiO2 and other materials co-doped with two or more dopants together have been studied. The greater complexity afforded by using multiple dopants offers opportunities for fine tuning of electronic, optical and chemical properties that may lead to the desired visible light active catalysts. However, greater complexity requires more detailed understanding of the physical and chemical effects of the dopants on the host material. An alternative to single phase catalysts is to couple photocatalysts together. By forming a heterojunction, photoexcited electrons and holes can be separated by migrating across the junction according to the relative energies of the valence and conduction bands of the two catalysts. A notable type of heterojunction currently receiving much attention is the Z-scheme.26 This mechanism mimics biological photosynthesis, which is based on two photosystems. Z-schemes have a number of advantages, chiefly that two narrow band gap semiconductors can be used to produce the same redox power as a single wide band gap semiconductor.
When selecting materials to combine to form a water splitting Z-scheme it is necessary to consider how the band edges of the two materials align with each other and the redox potentials of water. This can be achieved by comparing the band edges relative to the Fermi level (EF) for each material. To date, materials that naturally exhibit band edges at appropriate offsets have been used to construct Z-schemes, for example, Ta3N5/TaON.27
Overall, the trend in photocatalysis research is for greater complexity, either in codoped systems or through combination of two or more phases to create heterojunctions and Z-schemes. In both these approaches, there is a need for a good understanding of the catalysts themselves to allow rational design of effective materials. Here we seek to better understand the effects of a complex doping regime on the properties of TiO2. Rh/Sb codoped TiO2 and related materials, such as Y2Ti2O7, SrTiO3 and BaTiO3, have been recently developed as effective photocatalysts, both as single phase materials and as part of a Z-scheme.28–31 Domen et al. produced a Z-scheme combining Ta3N5 and Rh doped SrTiO3, achieving visible light water splitting, with H2 evolution at rates of up to 12 μmol h−1, when loaded with an Ir co-catalyst. Rosseinsky and co-workers have achieved >7 μmol−1 O2 evolution using visible light over a Rh-doped Y2Ti2O7 catalyst.25 Kudo and co-workers32 reported visible light photoactivity of Rh and Sb codoped TiO2 (where Sb/Rh = 0, 0.5, 1, 2 and 3 were investigated) and showed that, with Sb/Rh ≥ 2, O2 evolution exceeds 10 μmol h−1. Their Sb/Rh doped SrTiO3 system has also achieved sacrificial O2 and H2 evolution under visible light.29
It has been shown that doping TiO2 with Rh introduces 4d t2g states into the band gap.33,34 The occupancy of these Rh 4d states naturally depends on the oxidation state of the Rh dopant. It has been found that upon doping of TiO2 with Rh onto the Ti4+ site, Rh4+ is the most stable oxidation state, presumably in order to maintain overall charge neutrality. However, Rh4+ possesses a partially filled 4d t52g energy level, the inclusion of which causes the Fermi level to be positioned at a lower energy than that of pristine TiO2. The addition of electrons by introducing Sb(V), allows t62g Rh(III) to exist in the system and the n-type character of the material to be retained.32,34
Here we report a comprehensive study of the dopant chemistry of Rh/Sb doped TiO2. By using both ceramic powder samples and single crystals we are able to gain greater understanding of the effects of Rh/Sb codoping regimes in TiO2, which can allow us to tune the electronic properties, making possible the design and optimisation of the individual Z-scheme components. We demonstrate the use of these co-dopants to tune the Fermi level position to yield materials with the required band offsets to form a Z-scheme.
Where previous investigations have focussed on equimolar doping regimes, here a range of dopant ratios have been incorporated into the TiO2 lattice and comparisons between TiO2:Rh and TiO2:RhSb have been made. A comprehensive characterisation of the materials has been completed using XPS, XRD, UV-vis, EXAFS and XANES to provide information about the electronic structures of the materials in addition to the oxidation states, chemical environments and concentration of dopant ions. In order to profile the dopant concentration penetrating into the materials, a TiO2 single crystal was doped by high temperature diffusion, depth profiling used to determine the spatial distribution of dopants, and revealing that the spontaneous spatial arrangement of dopants leads to a built in electric field that may be highly beneficial for charge carrier separation in photocatalysis.
Experimental section
Rhodium doped TiO2 samples were synthesised by solid-state reaction of TiO2 powder (Aldrich 99.99%) with the appropriate amount of Rh2O3. The powders were ground using an agate pestle and mortar, calcined at 1100 °C for 10 hours, then re-ground and calcined at 1100 °C for a further 10 hours.Antimony co-doped TiO2 samples were produced by grinding Rh doped powders with the appropriate amount of Sb2O3, calcination at 900 °C for 10 hours, re-grinding and calcination at 900 °C for a further 10 hours.
For comparison an antimony doped sample was synthesised, again by solid stated reaction. TiO2 was ground with the appropriate amount of Sb2O3, calcined at 900 °C for 10 hours, then re-ground and calcined at 900 °C for a further 10 hours. Table 1 outlines the samples and their synthesis conditions.
| Sample | Calcination temperature (°C) | Total calcination time (h) |
|---|---|---|
| TiO2:Rh[1%] | 1100 | 20 |
| TiO2:Rh[3%] | 1100 | 20 |
| TiO2:Rh[5%] | 1100 | 20 |
| TiO2:Rh[7%] | 1100 | 20 |
| TiO2:Rh[9%] | 1100 | 20 |
| TiO2:Rh[5%]Sb[1%] | 1100 then 900 | 20 |
| TiO2:Rh[5%]Sb[3%] | 1100 then 900 | 20 |
| TiO2:Rh[5%]Sb[5%] | 1100 then 900 | 20 |
| TiO2:Rh[5%]Sb[7%] | 1100 then 900 | 20 |
| TiO2:Rh[5%]Sb[9%] | 1100 then 900 | 20 |
| TiO2:Sb[5%] | 900 | 20 |
A TiO2 single crystal was doped with Rh and Sb by solid state diffusion of dopants from the prepared doped powder into a TiO2 (110) single crystal, purchased from Alineason.
The phase composition of the resultant powders was confirmed by X-ray diffraction (XRD) on a Bruker D4 powder diffractometer, using Cu-Kα radiation (λ = 1.54 Å). The surface compositions of the powders were measured by X-ray photoelectron spectroscopy (XPS) using a Thermo K-alpha instrument utilising a 72 W monochromated Al Kα X-ray source (with photon energy of 1486.6 eV). A dual beam flood gun was used to compensate for sample charging, instrument specific relative sensitivity factors were used to normalise the data and the binding energy scale was referenced by setting the C 1s peak from adventitious carbon to 285.0 eV.
Sb and Rh K-edge X-ray Absorption Spectra (XAS) were taken at beamline (B18), Diamond Light Source, UK. The beamline is equipped with Si (111) double crystal monochromator, ion chambers for measuring incident and transmitted beam intensity and operates at 3 GeV and 300 mA. All measurements were carried out in absorption mode and typically 12 scans were averaged to produce the data. Spectra were processed using Athena software.
Results
X-ray diffraction
X-ray diffraction patterns of all doped TiO2 samples confirm single phase samples, with no detectable Rh2O3 or Sb2O3 phases present. Patterns were indexed in the rutile structure (tetragonal P42/mnm) and lattice parameters were determined by least squares refinement. Both the a and c lattice parameters of all TiO2:Rh powders increase upon incorporation of Rh into the lattice up to 5% doping (ESI Fig. S1a and b†). The cell volume of TiO2:Rh[5%] is 62.5775 Å3 compared with 62.1216 Å3 for pristine TiO2. Above 5% Rh doping lattice parameters increase slightly. Given the modest increase in cell volume upon doping, it is expected that the Rh dopant substitutes onto the Ti site rather than interstitially, a conclusion supported by XAS data presented below. However, the ionic radius of six coordinate Rh4+ is very similar to that of Ti4+ (0.60 Å and 0.605 Å, respectively35) and therefore the observed lattice parameter increase cannot be explained in terms of the size of the dopant ion. It may indicate the presence of some Rh3+, which has an ionic radius of 0.665 Å,35 in these samples.Incorporation of Sb affects both lattice parameters; again an increase in both a and c parameters is observed as nominal Sb doping increases. However, above 5% Sb doping the lattice parameters decrease slightly. Since the presence of Sb5+ is expected to encourage the formation of Rh3+, the initial lattice parameter increase could be caused by an increased proportion of Rh3+ ions. The presence of bulk Sb3+, which has an ionic radius of 0.76 Å (ref. 35) and could also account for increases in these lattice parameters, is eliminated by XAS data presented below. With excess Sb doping, the lattice parameters decrease. In this regime, excess Sb5+ may be compensated for by titanium vacancies, leading to a reduction in unit cell size.
X-ray absorption spectroscopy
Fig. 1 shows the EXAFS Fourier transform of TiO2:Rh[5%] measured at the Rh K-edge which is compared with TiO2 (measured at the Ti K-edge) and Rh2O3 (Rh K-edge) standard powders. Labelled peaks show the distances between nearest neighbours (at around 1.5 Å) and second nearest neighbours (2.3–3.5 Å); note these distances appears short, since they are not corrected for the phase shift. A detailed analysis of the Rh K-edge EXAFS data shows the nearest neighbour distances TiO2:Rh (Fig. 1b) suggest the Rh–O distance in the TiO2:Rh system is ca. 2.0 Å which is similar to Rh–O distances in Rh2O3; this may be due to the presence of Rh2O3 phase or substituted Rh3+ in TiO2 lattice with increase in distance due to its higher ionic radii. More convincing evidence for Rh substitution in TiO2 lattice comes from the analysis of higher neighbours. Ti–Ti distances in TiO2 are 2.96, 3.4 and 3.6 Å whereas in Rh2O3 Rh–Rh distances are at ca. 2.72, 2.99, 3.3 and 3.52 Å. Thus by comparing these FTs, it appears that Rh–Ti distances are similar to those observed for TiO2 structure rather than Rh2O3. Therefore, we conclude that Rh ions are substituted in TiO2 structure than present as RhOx secondary phase.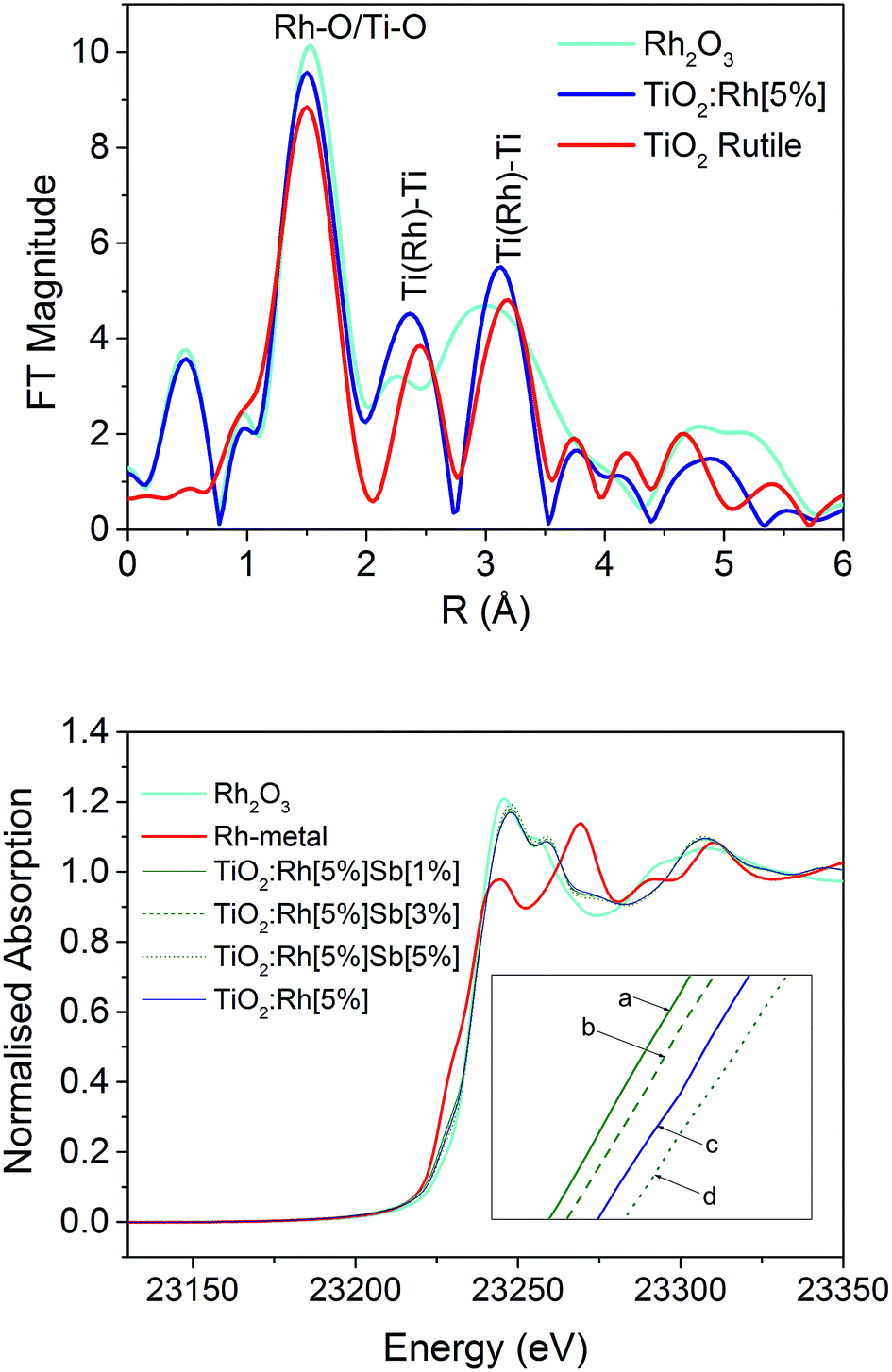 | ||
| Fig. 1 Fourier transform of the Rh K-edge EXAFS of TiO2:Rh[5%] by comparison with TiO2 (Ti K-edge EXAFS) and Rh2O3 (Rh K-edge EXAFS) standard powders (top), and Rh K-edge XANES of Rh2O3, Rh-metal, TiO2Rh[5%], TiO2Rh[5%]Sb[1%], TiO2Rh[5%]Sb[3%] and TiO2Rh[5%]Sb[5%] (bottom); note that the Fourier transform presented here are not phase shift corrected and therefore the distances appear shorter than expected values. Actual distances reported here are based on the full analysis of the EXAFS data which includes phase shift correction. | ||
Rh K-edge X-ray absorption near edge structure (XANES) data for Rh2O3, Rh-metal, and doped samples TiO2:Rh[5%], TiO2:Rh[5%]Sb[1%], TiO2:Rh[5%]Sb[3%] and TiO2:Rh[5%]Sb[5%] are also shown in Fig. 1 (bottom). It is clear that the XANES of all doped samples is closely comparable Rh2O3, and that there is minimal similarity with the absorption spectrum of Rh-metal. Between 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 215 eV and 23235 eV some distinction can be seen between doped samples (Fig. 1b inset), suggesting variance in Rh oxidation state across the series.
215 eV and 23235 eV some distinction can be seen between doped samples (Fig. 1b inset), suggesting variance in Rh oxidation state across the series.
The Ti/Sb-edge EXAFS Fourier transform data for TiO2, Sb2O3, Sb2O5 and doped samples TiO2:Rh[5%]Sb[1%], TiO2:Rh[5%]Sb[3%] and TiO2:Rh[5%]Sb[5%] (ESI, Fig. S2a†) demonstrates the lack of distinct position of Sb ions in the doped samples. It is unclear if Sb ions reside on the Ti4+ site, though second nearest neighbour distances suggest that as more Sb is incorporated into the material, the Sb environment is more Sb2O5-like. However, what is more clear is that X-ray absorption of the Sb K-edge (ESI Fig. S2b†) shows that all Sb atoms are likely Sb(V), as opposed to Sb(III).
X-ray photoelectron spectroscopy
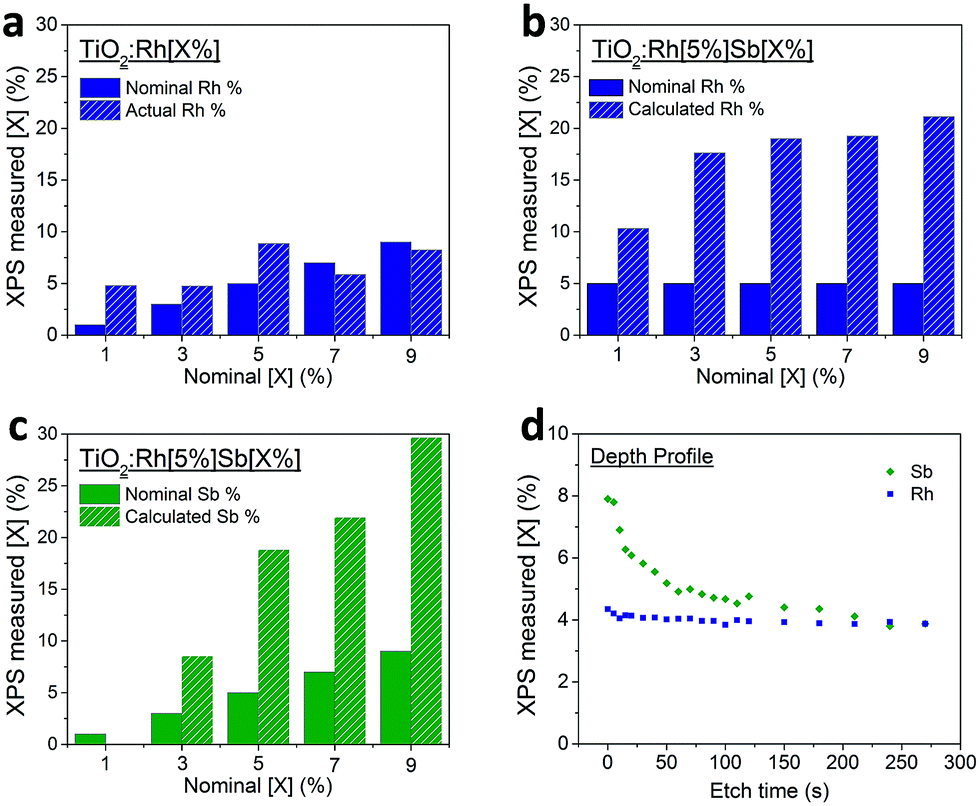 | ||
| Fig. 2 (a–d) Dopant percentage amount as determined from XPS; (a) Rh in TiO2:Rh, (b) Rh in TiO2:RhSb and (c) Sb in TiO2:RhSb, (d) depth profile showing atomic% of Rh and Sb as determined by XPS spectra taken after intervals of etching with an Ar+ ion gun. | ||
In the highest levels of doping studied here, TiO2:Rh[5%]Sb[9%], the Rh and Sb cations represent over half of the surface cations detected by XPS. Such a large change in surface composition will inevitably have effects on surface photochemical processes. It has been reported previously that Sb dopants surface segregate in Sb doped TiO2, although this has been speculated to be due to the presence of Sb3+ lone pair cations preferring surface sites where they may adopt a less symmetric coordination. The attraction of Rh dopants to Sb at the surface identified here is best explained by a defect clustering model where Sb5+ and Rh3+ dopants preferentially reside on neighbouring or closely spaced cation sites in order to maintain local charge neutrality. The preference of Sb dopants at surface sites is therefore seen to ‘pull’ Rh ions out of the bulk to the surface. This is evidence not only for electronic interaction between the dopants (i.e. reduction of Rh4+ to Rh3+ through Sb doping), but also significant interdependence of the spatial distribution of dopants within the TiO2 matrix.
To investigate further, a single crystal diffusion study was undertaken. The random orientation and small size of particulates in powder samples is not conducive to meaningful compositional depth profiling using XPS or similar techniques. Instead, by using a doped TiO2 single crystal as a model system, the relationship between bulk and surface dopants can be revealed more clearly. A TiO2 (110) polished single crystal was heated under a TiO2:Rh[5%]Sb[9%] powder at 1100 °C for 10 h to induce diffusion of the dopants into the crystal. Depth profiles were then recorded in XPS using Ar ion sputtering. Fig. 2d shows the amount of dopant determined by XPS at intervals between Ar+ ion etchings.
As with doped TiO2 powders, Sb in this doped TiO2 single crystal is much more highly concentrated at the surface than in the bulk. The depth profile reveals that relative Sb percentage diminishes further into the material. By comparison, Rh is evenly distributed throughout the depth studied. It is evident that Rh diffuses into the TiO2 (110) surface to a much greater degree than Sb under the conditions used. This could be due to either a higher diffusion coefficient of Rh – i.e. the Rh ions (of whatever oxidation state) can move through the TiO2 lattice more easily than Sb ions can, or alternatively, the Sb ions are thermodynamically more stable at the surface and do not diffuse extensively to the bulk. Surface enrichment of Rh in this material is less pronounced than in powder samples, though a small amount of enrichment is observed in the first few layers of the profile. This interesting contrast with the powder samples may indicate that the (110) TiO2 surface is atypical in not showing significant surface enrichment of Rh in the presence of Sb dopants.
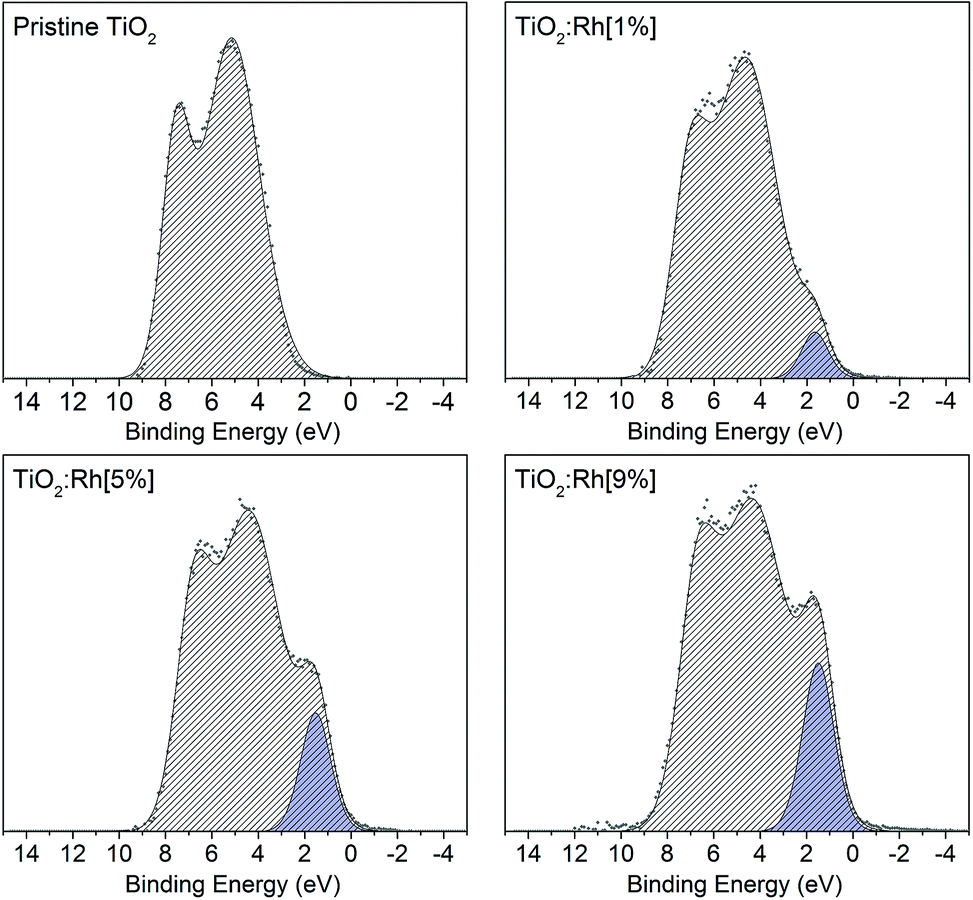 | ||
| Fig. 3 Valence band spectra of pristine TiO2 and 1, 5 and 9% Rh doped TiO2. As the nominal amount of Rh increases, the prominence of an in-gap feature due to Rh 4d t2g electrons also increases. | ||
The area of this component is proportional to the Rh content, and is assigned to the presence of filled Rh 4d t2g states. In samples codoped with Sb, this band gap feature becomes more prominent with increased Sb doping. While EXAFS results indicated that bulk Sb dopant ions are Sb5+, it is still possible that surface Sb3+ is present,34,35 which would be expected to contribute filled Sb 5s states to the valence band region. Such features were not identified here, which may suggest the amount of Sb3+ is small, or that an Sb 5s feature is masked by the more intense Rh 4d peak. Since codoped samples were synthesised with nominal 5% Rh, this increased prominence of the Rh valence band feature correlated with the increased surface segregation of Rh cause by the presence of Sb as a codopant.
Firstly, we can determine the extent of Fermi level shift by using O 1s peak position as a reference point. Since the EF of a material is calibrated as 0 eV in XPS, any shift of EF will be observed as a shift of all other core line peaks. Assuming as we do that O2− is not chemically changed by the doping process, the absolute position of the main O 1s peak and any shift that is observed by comparison to that of O 1s in pristine TiO2 can be assumed to be caused by movement of the Fermi level.
Secondly, the energy difference between the O 1s peak and a second core line will change only due to chemical shift of the second core line. This can be used to eliminate the effect of Fermi level shift from the Rh corelines and allow accurate assessment of any chemical shift.
The presence of antimony presents the complication that the Sb 3d5/2 core line lies under the O 1s core line and is masked by it completely. For quantification of dopants the Sb 3d3/2 peak, which lies 9.4 eV higher than Sb 3d5/2,39 was utilised along with the appropriate relative sensitivity factor determined from the spectrometer. However, for determination of the binding energy of the O 1s peak, a Sb 3d5/2 peak was fitted by constraining its area (2:3 ratio with Sb 3d3/2), full-width half-maximum (equal to Sb 3d3/2) and doublet separation (9.4 eV lower than Sb 3d3/2) to that of the Sb 3d3/2. Additional components to represent the oxide anions and another to account for surface hydroxyl groups, which manifest as a shoulder to the high binding energy side of the O 1s, were added. An example spectrum is shown in Fig. S3 of the ESI;† the O 1s positions quoted in Fig. 4 and Table 2 are those of the oxide anion generated from these fits.
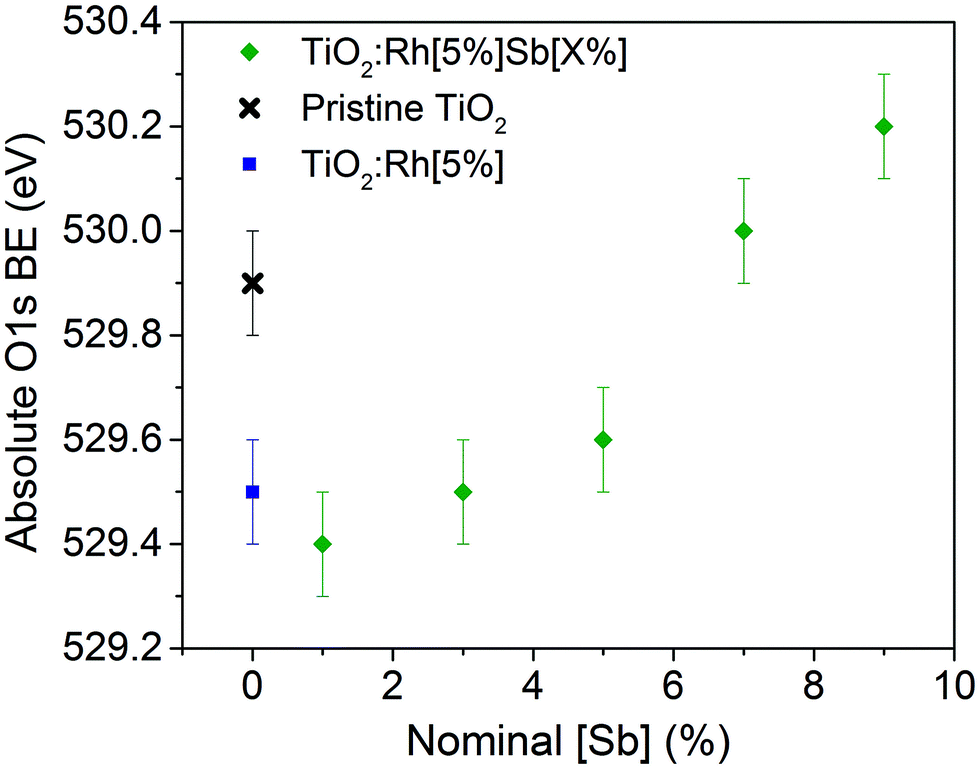 | ||
| Fig. 4 Oxide O 1s absolute binding energy vs. nominal amount of Sb dopant. As O2− is assumed chemically unchanged in the doped systems by comparison with pristine TiO2, the observed variation in oxide O 1s binding energy can be attributed to movement of the Fermi level, which provides the 0 eV binding energy reference in XPS. | ||
| Sample | O 1s position (eV) | Core line separation (O 1s–Rh 3d5/2) (eV) | Band gap energy (eV) |
|---|---|---|---|
| a Band edge obscured by Rh d–d transition. | |||
| TiO2:Sb[5%] | 530.1 | — | 3.1 |
| TiO2:Rh[5%] | 529.0 | 220.2 | |
| TiO2:Rh[5%]Sb[1%] | 528.4 | 220.2 | |
| TiO2:Rh[5%]Sb[3%] | 529.5 | 220.0 | |
| TiO2:Rh[5%]Sb[5%] | 529.6 | 220.2 | 2.25 |
| TiO2:Rh[5%]Sb[7%] | 530.0 | 220.6 | 2.24 |
| TiO2:Rh[5%]Sb[9%] | 530.2 | 220.8 | 2.23 |
TiO2:Rh[5%] exhibits an oxide O 1s shift of −0.4 eV by comparison to pristine TiO2. As can be seen in Fig. 4, codoping with Sb causes the oxide O 1s peak position to shift to higher binding energies as more Sb is doped into the sample. Undoped TiO2 is an n-type semiconductor, where the Fermi level is pinned close to the conduction band minimum by the presence of a small concentration of Ti3+ defects. The oxide O 1s core line shifts observed here are interpreted as follows. Upon Rh doping of TiO2, the Fermi level shifts downwards, towards the valence band, by 0.4 eV. This is due to the introduction of partially filled Rh 4d states within the band gap. When Sb is codoped into the structure in increasing amounts, the Fermi level (EF) rises. This is assigned to filling of the Rh states within the band gap through introduction of Sb5+, an electron dopant and reduction of Rh4+ to Rh3+, which has a filled t62g configuration in the low spin state. Since EF is the reference point for the binding energy scale in XPS, a rising EF leads to an apparent increase in binding energy for all core lines. Since the chemical shift of the oxide O 1s peak is assumed to be small, the shift in oxide O 1s shown in Fig. 4 is directly representative of the variation in EF induced by the cooping regime. Thus, there is a direct link between the Rh/Sb ratio of dopants in TiO2 and the EF position: more Rh rich materials will have more p-type character, whilst more Sb rich materials will be more n-type in character. Interestingly, at nominal equimolar Sb/Rh doping, the Fermi level is still significantly lower than in pristine TiO2, showing that not all of the Rh4+ has been reduced to Rh3+. It is not until Sb/Rh ≥ 1.4 that the Fermi level is situated at a higher energy even than that of pristine TiO2. This is in agreement with reported Electron Spin Resonance (ESR) measurements, which indicate that Sb/Rh must be greater than 1 to eliminate all Rh3+,32 and perhaps suggests that some Sb is present in the +3 charge state, or that some Sb is lost from the samples on annealing, making the nominal Sb doing values higher than the true value. The total variation seen in the O 1s peak across all samples is 0.8 eV. Thus variable levels Sb doping can be used to tune the Fermi level of these materials within a good proportion of the band gap, and this may be a useful method for designing photocatalytic materials with specific band edge relationships, for example, when constructing a Z-scheme.
The interpretation of Rh oxidation state from the Rh core line XPS peaks is also complicated by competing chemical shifts and Fermi level movement. Once again utilising the well-defined and chemically unaltered oxide O 1s core line as a reference point, the effect of Fermi level shift on the Rh core line can be removed, allowing determination of any chemical shift in the Rh 3d coreline.
As shown in Fig. 5, the absolute binding energy of Rh 3d5/2 electrons in TiO2:Rh[5%] and TiO2:Rh[5%]Sb[9%] is 309.3 eV for both samples. Ordinarily, absent Fermi level shifts, this would indicate that Rh in these samples is in the same oxidation state. Fig. 5 shows comparison between two samples with the same nominal Rh amount but different nominal Sb amounts, TiO2:Rh[5%] and TiO2:Rh[5%]Sb[9%]. The O 1s–Rh 3d5/2 core line separation is considerably larger (220.8 eV) in TiO2:Rh[5%]Sb[9%] than in TiO2:Rh[5%] (220.2 eV). Again, assuming that the oxide O 1s peak is unmoved by chemical shift, any increase in core line separation will be due to change in the chemical shift of the Rh 3d line. An increase in separation represents a movement of the Rh 3d coreline to lower binding energy, indicating that Rh in TiO2:Rh[5%]Sb[9%] is in a lower oxidation state than in TiO2:Rh[5%]. This correlates with the expected oxidation states of Rh in TiO2:Rh[5%] (+4) and TiO2:Rh[5%]Sb[9%] (+3). The O 1s–Rh 3d5/2 core line separation values for all codoped samples are shown in Table 2. By comparison all singly Rh doped samples exhibited a core line separation value of between 220.0 eV and 220.3 eV, indicating that only in samples where Sb/Rh ≥ 1.4 does it appear that all Rh is found in the +3 state.
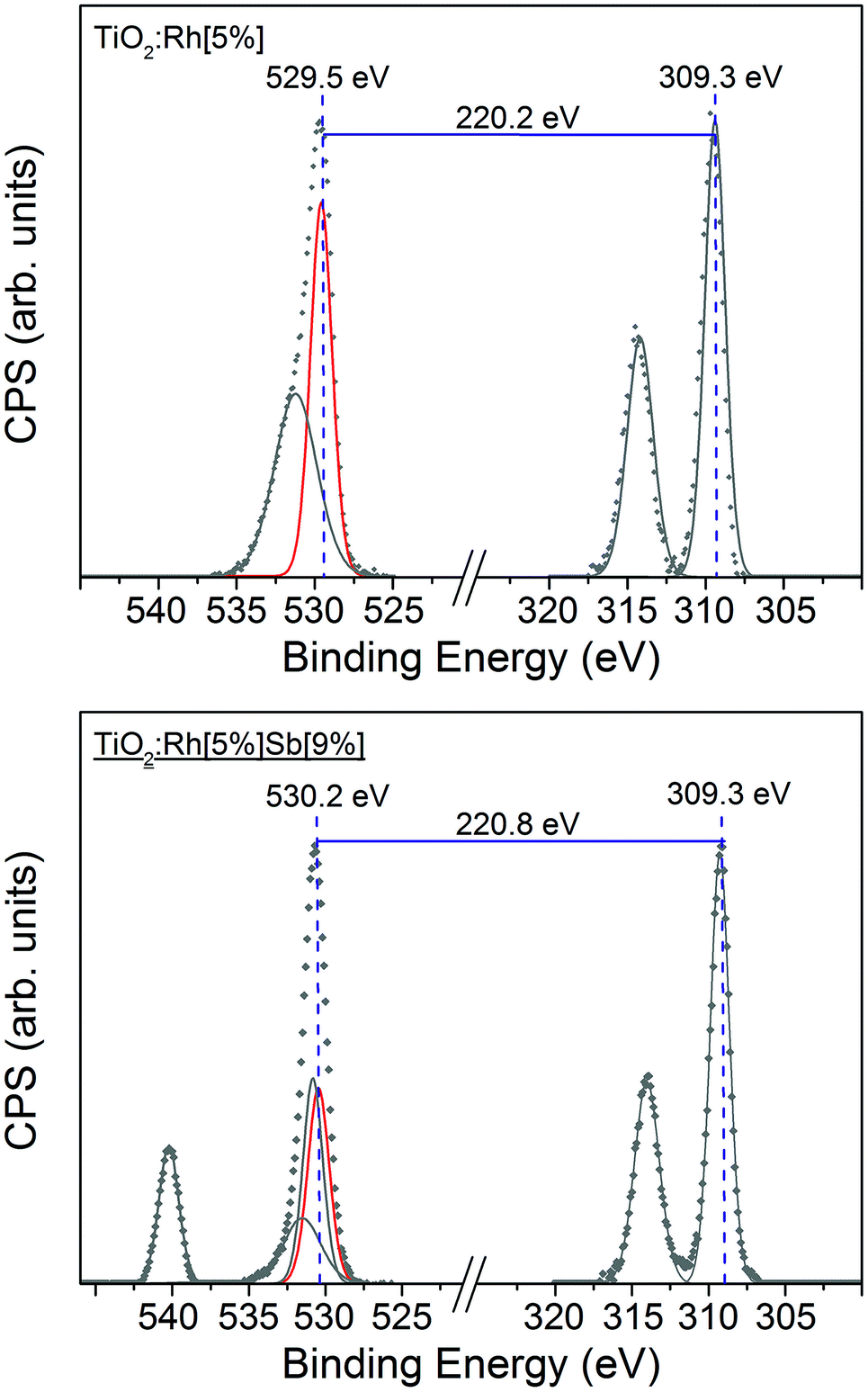 | ||
| Fig. 5 Binding energy separation between O 1s and Rh 3d peaks in TiO2:Rh[5%] (top) and TiO2:Rh[5%]Sb[9%] (bottom). The energy difference between the corelines increases with Sb doping. Assuming a static oxide O 1s peak with no chemical shift, the increasing energy difference upon Sb doping is attributed to a decrease in the binding energy of the Rh 3d coreline, corresponding to a less positive oxidation state for Rh. | ||
UV-vis analysis
Diffuse reflectance spectra transformed using the Kubelka–Munk function40 are shown in Fig. 6 in the range 0.6–3.5 eV for all samples. Two changes in the optical absorption spectra of TiO2 are seen upon Rh doping. Firstly, a broad absorption peak appears centred at 1.0 eV. Secondly, the band edge, which in TiO2 is around 3 eV, shifts to lower energy, corresponding to a band gap, calculated using the Tauc method, of 1.8 eV, although as discussed below, this is thought not to be a true interband transition but rather a d–d transition. Upon Rh/Sb doping, both these new features are modified. On increasing levels of Sb doping, the feature at 1.0 eV decreases in intensity, reaching a minimum at TiO2:Rh[5%]Sb[5%]. The band edge moves to higher energies, resulting in a band gap of 2.2 eV. These features can be interpreted in terms of the electronic configuration of the dopants. Rh4+ has a 4d5 configuration, and doping into oxide matrices has been reported to cause absorption band in the near infrared, as is observed here.41,42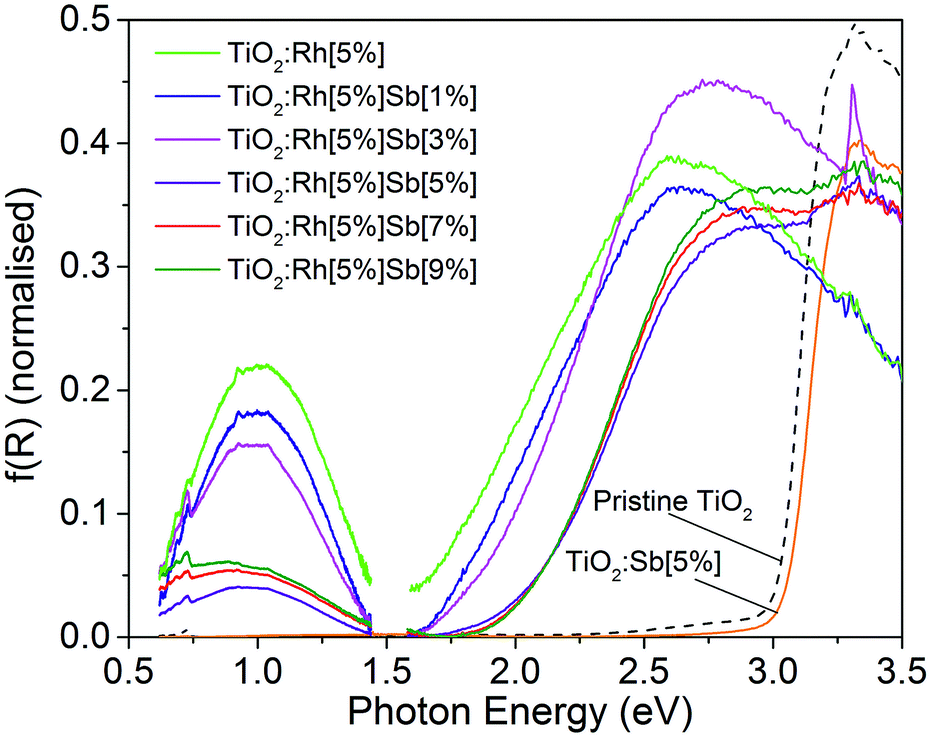 | ||
| Fig. 6 Absorption spectra for all TiO2:Rh[5%]Sb[X%] powders. A decrease in photon energy required for photoexcitation is exhibited by all Rh doped samples by comparison to pristine TiO2. An additional absorption around 1.0 eV, which can be attributed to a valence band to Rh4+ t2g transition, is prominent in samples containing Rh4+. The reduction in intensity of this feature is evidence of the suppression of Rh4+ in samples of higher Sb5+ concentration. | ||
This has been assigned to a O 2p → Rh t2g transition, i.e. a transition from the TiO2 VB to the partially filled interband gap Rh 4d state. The VB spectra in Fig. 3 shows that the Rh 4d states are situated approximately 1 eV above the TiO2 VB, which correlates with the optical transition observed here. Upon addition of Sb, the Rh4+ is reduced to Rh3+ which has 4d6 configuration. In a low spin state, the Rh t2g orbitals are filled, so the O 2p → Rh t2g transition does not occur. This correlates with the decrease in intensity of the 1.0 eV absorption feature with increasing Sb. Previously, the 1.0 eV transition in similar oxide systems has been assigned to Rh d–d transitions.43 However, in the few reported electronic spectra of Rh4+ compounds, the Rh4+ d orbital splitting is known to be significantly larger than 1.0 eV.44 The shift in band gap to lower energies upon Rh doping can be assigned to Rh 4d → conduction band (CB) transitions; similar transitions are seen in Rh doped oxides. Given this assignment, it is unexpected that the band gap should increase upon Sb doping: reduction of Rh should lead to raising of the Rh 4d energy levels toward the CB, which would result in a smaller band gap.
Another possibility is that in the Rh4+ doped samples, a Rh d–d transition occurs in the visible region. There are few reports of the electronic spectra of low spin Rh4+ compounds. [RhF6]2− displays its lowest energy d–d transition at 2.54 eV. Naturally for an oxide coordinated Rh ion the d orbital splitting will vary. Low spin Rh3+ d6 ions have no spin allowed d–d transitions. Therefore it is possible that he feature resembling a band edge at 1.8 eV may be a Rh4+ d–d transition, which is attenuated as the Rh is reduced upon addition of Sb codopant.
Discussion
It has been established by the results reported above that the electronic structure, specifically the Fermi level, of Rh/Sb co-doped TiO2 is strongly dependent upon the Rh/Sb ratio. In a semiconductor, the occupation of inter band states strongly affects the Fermi level. Here, EF depends upon the filling of Rh 4d orbitals that are positioned between the TiO2 VB and CB. The total EF shift possible in the range of dopant ratios studied here is 0.8 eV, between TiO2:Rh[1%]Sb[1%] and TiO2:Rh[5%]Sb[9%]. This suggests that combination of these two materials in a Z-scheme would be viable.It has also been shown that the spatial distribution of Sb and Rh cations within the TiO2 matrix is quite complex. In Rh doped TiO2, with no Sb co-dopant, there is modest surface segregation of the Rh. However, upon introduction of Sb, surface segregation of Rh is significantly enhanced – Rh is pulled from the bulk of TiO2 to the surface, presumably by favourable defect clustering. The single crystal study presented in Fig. 2d illustrates that Sb is strongly surface segregated in TiO2 (110) crystals, which serves as a model for the polycrystalline powders used in photocatalysis. In contrast, Rh is present throughout the single crystal in approximately even proportions. Due to this spontaneous spatial arrangement of the dopants, the Rh/Sb ratio changes with depth (Fig. 2d). As already stated, the Rh/Sb ratio strongly affects the Fermi level in this system. Therefore, it can be concluded that the Fermi level, relative to the band edges also changes with depth in the Rh/Sb doped single crystal studied here. The surface is more Sb rich, so will be more n-type, whilst the bulk is more Rh rich, so will be more p-type. Thus by the spatial arrangement of the dopants, a material with a built in electric field is produced – such a field will certainly affect the charge carrier migration necessary for photocatalysis, and the direction of the field is expected to enhance electron migration towards the surface whilst hindering hole movement in the same direction.
The Rh/Sb ratio can be calculated at each point along the depth profile of the co-doped TiO2 (110) crystal shown in Fig. 2d. The position of the Fermi level relative to the band edges can then be estimated at each point by matching the Rh/Sb ratio with a curve drawn through the experimental points shown in Fig. 4. Using this method, and assuming that the Fermi level is constant in the material (and so it is the band edges that change energy with depth), we have calculated the conduction band minimum (CBM) and valence band maximum (VBM) positions as a function of depth in our TiO2 (110) single crystal sample. This is shown in Fig. 7. It is clear that significant band bending, of around 0.5 eV, occurs due to the influence of the dopants spatial arrangement. It should be emphasised that only the effects of the dopants are shown in Fig. 7 – there will be other contributions to surface band bending, such as the presence of surface states, as occurs with all semiconductors.
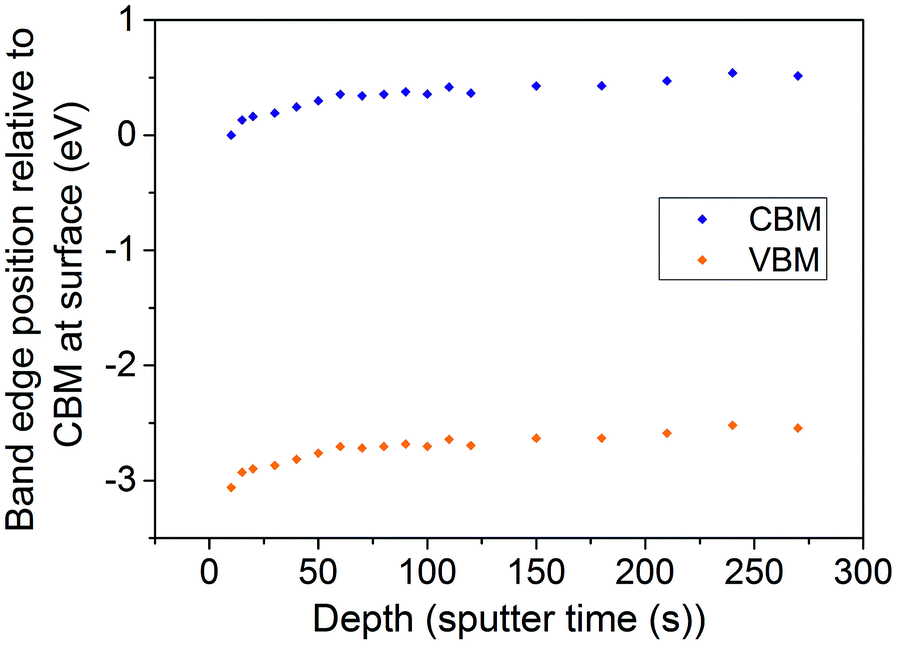 | ||
| Fig. 7 VBM and CBM variation with depth in a Sb/Rh codoped TiO2 (110) crystal. The CBM at the surface is placed at 0 eV and the band gap is assumed to be 3.06 eV as in bulk rutile TiO2. The Sb/Rh dopant ratio was measured by XPS depth profiling. This was correlated with the Fermi level shifts as described in the text. | ||
The band bending postulated for TiO2 (110) crystals may also apply to powders, and is likely to strongly affect charge carrier migration. The spatial confinement of dopants to affect (and effect) charge carrier migration is of fundamental importance in the field of silicon electronics. However, despite this the approach has not so far been intentionally taken in designing photocatalysts or photoelectrocatalysts. Our results indicate for the first time that a photocatalyst system exhibits spontaneous dopant segregation which is expected to cause significant band bending. This may contribute to the high photoactivity reported in this system; as previously stated Kudo and co-workers32 reported photoactivity of Rh and Sb codoped TiO2 (where Sb/Rh = 0, 0.5, 1, 2 and 3 were investigated) and showed that, with Sb/Rh ≥ 2, O2 evolution exceeds 10 μmol h−1. This increased knowledge of the electronic properties of these materials may serve as a starting point for design of more highly effective photocatalysts. It is important to note that the materials we have synthesised are model systems; the use of solid state synthesis results in low surface area powders due to the high temperatures used. This renders the samples unsuitable for photocatalysis, but provides a quick, simple and highly reproducible route to production of the materials, allowing a high throughput of varying doping ratios. As such we were not able to measure water splitting from these samples, but work is ongoing to translate the design principles derived here to high surface area nanomaterials.
Conclusions
This study investigates the effect of dopant ratio on the structural and electronic properties of Rh/Sb codoped materials. Issues with XPS chemical shift referencing were overcome by using the position of the well-defined and chemically unchanged oxide O 1s peak as a reference from which movement of EF (at 0 eV) and other core lines can be observed. The absolute position of oxide O 1s peaks across all codoped samples provided information about the position of EF within the bandgap, which is dependent of Rh oxidation state. It was observed that the O 1s peak appears to decrease in binding energy, which is indicative of a lowering of the Fermi level energy, when doped with rhodium. However, the O 1s binding energy is restored and even increased from that of pristine TiO2, with increasing levels of Sb codoping. It was determined that the Sb/Rh ratio must be atleast 1 to allow the Fermi level to revert to the position of pristine TiO2. These materials could provide the basis for Z-scheme photocatalysts, with highly tunable band alignments.Charge separation of O 1s and Rh 3d core lines provided further support that a reduction in oxidation state occurs when TiO2:Rh is codoped with Sb. A larger core-line separation for codoped powders indicates that Rh 3d electrons are held at a lower binding energy than Rh 3d electrons singly doped TiO2:Rh powders. This indicates, Rh3+ dominates in codoped powders whereas Rh4+ dominates in singly doped powders. This suppression of Rh4+ by Sb codoping was further corroborated by diffuse reflectance spectra, which show an additional absorption due to VB → Rh4+ 4d t2g transitions.
All samples showed evidence of surface enrichment of the dopant ions, a phenomenon that has previously been observed for Sb in TiO2 and SnO2.45,46 Sb incorporation was seen to cause migration of Rh from bulk to surface in all codoped powders, likely due to defect clustering of the Sb5+ and Rh3+ ions. A single crystal depth profile confirmed surface segregation of Sb in TiO2:RhSb. This spontaneous spatial arrangement of the co dopants is expected to cause a built in electric field in the region of the semiconductor surface, which may strongly affect the photogenerated charge carrier migration and hence photocatalytic and photoelectrocatalytic efficiency.
Detailed characterisation of these materials has revealed that they exhibit interesting structural and electrical properties in the context of photocatalytic water splitting. Ability to control the position of the Fermi level by dopant ratio, and surface segregation of dopants and its electronic effects, provide two interesting possibilities for optimising photocatalytic materials.
Acknowledgements
RGP acknowledges EPSRC grant EP/K014099/1. ENKG thanks the Department of Chemistry, UCL for funding a studentship. Dr Matthew S. Dyer is thanked for helpful discussions. The authors acknowledge Diamond B18 beamtime allocation number SP10306. Diego Gianolio is thanked for assistance with XAS measurements.Notes and references
- I. P. Parkin and R. G. Palgrave, J. Mater. Chem., 2005, 15, 1689–1695 RSC.
- K. Page, R. G. Palgrave, I. P. Parkin, M. Wilson, S. L. P. Savin and A. V. Chadwick, J. Mater. Chem., 2007, 17, 95–104 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. H. A. Fujishima, Nature, 1972, 238, 1 CrossRef.
- M. A. Henderson, Surf. Sci., 1996, 355, 151–166 CrossRef CAS.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- U. Diebold, J. Lehman, T. Mahmoud, M. Kuhn, G. Leonardelli, W. Hebenstreit, M. Schmid and P. Varga, Surf. Sci., 1998, 411, 137–153 CrossRef CAS.
- M. Pelaz, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 19 Search PubMed.
- M. J. Powell, C. W. Dunnill and I. P. Parkin, J. Photochem. Photobiol., A, 2014, 281, 27–34 CrossRef CAS.
- C. W. Dunnill, Z. A. Aiken, A. Kafizas, J. Pratten, M. Wilson, D. J. Morgan and I. P. Parkin, J. Mater. Chem., 2009, 19, 8747–8754 RSC.
- M. V. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS PubMed.
- H. Irie, Y. Watanabe and K. Hashimoto, Chem. Lett., 2003, 32, 772–773 CrossRef CAS.
- W. Ren, Z. Ai, F. Jia, L. Zhang, X. Fan and Z. Zou, Appl. Catal., B, 2007, 69, 138–144 CrossRef CAS.
- S. In, A. Orlov, R. Berg, F. García, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790–13791 CrossRef CAS PubMed.
- N. Lu, X. Quan, J. Li, S. Chen, H. Yu and G. Chen, J. Phys. Chem. C, 2007, 111, 11836–11842 CAS.
- Y. Matsumoto, T. Shimuzu and E. Sato, Electrochim. Acta, 1982, 27, 419–424 CrossRef CAS.
- I. Watanabe, Y. Matsumoto and E.-I. Sato, J. Electroanal. Chem. Interfacial Electrochem., 1982, 133, 359–366 CrossRef CAS.
- A. Kudo, R. Niishiro, A. Iwase and H. Kato, Chem. Phys., 2007, 339, 104–110 CrossRef CAS.
- Z. Zhang, A. Kladi and X. E. Verykios, J. Phys. Chem., 1994, 98, 6804–6811 CrossRef CAS.
- K. Maeda, D. Lu, K. Teramura and K. Domen, Energy Environ. Sci., 2010, 3, 470–477 Search PubMed.
- B. Kiss, C. Didier, T. Johnson, T. D. Manning, M. S. Dyer, A. J. Cowan, J. B. Claridge, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., 2014, 126, 14708–14712 CrossRef.
- H. J. Yun, H. Lee, N. D. Kim, D. M. Lee, S. Yu and J. Yi, ACS Nano, 2011, 5, 4084–4090 CrossRef CAS PubMed.
- W.-J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798–1803 CrossRef CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 150, 187–196 CrossRef.
- K. Furuhashi, Q. Jia, A. Kudo and H. Onishi, J. Phys. Chem. C, 2013, 117, 19101–19106 CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543–2546 RSC.
- R. Niishiro, R. Konta, H. Kato, W.-J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420–17426 CAS.
- S. Kitano, N. Murakami, T. Ohno, Y. Mitani, Y. Nosaka, H. Asakura, K. Teramura, T. Tanaka, H. Tada, K. Hashimoto and H. Kominami, J. Phys. Chem. C, 2013, 117, 11008–11016 CAS.
- F. E. Oropeza and R. G. Egdell, Chem. Phys. Lett., 2011, 515, 249–253 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. E. Castle, J. Adhes., 2008, 84, 368–388 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- M. V. Pessa, A. Vuoristo, M. Vulli, S. Aksela, J. Väyrynen, T. Rantala and H. Aksela, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20(8), 3115–3123 CrossRef CAS.
- V. Dzimbeg-Malcic, Z. Barbaric-Mikocevic and K. Itric, Tech. Gaz.–Univ. Osijek, 2011, 18, 7 Search PubMed.
- M. Kučera, J. Kuneš and R. Gerber, J. Appl. Phys., 1999, 85, 5986–5988 CrossRef.
- K. Shinagawa, E. Tobita, K. Ando, T. Saito and T. Tsushima, J. Appl. Phys., 1997, 81, 1368–1371 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- G. C. Allen, G. A. M. El-Sharkawy and K. D. Warren, Inorg. Chem., 1973, 12, 2231–2237 CrossRef CAS.
- P. A. Cox, R. G. Egdell, C. Harding, W. R. Patterson and P. J. Tavener, Surf. Sci., 1982, 123, 179–203 CrossRef CAS.
- A. Gulino, G. G. Condorelli, I. Fragalà and R. G. Egdell, Appl. Surf. Sci., 1995, 90, 289–295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta00293e |
| This journal is © The Royal Society of Chemistry 2016 |