
Ion specificities of artificial macromolecules
Lvdan
Liu
,
Ran
Kou
and
Guangming
Liu
*
Department of Chemical Physics, Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, P. R. China 230026. E-mail: gml@ustc.edu.cn
First published on 22nd August 2016
Abstract
Artificial macromolecules are well-defined synthetic polymers, with a relatively simple structure as compared to naturally occurring macromolecules. This review focuses on the ion specificities of artifical macromolecules. Ion specificities are influenced by solvent-mediated indirect ion–macromolecule interactions and also by direct ion–macromolecule interactions. In aqueous solutions, the role of water-mediated indirect ion–macromolecule interactions will be discussed. The addition of organic solvents to aqueous solutions significantly changes the ion specificities due to the formation of water–organic solvent complexes. For direct ion–macromolecule interactions, we will discuss specific ion-pairing interactions for charged macromolecules and specific ion–neutral site interactions for uncharged macromolecules. When the medium conditions change from dilute solutions to crowded environments, the ion specificities can be modified by either the volume exclusion effect, the variation of dielectric constant, or the interactions between ions, macromolecules, and crowding agents.
 Lvdan Liu | Lvdan Liu received her BS degree from the Department of Chemistry at Anhui Normal University in 2011. She received her PhD degree in physical chemistry from the University of Science and Technology of China in 2016. Her research interests focus on the ion specificities of thermosensitive polymers. |
 Ran Kou | Ran Kou received his BS degree from the Department of Chemistry at Anhui Normal University in 2013. He is currently pursuing his PhD degree at the Department of Chemical Physics at the University of Science and Technology of China. His research interests focus on the ion specificities of polyelectrolytes. |
 Guangming Liu | Guangming Liu is an associate professor in the Department of Chemical Physics at the University of Science and Technology of China (USTC). He received his PhD degree in physical chemistry from the USTC in 2007. From 2008 to 2010, he worked as a postdoctoral fellow at The Australian National University. His present research interests focus on the ion specificities of artificial macromolecules. |
1. Introduction
Ion specificities or Hofmeister effects are ubiquitous in biological and chemical systems.1–6 Ion specificities have attracted extensive attention since the Czech scientist Franz Hofmeister firstly discovered these effects in 1888 in the experiments on salt-induced protein precipitation.7–15 In general, the ions follow the series SO42− > HPO42− > CH3COO− > F− > Cl− > Br− > NO3− > ClO3− > I− > SCN− > ClO4− for anions and Mg2+ > Ca2+ > Li+ > Na+ > K+ > Rb+ > Cs+ > NH4+ > N(CH3)4+ > guanidinium+ for cations in terms of their ionic hydration strength.16,17The ions are usually categorized as chaotropes or kosmotropes based on their perceived influence on the water structure.18 The ions on the left side of the series, defined as kosmotropes, exhibit strong interactions with water molecules, whereas the ions on the right side of the series, defined as chaotropes, are weakly hydrated by water molecules.19 It has been widely recognized that chaotropes and kosmotropes exhibit distinct properties in aqueous solutions (Fig. 1).4,9,13,17,20 For example, in comparison with chaotropes, kosmotropes are more strongly hydrated by water molecules, generally leading to higher stability and lower solubility of proteins through the salting-out effect. Note that the ion sequence can reverse or partially alter in some cases depending on the surface charge and polarity (hydrophobicity/hydrophilicity),21–25 solution pH,26,27 salt concentration,28 buffer type,4,29 and counterion type.30,31
 | ||
| Fig. 1 Schematic illustration of the distinct properties between the kosmotropes and chaotropes in aqueous solutions. Adapted from ref. 17 by permission of Elsevier. | ||
Several models have been proposed to interpret the mechanism of specific ion effects.4,8,17,32,33 Collins proposed an empirical model in which oppositely charged ions with similar water affinities can form strong ion pairs, which dominates the specific ion–ion and ion–charged site interactions.8,32 Ninham et al. suggested that specific ion effects are due to the polarizability of ions and are manifested through ion dispersion forces.33,34 Kunz et al. suggested an alternative explanation to relate Hofmeister effects to the specific ion interactions with various surfaces such as hydrophobic solid–water and air–water interfaces, where kosmotropes are repelled from the surfaces but chaotropes are adsorbed onto the surfaces.35 Additionally, Jungwirth et al. performed a number of simulations to explore the mechanism of specific ion effects.36–39
During the past decades, much effort has been devoted to understanding the ion specificities of natural macromolecules (e.g., proteins, peptides, and DNA) and colloids. Ion specificities in biological and colloidal systems have been extensively reviewed.4,8,9,14,16,17,31,32,40,41 In analogy with the remarkable ion specificities of natural macromolecules, Hofmeister phenomena can also be observed in artificial macromolecules.13,20,42–53 Compared with that of natural macromolecules, the study on the ion specificities of artificial macromolecules has the following advantages:
(i) Artificial macromolecules usually have simple and well-defined chemical structures. Therefore, they may serve as effective model systems for understanding the ion specificities of natural macromolecules. For example, Cremer et al. employed poly(N-isopropylacrylamide) (PNIPAM) as a model system to study the specific ion effects on the solubility of macromolecules and attributed the observed ion specificities to the combined effect of three types of ion–PNIPAM interactions.20 As PNIPAM possesses a chemical structure similar to proteins, the proposed mechanism can be employed to interpret specific ion effects on proteins.20,28,54,55
(ii) With development in polymer synthesis, it is possible to precisely design the chemical structures of artificial macromolecules by introducing the desired groups into the backbone or the side group of polymers. This provides an opportunity to individually investigate the specific interactions between the ions and different kinds of chemical groups. This will be useful for revealing the role of each kind of specific ion interaction in the ion specificities of macromolecules.
(iii) Artificial macromolecules can be designed to have stimuli-responsive properties. This enables the ion specificities of artificial macromolecules to be investigated using a wide range of techniques such as light scattering, turbidity, fluorescence spectroscopy, quartz crystal microbalance, ellipsometry, and atomic force microscopy.20,56–60 This increases the potential range of studies of ion specificities of artificial macromolecules and should accelerate the understanding of the mechanisms behind the relevant Hofmeister phenomena.
(iv) Specific ion effects can be used to control various properties of artificial macromolecules such as the wettability of polymer surfaces,61–63 ice nucleation on polyelectrolyte brush surfaces,64 the mechanical properties of polymeric gels,65 the optical responses of ultrathin hydrogels,66 and the performance of polymeric organocatalysts.67 As artificial macromolecules have been applied in a very wide range of fields, studies of ion specificities of artificial macromolecules are important for uncovering new functions and applications of artificial macromolecules.
Therefore, rather than further discussing specific ion effects on naturally occurring macromolecules, we will focus on the ion specificities of artificial macromolecules in this review. We will review indirect and direct ion–macromolecule interactions in the second and third section, respectively. In the fourth section, we will discuss the ion specificities of artificial macromolecules in crowded environments. In the last section, we will discuss some current challenges and future directions in the field of ion-specific effects on artificial macromolecules.
2. Indirect solvent-mediated ion–macromolecule interactions
2.1 Aqueous solutions
There is a conventional concept that ions have an effect on the bulk water structure based on the variation in viscosity of salt solutions (eqn (1)):32,68| η/ηo = 1 + Ac1/2 + Bc | (1) |
Based on the Jones–Dole viscosity B coefficients, Collins conceptually divided the interfacial region near a solute (e.g., macromolecules) into three layers as shown in Fig. 2.32 The insertion of kosmotropic ions into the third water layer makes the bulk water a poorer solvent by decreasing the water accessible surface area of the solute, leading to a decrease in the solubility of the solute.32 In contrast, the insertion of chaotropic ions into the third water layer makes the bulk water a better solvent by increasing the water accessible surface area of the solute, resulting in an increase in the solubility of the solute, though the dominating factor for the increase of solute solubility should be direct ion–solute interactions.32
 | ||
| Fig. 2 Indirect water-mediated ion–solute interactions. Reproduced from ref. 32 by permission of Elsevier. | ||
Numerous studies have been conducted to explore the effect of indirect ion–macromolecule interactions on the ion specificities of artificial macromolecules.67,76–83 Freitag et al. investigated the ion-specific thermo-responsive behavior of PNIPAM and attributed the ion specificity to the structure making/breaking ability of the ions on the basis of viscosity B coefficients.76 Elaissari et al. showed that specific ion effects on the solubility of charged PNIPAM are dominated by the competition for water molecules between ions and PNIPAM.78 Additionally, indirect ion–macromolecule interactions also play an important role in the ion specificities of the swelling of polymeric hydrogels,79 the phase separation of poly(propylene oxide),80 the modulation of the catalytic efficiency of polymeric organocatalysts,67 the aggregation of thermo-sensitive charged microgels,81 and the conformational behavior of polymer brushes.82,83
Cremer et al. systematically studied specific anion effects on PNIPAM and on several other kinds of thermo-sensitive polymers in aqueous solutions.13,20,45,84,85 They proposed a model consisting of three types of interactions between anions, PNIPAM, and hydration water molecules to address anion specificity: specifically, (i) a salting-out effect caused by destabilization of hydrogen bonding between PNIPAM and hydration water molecules through anionic polarization (Fig. 3a), (ii) a salting-out effect caused by an increase in the PNIPAM/water interfacial tension due to the increase of salt concentration (Fig. 3b), and (iii) a salting-in effect caused by the direct binding of chaotropic anions on the PNIPAM surface (Fig. 3c).20 The respective effect can be analysed by modelling the salt concentration dependence of the lower critical solution temperature (LCST) of PNIPAM using eqn (2) including a constant term, a linear term and a Langmuir isotherm:20
 | (2) |
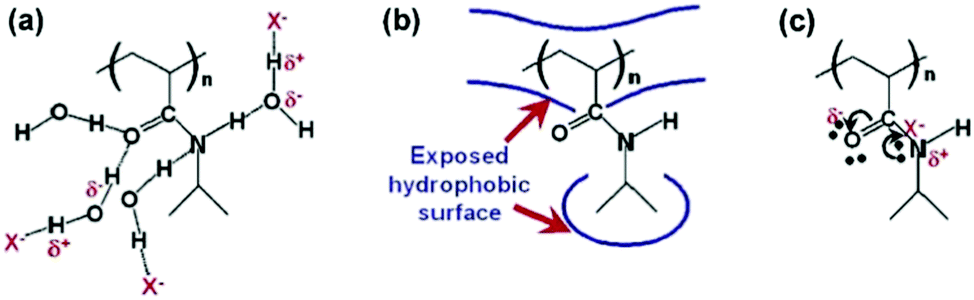 | ||
| Fig. 3 Interactions between anions, PNIPAM, and hydration water molecules. (a) The destabilization of hydrogen bonding between PNIPAM and the hydration water molecules through polarization by the anion, X−. (b) The hydrophobic hydration can be modulated by the anion through changing the surface tension. (c) Direct binding of the anion to the PNIPAM surface. Reproduced from ref. 20 by permission of the American Chemical Society. | ||
Similar to inorganic ions, organic ions, e.g., ionic liquids, also exhibit strong ion-specific effects on the phase transition behavior of PNIPAM mediated by indirect ion–PNIPAM interactions.88,89 Venkatesu and Reddy suggested that specific ion effects on the phase transition of PNIPAM are attributed to weakening of hydrogen bonds between the polymer and water molecules induced by the competition of organic ions with water molecules from the hydration shell.88 Byrne et al. also suggested that the interactions between water molecules and ionic liquids are responsible for the ion-specific LCST behavior of PNIPAM.89 Chaotrope-type ionic liquids enhance the hydration of PNIPAM caused by unfavourable water–anion interactions, whereas kosmotrope-type ionic liquids destabilize the hydration of PNIPAM due to strong interactions between water and anions.89
2.2 Aqueous–nonaqueous solvent mixtures
As water molecules can form solvent complexes with organic solvent molecules via hydrogen bonds,90,91 indirect water-mediated ion–macromolecule interactions should be affected by the addition of organic solvents. Thus, the ion specificities of artificial macromolecules in water–nonaqueous solvent mixtures should be different from those in water.48,49,53,58,92 There are many open questions regarding the ion specificities in water–nonaqueous solvent mixtures. For example, how is the relative strength of specific ion effects influenced by the addition of organic solvents?Recently, our group has performed systematic studies on the ion specificities of artificial macromolecules in water–nonaqueous solvent mixtures.48,49,53,58,92Fig. 4 shows the ion-specific reentrant behavior of PNIPAM in water–methanol mixtures.58 SCN− and ClO4− depress the reentrant transition, whereas other anions enhance the transition. Water and methanol are good solvents for PNIPAM, but water/methanol complexes are poor solvents for PNIPAM due to the strong hydrogen bonding interactions between water and methanol molecules.93,94 Thus, the anion-specific reentrant behavior is induced by the difference in the anionic structure making/breaking ability of water/methanol complexes. As more water/methanol complexes are formed with the addition of methanol, amplification of anion specificity results, induced by the anion-modulated shift of the equilibrium between solvent complexes and free solvent molecules.58 López-León et al. also reported a similar anion-specific reentrant behavior of PNIPAM microgels in water–ethanol mixtures.95
 | ||
| Fig. 4 Dependence of LCST on the volume fraction of methanol (XM) of PNIPAM for anions with Na+ as the common cation in the water–methanol mixtures. The top axis is the change in molar fraction of methanol (xM). Adapted from ref. 58 by permission of the American Chemical Society. | ||
The relative strength of specific ion effects can be amplified by the addition of ethylene glycol (EG) but is independent of the addition of hydrogen peroxide (H2O2), even though both additives can act as hydrogen bond donors and acceptors due to the presence of two hydroxyl groups.48 This is because solvent complexes formed in H2O–EG mixtures form stronger hydrogen bonds that more effectively differentiate the interactions between the anions and solvent complexes than those formed in H2O–H2O2 mixtures.48 The amplification of ion specificity is also influenced by the chemical structures of alcohols.92 Alcohols amplify the anion specificity following the series methanol (MeOH) < ethanol (EtOH) < 1-propanol (1-PrOH) < 2-propanol (2-PrOH) for the monohydric alcohols (Fig. 5a) and following the series D-sorbitol (D-SOR) ≈ xylitol (XYL) ≈ meso-erythritol (m-ERY) < glycerol (GLY) < EG < MeOH for polyhydric alcohols (Fig. 5b). The relative extent of amplification of specific anion effects is determined by the stability of water/alcohol complexes, which is correlated with the chemical structures of alcohols. The more stable solvent complexes formed via stronger hydrogen bonds more effectively differentiate the anions through indirect solvent-mediated anion–polymer interactions, resulting in a stronger amplification of specific ion effects.
 | ||
| Fig. 5 (a) ΔLCST of PNIPAM as a function of the molar fraction of monohydric alcohols (x) for the anions with Na+ as the common cation. (b) ΔLCST of PNIPAM as a function of the molar fraction of polyhydric alcohols (x) for the anions with Na+ as the common cation. The values of ΔLCST are obtained using the LCST for SCN− as the reference. Reproduced from ref. 92 by permission of the American Chemical Society. | ||
Alcohols are protic organic solvents, which can act as both hydrogen bond donors and hydrogen bond acceptors. In comparison with alcohols, aprotic organic solvents such as dimethylsulfoxide (DMSO) are only hydrogen bond acceptors. Thus, the addition of aprotic organic solvents may have different effects on the ion specificities of artificial macromolecules compared with that of protic organic solvents. An inverted V-shaped anion series is observed for the anion-specific upper critical solution temperature (UCST) behavior of PNIPAM in a H2O–DMSO mixture.53 This is different from that observed in a H2O–EtOH mixture, where the ordering of anions follows the classical Hofmeister series.53 The combination of the two opposing effects of anion adsorption on the PNIPAM surface and the anionic polarization of hydrogen bonding between DMSO and PNIPAM leads to the inverted V-shaped anion series.53
3. Direct ion–macromolecule interactions
3.1 Ion pairing
Ion pairing means that the interaction between a cation and an anion is strong enough to form a temporary neutral species.96 It is anticipated that the tendency to form ion pairs is ion specific as the formation of ion pairs involves ion–ion, ion–water, and water–water interactions. Collins proposed an empirical rule, called the law of matching water affinities (LMWA), to explain the ion-specific pairing between cations and anions, as shown in Fig. 6a.9,32 The LMWA is basically depicted within an electrostatic framework by considering ion–ion, ion–water and water–water interactions, which can be deduced from the “volcano plot” (Fig. 6b).32 In the volcano plot, a small difference in the heats of hydration between kosmotropic (chaotropic) cations and kosmotropic (chaotropic) anions leads to a high tendency of the ions to form a contact ion pair, in an endothermic process, whereas the opposite phenomena are observed between the kosmotropic cation (anion) and the chaotropic anion (cation).32 More specifically, the LMWA implies that two strongly hydrated oppositely charged ions (kosmotropes) can form a contact ion pair because the electrostatic interactions between cations and anions are sufficiently strong to remove their ionic hydration shells.32 For the cases of weakly hydrated chaotropes, cations and anions can also form a contact ion pair even though electrostatic attractions are weak between them. This is because ion–water interactions are weaker than water–water interactions and the energetic cost of dehydration of the ions is more than compensated by the energetic benefit of forming additional water–water bonds.32 For the small (kosmotrope) and big (chaotrope) ions, the electrostatic attractions between them are insufficient to remove the hydration shell of kosmotropic ions. Consequently, oppositely charged kosmotropes and chaotropes do not form contact ion pairs.32 | ||
| Fig. 6 (a) The law of matching water affinities. The tendency of oppositely charged ions to form contact ion pairs depends on the matching in ion size between the cation and the anion. (b) The volcano plot describes the relationship between the standard heat (q) of solution of a crystalline salt at infinite dilution and the difference between the absolute heats (WX− − WM+) of hydration of the corresponding individual gaseous anions and cations. Reproduced from ref. 32 by permission of Elsevier. | ||
The LMWA can be used to explain the ion specificities of natural macromolecules,5,15,28,97 for example, the interactions between Hofmeister anions and a protein binding pocket.15 Similarly, the concept of matching water affinities can also be employed to understand the ion specificities of artificial charged macromolecules. In previous studies, Strauss et al. found that polyelectrolytes containing sulfonate groups exhibit an opposite series of counterion binding with those containing carboxylate or phosphonate groups.98,99 This is understandable because alkyl sulfonate is regarded as a weakly hydrated group, whereas alkyl carboxylate and phosphonate groups are considered as strongly hydrated groups.100 Similar results are also observed in other studies in terms of specific interactions of alkali cations with polyacrylates and polysulfonates.101,102 For the case of cationic polyelectrolytes with linked trimethylammonium groups, the strength of interaction between the polyelectrolyte and counter-anions decreases following the series I− > Br− > Cl− > F− because trimethylammonium is considered to be a weakly hydrated group.103 Likewise, the more weakly hydrated chaotropic anions tend to exhibit stronger interactions with weakly hydrated positively charged groups associated with poly(3-alkoxy-4-methylthiophene).104 The doping level of Hofmeister anions in polyelectrolyte complexes can be understood by the LMWA as well.105
Our recent studies have shown that specific ion pairing interactions play an important role in determining the properties of polyelectrolytes.43,51,56 The ion-specific pairing between the weakly hydrated dimethylamino group and counterions strongly influences the fluorescence properties of the star weak polyelectrolytes containing a triphenylene core.56 The ion-specific collapse of poly[2-(methacryloyloxy)ethyltrimethylammonium chloride] (PMETAC) brushes in the presence of chaotropic anions is determined by the specific ion pairing interactions between the anions and the positively charged quaternary ammonium groups associated with the strong polyelectrolyte chains.43 For polyzwitterionic brushes, the hydration of the brushes is related to the inter/intrachain association, which can be modulated by specific ion pairing interactions between the ions and the charged groups associated with the grafted chains.51 This has important implications for the protein adsorption resisting properties of the polyzwitterionic brushes.51
Using the fluorescence resonance energy transfer (FRET) method, Gao et al. found that strongly hydrated kosmotropic anions cannot form stable ion pairs with weakly hydrated tertiary amine groups associated with the block copolymer of poly(ethylene oxide)-block-poly(tertiary amine); thus, stable polymer micelles cannot be formed (Fig. 7a).57 In contrast, weakly hydrated chaotropic anions can induce micellization of the block copolymer through the formation of stable ion pairs.57 Micelle formation shortens the distance between the donor and the acceptor, thereby improving the energy transfer efficiency (Fig. 7b and c).57
 | ||
| Fig. 7 (a) Anion-induced micellization of poly(ethylene oxide)-block-poly(tertiary amine) copolymers. (b) Addition of chaotropic anions results in the polymer micelle formation and the efficient energy transfer from the donor to the acceptor. (c) Hofmeister effects in the anion-induced micellization of the block copolymer. Reproduced from ref. 57 by permission of John Wiley & Sons Ltd. | ||
Huck et al. employed PMETAC brushes as a platform to tune the surface wettability via the hydrophobic/hydrophilic character of the ion pairs (Fig. 8).61 As the strength of interactions between the anions and the weakly hydrated quaternary ammonium group increases following the series PO43− < Cl− < SCN− < ClO4−, the contact angle on the surface of the brushes increases, following the same anion series.61 On the other hand, Salomäki et al. exploited anion specificity to control the growth of polyelectrolyte multilayers via specific ion paring interactions.106 The more chaotropic anions screen the polyelectrolyte charges more effectively through stronger ion pairing interactions, leading to a looser chain conformation and a thicker multilayer.106
 | ||
| Fig. 8 Change in water wettability on the surface of the PMETAC brushes with the corresponding counterions: (a) PO43−, (b) Cl−, (c) SCN−, and (d) ClO4−. Reproduced from ref. 61 by permission of John Wiley & Sons Ltd. | ||
3.2 Direct binding of ions to neutral macromolecules
Chaotropic ions have a propensity to adsorb at the air–water interface, whereas kosmotropic ions are repelled from the air–water interface.3,14,107–109 Similarly, poorly solvated nonpolar protein surface patches have hydrophobic-like attraction for large chaotropic ions but repel small kosmotropic ions.35–37 Based on the same underlying mechanism, ions can also bind to the neutral surface of artificial macromolecules. Theoretically, on the basis of molecular dynamics simulations, Jungwirth et al. suggested that the weakly hydrated iodide ion is attracted to the neutral macromolecule surface, while the highly hydrated fluoride ion is repelled.110,111 Experimentally, Cremer et al. demonstrated that the specific anion effects on the phase transition of PNIPAM can be described by a model including a linear term and a Langmuir-type adsorption isotherm (see Section 2.1).20 The Langmuir-type binding term represents the salting-in effect induced by the direct binding of chaotropic anions to the PNIPAM surface.20 The apparent binding constant of the chaotropic anions increases with increasing PNIPAM molecular weight and concentration.45The binding of chaotropic anions (e.g., SCN−) to the PNIPAM surface is directly confirmed by isothermal titration calorimetry (ITC) measurements, as shown in Fig. 9.47 The endothermic binding of SCN− is explained by the electrostatic perturbation of hydrophobic hydration of PNIPAM. Antunes et al. suggested that the adsorption of the chaotropic SCN− anion to the PNIPAM surface leads to the formation of a charged complex, accompanied by an increase in the solubility of PNIPAM.112 You et al. confirmed the binding of SCN− to the PNIPAM surface using Zeta potential measurements and showed that the interactions between SCN− and PNIPAM can induce the self-assembly of homopolymers into multiple nanostructures such as nanoparticles, nanorods and nanovesicles.113,114
 | ||
| Fig. 9 ITC titration curve of the binding of SCN− to the PNIPAM surface. Reproduced from ref. 47 by permission of the American Chemical Society. | ||
It has been reported that anions can also bind to other types of neutral macromolecules such as poly(acrylamide) and poly(vinylpyrrolidone).115–118 For instance, Mizrahi and co-workers suggested that specific anion effects on the osmotic pressure of poly(acrylamide) in aqueous solutions are related to the preferential binding of large anions to the polymer surface induced by specific anion–water–polymer interactions.117 More specifically, large anions (e.g., I−) are preferentially adsorbed onto the polymer surface because the formation of bulk water structures is energetically favorable over the hydration of both the anions and the polymer surface.117
For cations, it is suggested that the cation–polymer binding affinity is inversely correlated with the cation–anion ion-pairing interactions in aqueous solutions.46 The hydration strength of cations also influences binding, because the direct binding of cations requires partial desolvation of the cations which is energetically unfavorable.119 Zhang et al. investigated the cation-specific effects on the phase transition of PEO–PPO–PEO and found that both direct ion–macromolecule interactions and water-mediated indirect ion–macromolecule interactions strongly influence the phase transition behavior.50 They have shown that the monovalent cations Na+, K+, Rb+, and Cs+ do not bind to the polymer surface, while Li+, NH4+ and divalent cations including Mg2+, Ca2+, Sr2+, Ba2+, Co2+, Ni2+, Cu2+, and Cd2+ do bind to the polymer surface.50
3.3 The theory of ion dispersion forces versus the LMWA
It is important to note that ion dispersion interactions may contribute to all the types of direct ion–macromolecule interactions.16,33,34 As the ion polarizability is related to the size of ions, specific ion effects will arise due to the different sizes of ions.34 Ninham et al. suggested that the ion-specific increment of surface tension of the air–water interface can be explained by the theory of ion dispersion forces, though the repulsive electrostatic image potential between the aqueous ion and the image charge in air also contributes to the increase of surface tension.120 Likewise, the ions exhibit ion-specific positive adsorption at the oil–water interface driven by dispersion forces.120 Therefore, similar to the nonpolar oil–water interface, the direct binding of ions to the nonpolar surface of artificial macromolecules could be driven by ion dispersion forces.31 Ion dispersion forces could also contribute to the interactions between ions and the polar groups of artificial macromolecules.31 For polyelectrolytes, Ninham et al. showed that ion dispersion forces have important influences on the counterion condensation of polyelectrolytes.121As discussed in Section 3.1, the LMWA can qualitatively predict the ion sequence related to specific ion–charged site interactions.15,17,122 Meanwhile, it is recognized that the theory of ion dispersion forces could describe ion–charged site interactions as well.31 Although the ion sequence in some specific cases, e.g., the interactions between cations and a negatively charged kosmotropic group, predicted by the LMWA appears to conflict with that predicted by the theory of ion dispersion forces, Ninham and Salis have attempted to reconcile these two approaches in their recently published review paper.31 They suggested that the driving forces for the formation of ion pairs depicted in the LMWA are in fact due to the cooperation of electrostatic and quantum mechanical dispersion forces.31 In comparison with the LMWA, the different results obtained from the theory of ion dispersion forces in some cases are attributed to the fact that the surface with discrete charged sites is considered to be a uniformly charged surface with a constant dielectric function without taking into account the chemical nature of the surface charged sites.31 Therefore, it is thought that these two approaches are in fact the same if the polarizability, size, and hydration of the surface charged sites are included in the modelling of the theory of ion dispersion forces.31 In addition, the interactions between the ions and the surface neutral sites are neglected in the LMWA but are considered in the theory of ion dispersion forces.31
Theoretically, the dispersion interaction between an ion and a macromolecular surface should be proportional to the dispersion B coefficient.123 The dispersion B coefficient can be calculated from the effective polarizability of the ion in solution and the dielectric difference between the solvent and the macromolecular surface, both of which are related to the dielectric function of the solvent.123 As the dielectric constant of the water–organic solvent mixtures is strongly dependent on solvent composition,124–126 the addition of organic solvents to aqueous solutions would affect the ion specificities of artificial macromolecules through ion dispersion forces, in addition to the formation of water–organic solvent complexes mentioned in Section 2.2.
4. Effect of crowding on the ion specificities of macromolecules
4.1 Macromolecular crowding
The intracellular environment where protein molecules undergo folding and unfolding contains not only various ions but also a high concentration of biomacromolecules such as nucleic acids and polysaccharides.127–129 The local concentration or the activity coefficient of ions is enhanced in crowded environments due to the volume exclusion effect.130,131 This effect is expected to strongly influence the ion specificities of macromolecules in crowded environments because specific ion effects are closely related to the ion concentration.20,132 On the other hand, the volume exclusion effect generated by the crowding agents decreases the diffusion rate of the macromolecules but increases the re-collision probability of macromolecules within the cage of crowding agents.133 Shiraki et al. investigated specific ion effects in the thermal aggregation of hen egg white proteins in a crowded environment of Ficoll 70.134 They found that chaotropic SCN− unexpectedly promotes the aggregation of proteins in the crowded environment and suggested that the increase of protein aggregation induced by SCN− may be because the chaotropic anion increases reaction-limited protein aggregation in a crowded environment.134For artificial macromolecules, it is reported that ions can interact with both PNIPAM and macromolecular crowding agents (Fig. 10a).135 As a consequence, there is a competition between PNIPAM and the crowding agents in terms of their interactions with the ions. Furthermore, the crowding agents can interact with PNIPAM as well, which interferes with the ion–PNIPAM interactions (Fig. 10b). As a result, the salting-in effect exerted by SCN− on PNIPAM solubility is weakened in a crowded environment of dextran but is strengthened in a crowded environment of PEG, due to different anion–crowder interactions. Meanwhile, the salting-out effect generated by Cl− and CH3COO− on PNIPAM solubility is weakened by the crowding of dextran but is strengthened by the crowding of PEG because of the different PNIPAM–crowder interactions.135
 | ||
| Fig. 10 Schematic illustration of the influence of crowding on the ion specificities of artificial macromolecules. (a) Competition between PNIPAM and crowding agents in terms of their interactions with the ions. (b) PEG interacts with PNIPAM as a hydrogen bond acceptor, whereas dextran interacts with PNIPAM as hydrogen bond donors. Adapted from ref. 135 by permission of The Royal Society of Chemistry. | ||
Besides the volume exclusion effect and the interactions between the crowding agents, macromolecules and ions, the solvent dielectric constant is also changed in crowded environments.136,137 It has been reported that the dielectric constant of water decreases as the extent of crowding increases due to constraints on water mobility.137 As the dispersion interactions between ions and macromolecules are dependent on the solvent dielectric constant, the ion specificities of artificial macromolecules should be altered in crowded environments due to the variation of the solvent dielectric constant.123 Sekiya et al. found that macromolecular crowding significantly modifies specific ion effects on the phase transition of PNIPAM for chaotropic anions.55 As the water dielectric constant is decreased in the crowded environment of PEG, the specific ion effects are altered induced by the increasing electrostatic repulsion between the chaotropic anion bound PNIPAM chains and the decreasing affinity of the chaotropic anions to the PNIPAM surface.55
4.2 Small-molecule crowding
In addition to the macromolecular crowders, small molecules, such as sugars, urea, amino acids, methylamines, and methylsulfonium compounds are also highly concentrated in the cellular environment.129 Under the conditions of small-molecule crowding, the volume exclusion effect exerted by small molecules is relatively weak, but the solvent properties may change significantly as the hydration of small hydrophilic molecules consumes a large fraction of the water molecules.129 Moreover, small molecules also have favorable or unfavorable interactions with macromolecules.129 Therefore, similar to macromolecular crowding, the ion specificities of macromolecules are also expected to be modified by small-molecule crowding.Fig. 11 shows the specific anion effect on the LCST behavior of PNIPAM in a crowded environment of methylated urea or glucose.138 Generally, chaotropic osmolytes, such as urea and its methylated derivatives, can bind to the surface of macromolecules, generating a salting-in effect, whereas kosmotropic osmolytes, such as glucose, are excluded from the surface of macromolecules, giving rise to a salting-out effect.139–141 The specific anion effect can be amplified by the methylated urea (Fig. 11a) but not by glucose (Fig. 11b). The amplification of the anion specificity is attributed to the increased difference in the anion-specific polarization of the hydrogen bonding between water and the polymer, induced by the formation of PNIPAM/methylated urea complexes via hydrophobic interactions.138
 | ||
| Fig. 11 (a) Change in the LCST of PNIPAM as a function of the concentration of tetramethylurea (CTMU) in the presence of different types of anions with Na+ as the common cation. (b) Change in the LCST of PNIPAM as a function of the concentration of glucose (CGlu) in the presence of different types of anions with Na+ as the common cation. Reproduced from ref. 138 by permission of The Royal Society of Chemistry. | ||
5. Conclusions and outlook
Although the ion specificities of artificial macromolecules have been investigated extensively, a widely accepted theory to fully understand the exact mechanisms is still lacking. It is anticipated that direct specific ion–macromolecule interactions could be described by the theory of ion dispersion forces if the polarizability, size, and hydration of the ions and the surface sites of macromolecules are considered in the modelling.31 On the other hand, no real theories have been developed to understand solvent-mediated indirect specific ion–macromolecule interactions, though some empirical models have been proposed to explain these interactions. Thus, further development of appropriate theories to fully rationalize both direct and indirect ion–macromolecule interactions is important.In aqueous solutions, it is recognized that both water-mediated indirect ion–macromolecule interactions and direct ion–macromolecule interactions play a significant role in the ion specificities of artificial macromolecules. For indirect ion–macromolecule interactions, the ions can affect the solubility of artificial macromolecules through either the competition for water molecules in the hydration shell of the macromolecules or the ionic polarization of hydrogen bonding between the hydration water molecules and the macromolecules.20,88
The relative strength of specific ion effects on artificial macromolecules can be amplified by the addition of alcohols to aqueous solutions. The amplification of ion specificities is induced by the formation of water–organic solvent complexes, which is correlated with the chemical structures of the alcohols. Besides the formation of solvent complexes, the solvent dielectric constant is also dependent on solvent composition in the water–organic solvent mixtures. As the dispersion interactions between ions and macromolecules are influenced by the variation of the solvent dielectric constant, the ion specificities of artificial macromolecules should also be influenced by the addition of organic solvents through ion dispersion forces. This has been overlooked previously and is desirable to investigate in future studies.
For direct ion–macromolecule interactions, the ion specificities of charged macromolecules are dominated by specific ion pairing interactions, which can be understood by either the LMWA or the theory of ion dispersion forces. For neutral polymers, both anions and cations can bind to the polymer surface. Because the cation and the anion are always added to solutions together, how the ion specificities of artificial macromolecules are influenced by the interactions between the cation and the anion themselves is an unsolved problem.5,38,142 For example, it is still unclear how specific ion effects are influenced by the competition or cooperation between the cation and the anion in terms of their interactions with artificial macromolecules. The determination of the ion-binding sites associated with the artificial macromolecules and the interaction modes between the ions and the binding sites is a key step to solve this problem.39,87,143,144
When the medium environments change from dilute solutions to crowded conditions, the rules for the ion specificities of artificial macromolecules will be changed. It has been reported that specific ion effects are influenced by volume exclusion, the variation of the solvent dielectric constant and the interactions between ions, macromolecules and crowding agents.55,134,135 However, these factors have been investigated separately previously and, in reality, specific ion effects should be determined by the combined effect of these factors, which should be considered in future studies. Moreover, relevant studies should be extended to the field of polymer modified therapeutic proteins in crowded environments to clarify the mechanism of ion specificities in the delivery of polymer conjugated therapeutic proteins in cellular environments.145–148
In addition to the ion-specific interactions between artificial macromolecules and the ions in solutions, it is also interesting to investigate the inter/intrachain specific ion interactions, because the macromolecular properties are closely related to these kinds of interactions. For example, poly(sulfobetaine) has many inter/intrachain self-associations due to the similarity in the hydration strength of the sulfonate and quaternary ammonium groups, whereas poly(carboxybetaine) has few self-associations because of the large difference in hydration strength between the carboxylate and quaternary ammonium groups.149 As a result, the poly(carboxybetaine) film exhibits better protein resistance properties than the poly(sulfobetaine) film.149
Additionally, studies on the ion specificities of artificial macromolecules should be extended to the field of energy storage devices, as the performances of energy storage devices (e.g., supercapacitors and batteries) are dependent on the specific interactions between the ions and the polymeric electrolytes.150,151 Investigations should also be extended to mixed electrolyte systems as specific ion effects are observed in real systems where different kinds of electrolytes usually coexist.42,152,153
Acknowledgements
We thank Prof. Vincent S. J. Craig and Dr Drew F. Parsons for helpful discussions. The financial support of National Program on Key Basic Research Project (2012CB933802), the National Natural Science Foundation of China (21374110, 21574121, 21622405), Users with Excellence of Hefei Science Center (2015HSC-UE008), the Youth Innovation Promotion Association of CAS (2013290), and the Fundamental Research Funds for the Central Universities (WK2340000066) is acknowledged.Notes and references
- V. A. Parsegian, Nature, 1995, 378, 335–336 CrossRef CAS.
- W. Kunz, P. Lo Nostro and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 1–18 CrossRef CAS.
- D. J. Tobias and J. C. Hemminger, Science, 2008, 319, 1197–1198 CrossRef CAS PubMed.
- P. Lo Nostro and B. W. Ninham, Chem. Rev., 2012, 112, 2286–2322 CrossRef CAS PubMed.
- P. Jungwirth and P. S. Cremer, Nat. Chem., 2014, 6, 261–263 CrossRef CAS PubMed.
- V. Mazzini and V. S. J. Craig, Curr. Opin. Colloid Interface Sci., 2016, 23, 82–93 CrossRef CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- K. D. Collins, Biophys. Chem., 2006, 119, 271–281 CrossRef CAS PubMed.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Biophys. Chem., 2007, 128, 95–104 CrossRef CAS PubMed.
- M. G. Cacace, E. M. Landau and J. J. Ramsden, Q. Rev. Biophys., 1997, 30, 241–277 CrossRef CAS PubMed.
- W. J. Xie and Y. Q. Gao, J. Phys. Chem. Lett., 2013, 4, 4247–4252 CrossRef CAS PubMed.
- P. Jungwirth and B. Winter, Annu. Rev. Phys. Chem., 2008, 59, 343–366 CrossRef CAS PubMed.
- Y. J. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS PubMed.
- P. Jungwirth and D. J. Tobias, Chem. Rev., 2006, 106, 1259–1281 CrossRef CAS PubMed.
- J. M. Fox, K. Kang, W. Sherman, A. Heroux, G. M. Sastry, M. Baghbanzadeh, M. R. Lockett and G. M. Whitesides, J. Am. Chem. Soc., 2015, 137, 3859–3866 CrossRef CAS PubMed.
- D. F. Parsons, M. Boström, P. Lo Nostro and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- W. Kunz, Curr. Opin. Colloid Interface Sci., 2010, 15, 34–39 CrossRef CAS.
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef CAS PubMed.
- K. D. Collins and M. W. Washabaugh, Q. Rev. Biophys., 1985, 18, 323–422 CrossRef CAS PubMed.
- Y. J. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- N. Schwierz, D. Horinek and R. R. Netz, Langmuir, 2010, 26, 7370–7379 CrossRef CAS PubMed.
- N. Schwierz, D. Horinek and R. R. Netz, Langmuir, 2013, 29, 2602–2614 CrossRef CAS PubMed.
- S. C. Flores, J. Kherb and P. S. Cremer, J. Phys. Chem. C, 2012, 116, 14408–14413 CAS.
- T. López-León, A. B. Jodar-Reyes, D. Bastos-González and J. L. Ortega-Vinuesa, J. Phys. Chem. B, 2003, 107, 5696–5708 CrossRef.
- T. López-León, M. J. Santander-Ortega, J. L. Ortega-Vinuesa and D. Bastos-González, J. Phys. Chem. C, 2008, 112, 16060–16069 Search PubMed.
- M. Boström, F. W. Tavares, S. Finet, F. Skouri-Panet, A. Tardieu and B. W. Ninham, Biophys. Chem., 2005, 117, 217–224 CrossRef PubMed.
- M. Boström, D. F. Parsons, A. Salis, B. W. Ninham and M. Monduzzi, Langmuir, 2011, 27, 9504–9511 CrossRef PubMed.
- Y. J. Zhang and P. S. Cremer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15249–15253 CrossRef CAS PubMed.
- A. Salis, M. C. Pinna, D. Bilanicova, M. Monduzzi, P. Lo Nostro and B. W. Ninham, J. Phys. Chem. B, 2006, 110, 2949–2956 CrossRef CAS PubMed.
- R. A. Robinson and R. H. Stokes, Electrolyte Solutions, Butterworth, London, 2nd edn, 1970 Search PubMed.
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- K. D. Collins, Methods, 2004, 34, 300–311 CrossRef CAS PubMed.
- D. F. Parsons and B. W. Ninham, Langmuir, 2010, 26, 6430–6436 CrossRef CAS PubMed.
- B. W. Ninham and P. Lo Nostro, Molecular Forces and Self Assembly in Colloid, Nano Sciences and Biology, Cambridge University Press, London, 2010 Search PubMed.
- W. Kunz, Specific Ion Effects, World Scientific Publishing Company, River Edge, NJ, 2010 Search PubMed.
- M. Lund, L. Vrbka and P. Jungwirth, J. Am. Chem. Soc., 2008, 130, 11582–11583 CrossRef CAS PubMed.
- M. Lund, P. Jungwirth and C. E. Woodward, Phys. Rev. Lett., 2008, 100, 258105 CrossRef PubMed.
- C. E. Dempsey, P. E. Mason and P. Jungwirth, J. Am. Chem. Soc., 2011, 133, 7300–7303 CrossRef CAS PubMed.
- K. B. Rembert, J. Paterova, J. Heyda, C. Hilty, P. Jungwirth and P. S. Cremer, J. Am. Chem. Soc., 2012, 134, 10039–10046 CrossRef CAS PubMed.
- P. Lo Nostro, A. Lo Nostro, B. W. Ninham, G. Pesavento, L. Fratoni and P. Baglioni, Curr. Opin. Colloid Interface Sci., 2004, 9, 97–101 CrossRef CAS.
- M. Fortini, D. Berti, P. Baglioni and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 168–172 CrossRef CAS.
- S. Z. Moghaddam and E. Thormann, Phys. Chem. Chem. Phys., 2015, 17, 6359–6366 RSC.
- R. Kou, J. Zhang, T. Wang and G. M. Liu, Langmuir, 2015, 31, 10461–10468 CrossRef CAS PubMed.
- A. Pica and G. Graziano, Phys. Chem. Chem. Phys., 2015, 17, 27750–27757 RSC.
- Y. J. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CAS.
- H. B. Du, R. Wickramasinghe and X. H. Qian, J. Phys. Chem. B, 2010, 114, 16594–16604 CrossRef CAS PubMed.
- I. Shechter, O. Ramon, I. Portnaya, Y. Paz and Y. D. Livney, Macromolecules, 2010, 43, 480–487 CrossRef CAS.
- L. D. Liu, T. Wang, C. Liu, K. Lin, Y. W. Ding, G. M. Liu and G. Z. Zhang, J. Phys. Chem. B, 2013, 117, 2535–2544 CrossRef CAS PubMed.
- Y. C. Long, T. Wang, L. D. Liu, G. M. Liu and G. Z. Zhang, Langmuir, 2013, 29, 3645–3653 CrossRef CAS PubMed.
- J. C. Lutter, T. Y. Wu and Y. J. Zhang, J. Phys. Chem. B, 2013, 117, 10132–10141 CrossRef CAS PubMed.
- T. Wang, X. W. Wang, Y. C. Long, G. M. Liu and G. Z. Zhang, Langmuir, 2013, 29, 6588–6596 CrossRef CAS PubMed.
- T. Wang, Y. C. Long, L. D. Liu, X. W. Wang, V. S. J. Craig, G. Z. Zhang and G. M. Liu, Langmuir, 2014, 30, 12850–12859 CrossRef CAS PubMed.
- L. D. Liu, T. Wang, C. Liu, K. Lin, G. M. Liu and G. Z. Zhang, J. Phys. Chem. B, 2013, 117, 10936–10943 CrossRef CAS PubMed.
- C. Wu and S. Q. Zhou, Phys. Rev. Lett., 1996, 77, 3053–3055 CrossRef CAS PubMed.
- K. Sakota, D. Tabata and H. Sekiya, J. Phys. Chem. B, 2015, 119, 10334–10340 CrossRef CAS PubMed.
- F. G. Chen, C. L. Li, X. Y. Wang, G. M. Liu and G. Z. Zhang, Soft Matter, 2012, 8, 6364–6370 RSC.
- Y. Li, Y. G. Wang, G. Huang, X. P. Ma, K. J. Zhou and J. M. Gao, Angew. Chem., Int. Ed., 2014, 53, 8074–8078 CrossRef CAS PubMed.
- T. Wang, G. M. Liu, G. Z. Zhang and V. S. J. Craig, Langmuir, 2012, 28, 1893–1899 CrossRef CAS PubMed.
- B. A. Humphreys, J. D. Willott, T. J. Murdoch, G. B. Webber and E. J. Wanless, Phys. Chem. Chem. Phys., 2016, 18, 6037–6046 RSC.
- J. D. Willott, T. J. Murdoch, G. B. Webber and E. J. Wanless, Macromolecules, 2016, 49, 2327–2338 CrossRef CAS.
- O. Azzaroni, A. A. Brown and W. T. S. Huck, Adv. Mater., 2007, 19, 151–154 CrossRef CAS.
- C. J. Huang, Y. S. Chen and Y. Chang, ACS Appl. Mater. Interfaces, 2015, 7, 2415–2423 CAS.
- D. A. Garrec and I. T. Norton, J. Colloid Interface Sci., 2012, 379, 33–40 CrossRef CAS PubMed.
- Z. Y. He, W. J. Xie, Z. Q. Liu, G. M. Liu, Z. W. Wang, Y. Q. Gao and J. J. Wang, Sci. Adv., 2016, 2, e1600345 Search PubMed.
- M. Jaspers, A. E. Rowan and P. H. J. Kouwer, Adv. Funct. Mater., 2015, 25, 6503–6510 CrossRef CAS.
- O. Zavgorodnya, V. Kozlovskaya, X. Liang, N. Kothalawala, S. A. Catledge, A. Dass and E. Kharlampieva, Mater. Res. Express, 2014, 1, 035039 CrossRef.
- Y. Xu, Z. Hua, J. Zhang, J. Yang, Z. L. Cao, D. Y. Zhang, L. X. He, V. S. J. Craig, G. Z. Zhang and G. M. Liu, Chem. Commun., 2016, 52, 3392–3395 RSC.
- G. Jones and M. Dole, J. Am. Chem. Soc., 1929, 51, 2950–2964 CrossRef CAS.
- H. D. B. Jenkins and Y. Marcus, Chem. Rev., 1995, 95, 2695–2724 CrossRef CAS.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347–349 CrossRef CAS PubMed.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, J. Chem. Phys., 2003, 119, 12457–12461 CrossRef CAS.
- R. Mancinelli, A. Botti, F. Bruni, M. A. Ricci and A. K. Soper, Phys. Chem. Chem. Phys., 2007, 9, 2959–2967 RSC.
- J. T. O'Brien, J. S. Prell, M. F. Bush and E. R. Williams, J. Am. Chem. Soc., 2010, 132, 8248–8249 CrossRef PubMed.
- J. Holzmann, R. Ludwig, A. Geiger and D. Paschek, Angew. Chem., Int. Ed., 2007, 46, 8907–8911 CrossRef CAS PubMed.
- Y. X. Chen, H. I. Okur, N. Gomopoulos, C. Macias-Romero, P. S. Cremer, P. B. Petersen, G. Tocci, D. M. Wilkins, C. W. Liang, M. Ceriotti and S. Roke, Sci. Adv., 2016, 2, e1501891 Search PubMed.
- R. Freitag and F. Garret-Flaudy, Langmuir, 2002, 18, 3434–3440 CrossRef CAS.
- J. Bunsow and D. Johannsmann, J. Colloid Interface Sci., 2008, 326, 61–65 CrossRef PubMed.
- T. López-León, J. L. Ortega-Vinuesa, D. Bastos-González and A. Elaissari, J. Colloid Interface Sci., 2014, 426, 300–307 CrossRef PubMed.
- J. M. G. Swann, W. Bras, P. D. Topham, J. R. Howse and A. J. Ryan, Langmuir, 2010, 26, 10191–10197 CrossRef CAS PubMed.
- E. Thormann, RSC Adv., 2012, 2, 8297–8305 RSC.
- Y. Hou, C. Q. Yu, G. M. Liu, T. Ngai and G. Z. Zhang, J. Phys. Chem. B, 2010, 114, 3799–3803 CrossRef CAS PubMed.
- Y. K. Jhon, R. R. Bhat, C. Jeong, O. J. Rojas, I. Szleifer and J. Genzer, Macromol. Rapid Commun., 2006, 27, 697–701 CrossRef CAS.
- C. A. Naini, M. Thomas, S. Franzka, S. Frost, M. Ulbricht and N. Hartmann, Macromol. Rapid Commun., 2013, 34, 417–422 CrossRef CAS PubMed.
- B. A. Deyerle and Y. J. Zhang, Langmuir, 2011, 27, 9203–9210 CrossRef CAS PubMed.
- Y. H. Cho, Y. J. Zhang, T. Christensen, L. B. Sagle, A. Chilkoti and P. S. Cremer, J. Phys. Chem. B, 2008, 112, 13765–13771 CrossRef CAS PubMed.
- P. T. Guner and A. L. Demirel, J. Phys. Chem. B, 2012, 116, 14510–14514 CrossRef PubMed.
- K. B. Rembert, H. I. Okur, C. Hilty and P. S. Cremer, Langmuir, 2015, 31, 3459–3464 CrossRef CAS PubMed.
- P. M. Reddy and P. Venkatesu, J. Phys. Chem. B, 2011, 115, 4752–4757 CrossRef CAS PubMed.
- N. J. Debeljuh, A. Sutti, C. J. Barrow and N. Byrne, J. Phys. Chem. B, 2013, 117, 8430–8435 CrossRef CAS PubMed.
- R. Politi, L. Sapir and D. Harries, J. Phys. Chem. A, 2009, 113, 7548–7555 CrossRef CAS PubMed.
- A. Bertoluzza, S. Bonora, M. A. Battaglia and P. Monti, J. Raman Spectrosc., 1979, 8, 231–235 CrossRef CAS.
- Y. Xu and G. M. Liu, J. Phys. Chem. B, 2014, 118, 7450–7456 CrossRef CAS PubMed.
- G. Z. Zhang and C. Wu, Phys. Rev. Lett., 2001, 86, 822–825 CrossRef CAS PubMed.
- G. Z. Zhang and C. Wu, J. Am. Chem. Soc., 2001, 123, 1376–1380 CrossRef CAS.
- T. López-León, D. Bastos-González, J. L. Ortega-Vinuesa and A. Elaissari, ChemPhysChem, 2010, 11, 188–194 CrossRef PubMed.
- N. Bjerrum, Z. Phys. Chem., Stoechiom. Verwandtschaftsl., 1926, 119, 145–160 CAS.
- J. Heyda, J. Pokorna, L. Vrbka, R. Vacha, B. Jagoda-Cwiklik, J. Konvalinka, P. Jungwirth and J. Vondrasek, Phys. Chem. Chem. Phys., 2009, 11, 7599–7604 RSC.
- U. P. Strauss and Y. P. Leung, J. Am. Chem. Soc., 1965, 87, 1476–1480 CrossRef CAS.
- J. Hen and U. P. Strauss, J. Phys. Chem., 1974, 78, 1013–1017 CrossRef CAS.
- N. Vlachy, B. Jagoda-Cwiklik, R. Vacha, D. Touraud, P. Jungwirth and W. Kunz, Adv. Colloid Interface Sci., 2009, 146, 42–47 CrossRef CAS PubMed.
- I. Lipar, P. Zalar, C. Pohar and V. Vlachy, J. Phys. Chem. B, 2007, 111, 10130–10136 CrossRef CAS PubMed.
- H. Daoust and M. A. Chabot, Macromolecules, 1980, 13, 616–619 CrossRef CAS.
- M. Druchok, B. Hribar-Lee, H. Krienke and V. Vlachy, Chem. Phys. Lett., 2008, 450, 281–285 CrossRef CAS.
- M. Qiu, S. R. Long, B. X. Li, L. Y. Yan, W. W. Xie, Y. L. Niu, X. F. Wang, Q. J. Guo and A. D. Xia, J. Phys. Chem. C, 2013, 117, 21870–21878 CAS.
- R. A. Ghostine, R. F. Shamoun and J. B. Schlenoff, Macromolecules, 2013, 46, 4089–4094 CrossRef CAS.
- M. Salomäki, P. Tervasmaki, S. Areva and J. Kankare, Langmuir, 2004, 20, 3679–3683 CrossRef.
- S. Gopalakrishnan, D. F. Liu, H. C. Allen, M. Kuo and M. J. Shultz, Chem. Rev., 2006, 106, 1155–1175 CrossRef CAS PubMed.
- P. B. Petersen and R. J. Saykally, Annu. Rev. Phys. Chem., 2006, 57, 333–364 CrossRef CAS PubMed.
- S. Ghosal, J. C. Hemminger, H. Bluhm, B. S. Mun, E. L. D. Hebenstreit, G. Ketteler, D. F. Ogletree, F. G. Requejo and M. Salmeron, Science, 2005, 307, 563–566 CrossRef CAS PubMed.
- M. Lund, R. Vacha and P. Jungwirth, Langmuir, 2008, 24, 3387–3391 CrossRef CAS PubMed.
- M. Lund and P. Jungwirth, J. Phys.: Condens. Matter, 2008, 20, 494218 CrossRef.
- M. C. M. Costa, S. M. C. Silva and F. E. Antunes, J. Mol. Liq., 2015, 210, 113–118 CrossRef CAS.
- L. H. Wang, Z. D. Zhang, C. Y. Hong, X. H. He, W. You and Y. Z. You, Adv. Mater., 2015, 27, 3202–3207 CrossRef CAS PubMed.
- L. H. Wang, T. Wu, Z. Zhang and Y. Z. You, Macromolecules, 2016, 49, 362–366 CrossRef CAS.
- J. D. Song, R. Ryoo and M. S. Jhon, Macromolecules, 1991, 24, 1727–1730 CrossRef CAS.
- P. H. Von Hippel, V. Peticolas, L. Schack and L. Karlson, Biochemistry, 1973, 12, 1256–1264 CrossRef CAS PubMed.
- Y. D. Livney, I. Portnaya, B. Faupin, O. Ramon, Y. Cohen, U. Cogan and S. Mizrahi, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 508–519 CrossRef CAS.
- J. Xia, L. X. Song, W. Liu and Y. Teng, Soft Matter, 2013, 9, 9714–9722 RSC.
- H. B. Du, S. R. Wickramasinghe and X. H. Qian, J. Phys. Chem. B, 2013, 117, 5090–5101 CrossRef CAS PubMed.
- M. Boström, D. R. M. Williams and B. W. Ninham, Langmuir, 2001, 17, 4475–4478 CrossRef.
- M. Boström, D. R. M. Williams and B. W. Ninham, J. Phys. Chem. B, 2002, 106, 7908–7912 CrossRef.
- J. Kherb, S. C. Flores and P. S. Cremer, J. Phys. Chem. B, 2012, 116, 7389–7397 CrossRef CAS PubMed.
- D. F. Parsons and B. W. Ninham, Langmuir, 2010, 26, 1816–1823 CrossRef CAS PubMed.
- P. S. Albright and L. J. Gosting, J. Am. Chem. Soc., 1946, 68, 1061–1063 CrossRef CAS PubMed.
- D. I. Donato, A. D'Aprano and E. Caponetti, J. Solution Chem., 1979, 8, 135–146 CrossRef.
- L. J. Yang, X. Q. Yang, K. M. Huang, G. Z. Jia and H. Shang, Int. J. Mol. Sci., 2009, 10, 1261–1270 CrossRef CAS PubMed.
- M. Senske, L. Tork, B. Born, M. Havenith, C. Herrmann and S. Ebbinghaus, J. Am. Chem. Soc., 2014, 136, 9036–9041 CrossRef CAS PubMed.
- R. J. Ellis and A. P. Minton, Nature, 2003, 425, 27–28 CrossRef CAS PubMed.
- S. Nakano, D. Miyoshi and N. Sugimoto, Chem. Rev., 2014, 114, 2733–2758 CrossRef CAS PubMed.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed.
- J. R. Wenner and V. A. Bloomfield, Biophys. J., 1999, 77, 3234–3241 CrossRef CAS PubMed.
- M. Boström, D. R. M. Williams and B. W. Ninham, Biophys. J., 2003, 85, 686–694 CrossRef.
- J. S. Kim and A. Yethiraj, Biophys. J., 2009, 96, 1333–1340 CrossRef CAS PubMed.
- K. Iwashita, N. Inoue, A. Handa and K. Shiraki, Protein J., 2015, 34, 212–219 CrossRef CAS PubMed.
- W. Q. Song, L. D. Liu and G. M. Liu, Soft Matter, 2015, 11, 5940–5946 RSC.
- F. Despa, A. Fernandez and R. S. Berry, Phys. Rev. Lett., 2004, 93, 228104 CrossRef PubMed.
- R. Harada, Y. Sugita and M. Feig, J. Am. Chem. Soc., 2012, 134, 4842–4849 CrossRef CAS PubMed.
- L. D. Liu, Y. Shi, C. Liu, T. Wang, G. M. Liu and G. Z. Zhang, Soft Matter, 2014, 10, 2856–2862 RSC.
- H. Y. Wei, Y. B. Fan and Y. Q. Gao, J. Phys. Chem. B, 2010, 114, 557–568 CrossRef CAS PubMed.
- J. R. D. Oro, J. Biol. Phys., 2001, 27, 73–79 CrossRef PubMed.
- S. Moelbert, B. Normand and P. D. Rios, Biophys. Chem., 2004, 112, 45–57 CrossRef CAS PubMed.
- P. E. Mason, C. E. Dempsey, L. Vrbka, J. Heyda, J. W. Brady and P. Jungwirth, J. Phys. Chem. B, 2009, 113, 3227–3234 CrossRef CAS PubMed.
- H. I. Okur, J. Kherb and P. S. Cremer, J. Am. Chem. Soc., 2013, 135, 5062–5067 CrossRef CAS PubMed.
- V. Balos, H. Kim, M. Bonn and J. Hunger, Angew. Chem., Int. Ed., 2016, 55, 8125–8128 CrossRef CAS PubMed.
- A. J. Keefe and S. Y. Jiang, Nat. Chem., 2012, 4, 59–63 CrossRef CAS PubMed.
- P. Zhang, F. Sun, C. Tsao, S. J. Liu, P. Jain, A. Sinclair, H. C. Hung, T. Bai, K. Wu and S. Y. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12046–12051 CrossRef CAS PubMed.
- A. L. Fu, Z. Z. Zhao, F. Y. Gao and M. M. Zhang, Pharm. Res., 2013, 30, 2108–2117 CrossRef CAS PubMed.
- S. Guillard, R. R. Minter and R. H. Jackson, Trends Biotechnol., 2015, 33, 163–171 CrossRef CAS PubMed.
- Q. Shao and S. Y. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- X. Peng, H. L. Liu, Q. Yin, J. C. Wu, P. Z. Chen, G. Z. Zhang, G. M. Liu, C. Z. Wu and Y. Xie, Nat. Commun., 2016, 7, 11782 CrossRef CAS PubMed.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- G. M. Liu, Y. Hou, X. A. Xiao and G. Z. Zhang, J. Phys. Chem. B, 2010, 114, 9987–9993 CrossRef CAS PubMed.
- C. L. Henry, C. N. Dalton, L. Scruton and V. S. J. Craig, J. Phys. Chem. C, 2007, 111, 1015–1023 CAS.
| This journal is © The Royal Society of Chemistry 2017 |