
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Force spectroscopy predicts thermal stability of immobilized proteins by measuring microbead mechanics†
Danijela
Gregurec
ab,
Susana
Velasco-Lozano
c,
Sergio E.
Moya
a,
Luis
Vázquez
d and
Fernando
López-Gallego
*ce
aSoft Matter Nanotechnology Group, CIC BiomaGUNE, Paseo Miramon 182, San Sebasitan-Donostia, 20009, Spain
bRLE-Bioelectronics Research Group, Massachusetts Institute of Technology, 77 Massachusetts Ave 8-031, Cambridge, MA 02139, USA
cHeterogeneus Biocatalysis Group, CIC BiomaGUNE, Paseo Miramon 182, San Sebasitan-Donostia, 20009, Spain. E-mail: flopez.ikerbasque@cicbiomagune.es
dInstituto de Ciencia de Materiales de Madrid (CSIC), Campus de Cantoblanco, C\Sor Juana Inés de la Cruz 3, Madrid, 28049, Spain
eIKERBASQUE, Basque Foundation for Science, Bilbao, Spain
First published on 29th September 2016
Abstract
Optimal immobilization of enzymes on porous microbeads enables the fabrication of highly active and stable heterogeneous biocatalysts to implement biocatalysis in synthetic and analytical chemistry. However, empirical procedures for enzyme immobilization still prevail over rational ones because there is an unmet need for more comprehensive characterization techniques that aid to understand and trace the immobilization process. Here, we present the use of atomic force spectroscopy (AFS) as an innovative solution to indirectly characterize immobilized proteins on porous materials and monitor the immobilization process in real time. We investigate the mechanical properties of porous agarose microbeads immobilizing proteins by indenting a colloidal probe (silica microparticle) into a single bead. AFS demonstrates that the binding of proteins to the solid matrix of an agarose microbead alters its stiffness. Interestingly, we discovered that irreversible and multivalent immobilizations that make microbeads stiffer also stabilize the immobilized proteins against the temperature. Hence, we propose atomic force spectroscopy as a useful technique to indirectly unravel the stability of the immobilized enzymes investigating the mechanics of the heterogenous biocatalysts as a solid biomaterial beyond the intrinsic mechanics of the proteins.
Introduction
Protein immobilization is an attractive technique to implement proteins in industrial biocatalysis and biosensing. Heterogeneous proteins, in particular enzymes, can efficiently perform chemical processes in the solid phase facilitating their integration into flow systems and microdevices.1 In this context, protein technologists are encouraged to develop operationally efficient and stable heterogeneous proteins,2,3 although the comprehensive characterization of such supported bioconjugates is still rather limited. Atomic force microscopy (AFM) has been successfully exploited to study the immobilization of proteins majorly on 2D-planar surfaces,4–6 while atomic force spectroscopy (AFS) has allowed the indentation of a probe into a solid sample to characterize the mechanical properties of the material.7 Particularly, AFS can provide valuable information about how the interactions between the proteins and the surfaces affect the mechanical properties of both solid materials and biomolecules.8,9 This technique is challenging for 3D-porous microbeads formed by soft materials like hydrogels, because they might experience a plastic (permanent) deformation upon indentation. Consequently, proteins immobilized on such soft beads have been rarely characterized by either AFM imaging or AFS, despite those porous microbeads being revealed as useful supports for chromatography and biocatalysis,10 and lastly they have been successfully applied for point-of-care biosensors.11Spatially resolved force maps in the micrometric areas of porous beads will aid the understanding of the global effects promoted by the protein immobilization on the elastic properties of a single microbead. Hence, innovative solutions for AFM imaging and AFS of porous microbeads immobilizing proteins are paramount for advancing the characterization of the solids that immobilize proteins as “ready-to-use” biomaterials for technological purposes. Inspired by colloidal probe-AFS studies on cells,12–14 we envision this technique as a highly promising tool to characterize the mechanical properties of hydrogel porous microbeads upon protein immobilization. The colloidal probe-AFS usually indents relatively larger material areas than standard sharp pyramidal tips, providing mechanical information about the global mechanics of the indented material. In living cells, colloidal probe-AFS has served to elucidate the cellular stiffness and how such a parameter relies on the types of proteins integrated into the lipid bilayer forming the membrane.12 The stiffness of the cells is quantified by Young's modulus calculated from the force–displacement (F–d) curves obtained from indentation studies using AFM. Recently, colloidal probe-AFS has also been used to measure the stiffness of soft polymeric particles loaded with fluorescence dyes to demonstrate that the mechanical properties of such particles determine their uptake into the cells.15
Agarose microbeads are broadly used for protein purification and immobilization because they are biocompatible, tunable and versatile carriers.16 A large number of proteins have been immobilized on agarose microbeads through a variety of immobilization chemistries determining their functional properties.17 The immobilization chemistry controls two parameters that define the functionality of the immobilized proteins; (1) the number of interactions between the protein and the solid surface,18 and (2) the protein orientation on the solid surface.19 However, the effect of these two parameters on the mechanics of a single microbead immobilizing proteins remains unknown.
In this work, we have used AFS to understand how the nature of the protein–surface interactions affects the mechanical properties of the agarose microbeads upon protein immobilization. We have monitored the immobilization of a superfolded green fluorescent protein (sGFP)20 on porous agarose microbeads by indenting a colloidal probe into those beads. We have observed how the stiffness of the microbeads increases as protein immobilization progresses. Interestingly, we have also found a correlation between the stiffness of the agarose microbeads upon immobilization and the thermal stability of the immobilized proteins. Based on such correlation we have been able to predict the optimal immobilization chemistry to stabilize several oxidoreductases.
Experimental methods
Materials
Agarose 6BCL and agarose activated with cobalt were purchased from Agarose Bead Technologies (Madrid, Spain). Cyanogen bromide activated agarose (Ag-CB), Epichlorohydrine, sodium metaperiodate, CoSO4, iminodiacetic acid were purchased from Sigma Chem. Co (St. Louis, USA). Other reagents were of analytical grade.Immobilization
Proteins were recombinantly expressed (see ESI†) and immobilized on agarose microbeads 6BCL (ABT, Spain) activated with different reactive groups and under different conditions (see ESI†). 0.1 g of wet agarose beads were incubated with 1 mL of 1 mg mL−1 sGFP. In the case of FDH, GlyDH or NOX, 1 mg mL−1 of such proteins were incubated with 0.1 g of the corresponding agarose beads. The immobilization conditions varied according to the immobilization protocol. The immobilization on Ag-G was carried out with 100 mM sodium carbonate at pH 10 for 1–24 h and then the immobilized proteins were reduced with 1 mg mL−1 of sodium borohydride for 30 minutes at room temperature. Immobilization on Ag-Co2+ and Ag-CB were carried out in 25 mM sodium phosphate buffer at pH 7 for 1–3 hours. After the immobilization on Ag-CB, the microbeads loading the proteins were blocked with 1 M hydroxylamine in 25 mM sodium phosphate at pH 8. After all immobilization and post-immobilization process the matrices were washed with an excess of distilled water and stored at 4 °C.Thermal denaturation and inactivation studies
For thermal denaturation of sGFP, 10 mg of immobilized sGFP were incubated with 100 μL of 25 mM sodium phosphate at pH 7 in a PCR tube. The samples were denatured with a dynamic temperature ramp (20–99 °C; rate: 0.5 °C min−1) monitoring their fluorescence in a RT-PCR machine. For thermal inactivation, 0.1 g of different immobilized enzymes and proteins were incubated in 1 mL of 25 mM sodium phosphate buffer at pH 7 at different temperatures and samples were withdrawn after different times and their enzymatic activities were measured (see ESI†). FDH was incubated at 60 °C for 1 hour, GlyDH was inactivated at 65 °C for 1 hour and NOX was inactivated at 83 °C for 11 hours.Sample preparation for AFM and AFS studies
In order to stabilize the spheres upon the contact of the cantilever during microscopy and spectroscopy experiments, glass coverslips (thickness 0.13–0.16 mm, Thermo Fisher Scietific, Madrid, Spain) were coated with three polyelectrolyte layers (PEM) consisting of PDADMAC/PSS/PDADMAC using the layer by layer electrostatic assembly.21 This coating stabilizes repulsive interactions between the surface and a bead thus enabling steady imaging regarding lateral movements. The thin polyelectrolyte coating did not influence the mechanical data since we did not observe differences in the sensitivity of the colloidal probe between glass and PEM coated glass. PEM coated glass was assembled in a custom made JPK holder. Imaging and spectroscopy were performed in HEPES/NaCl 10/150 mM buffer at pH 7.4. We carried out the measurements using AFM by adding 800 μL of a suspension of 1.25 mg mL−1 of beads immobilizing the proteins (10 mgprotein gbeads−1). To monitor the immobilization in real time we added into the AFM chamber 1 mg of Ag-G particles and 800 μL of 12.5 μg mL−1 sGFP in 100 mM of sodium carbonate 100 mM pH 10.Atomic force spectroscopy (AFS)
Sample stiffness and elasticity were measured through the determination of Young's modulus from the nanoindentation experiments performed using a NanoWizard II AFM (JPK, Berlin, Germany) by acquiring force–displacement (F–d) curves in liquid medium. Measurements were performed using a 1 μm radius borosilicate colloidal probe attached to the cantilever (Novascan Technologies, USA). The spring constant of the cantilever was calibrated through the thermal noise method in the 10/150 mM HEPES/NaCl buffer adjusted to pH 7.4 where measurements were performed. The cantilever spring constant was 0.070 N m−1. We have employed as reference the glass surface of the sample holder activated with the corresponding polyelectrolyte layers. ∼200 F–d curves were acquired at a maximum applied load of 5 nN over a sample area of 3 μm × 3 μm. The cantilever was approached to the apex of each bead of radius ∼60 μm. In this way a relatively flat area at the apex of the bead (never larger than 9 μm2) was sampled for collecting F–d curves. In this set-up, we avoid the possible lateral shift of the microbead during the nanoidentation experiments, which could affect the measured force curves. The approach–retract curves were made at a speed of 1 μm s−1, with the total extension of the z-piezo of just 1 μm. Under these conditions, the characteristic contact time of the tip and the sample was about 300 ms. Therefore, we can assume that the bead behaves like an incompressible elastic solid22 and, consequently, the Hertzian approach (see above) can be applied. In addition, the maximum indentation value obtained for the softest system, i.e., the bare agarose beads, was in the 50–60 nm range, which is considerably smaller that the bead diameter. Thus, the underlying substrate does not play any role in the measured properties. These indentation values imply, from simple geometrical considerations, that the maximum area and volume sampled during a force spectroscopy experiment are ∼0.3 μm2 and ∼0.4 μm3, respectively. These figures guarantee that we are sampling the mechanical properties of the most outer part of the beads. For each sample, at least 5 different beads were probed and the resulting data were screened and processed using the JPKSPM Data Processing software. In order to obtain Young's elastic modulus (E) of each sample avoiding the possible viscoelastic effects we followed the procedure described by Wiedemair et al. or Best et al. by fitting the approach curves23,24 to the Hertz model (eqn (1)) for a spherical indenter, assuming a Poisson ratio (ν) of 0.50 applied for the mechanical determination of agarose hydrogels.25,26 Further statistical analysis of the resulting E values was performed using OriginPro 2015 software. Only curves showing a clear contact point in the approach–retract cycle are considered for analysis; the results are shown as the mean E value with a standard error that by the definition considers the number of analyzed curves.Results and discussion
Real-time immobilization of sGFP on agarose microbeads monitored by colloidal probe-AFS
sGFP was immobilized on agarose microbeads activated with glyoxyl groups (Ag-G). These porous microbeads presented a fiber size range of 25–40 nm, a reactive group density of 75 μmol g−1, and a pore size range of 100–150 nm.27–29 We carried out the immobilization process inside an AFM sample holder that was coated by a polyelectrolyte multilayer to fix the beads during experiment (see ESI†). The immobilization was performed under alkaline conditions to promote the reaction between the primary amine groups from the lysine residues found on the sGFP surface and the glyoxyl groups located at the Ag-G surface, forming Schiff's bases.30 The changes in the mechanical properties of single agarose microbeads during the immobilization process were investigated in real-time by nanoindentation measurements in order to determine Young's modulus (E). In the design of a nanoindentation experiment, both the shape and the stiffness of the probe play a major role in guaranteeing the accurate values of the elasticity modulus (Fig. 1A).31–33 Colloidal probes are highly useful to sample surfaces or materials with a high mechanical heterogeneity across their sub-micrometric structure where pyramidal tips may lead to unreliable and noisy mechanical data. | ||
| Fig. 1 (A) Atomic force spectroscopy of agarose microbeads upon protein immobilization. Nanoindentation of a colloidal probe (r = 1 μm) into the apex of a hydrogel bead (r = 60 μm). (B) Force–displacement (F–d) curve of Ag-G microbeads where sGFP molecules are immobilized. Colloidal probe approaches to the surface (red curve) until the contact point (green dot) where the cantilever starts to deflect. Approach half-cycle is followed by indentation of the agarose bead for the δ value. After indentation, the probe is retracted to its starting position (blue curve). The dimensions of the protein do not correspond to its real scale (sGFP = 2.4 × 4.2 nm), they are magnified for figure clarity. | ||
Fig. 1A illustrates the AFM set-up to measure the indentation of a silica sphere (1 μm radius) into the apex of the microbead (a quasi-planar area) as a function of the applied force, resulting in a force displacement (F–d) curve. A typical F–d curve from sGFP immobilized on the Ag-G agarose bead is shown in Fig. 1B, where the microbead is indented tens of nanometers by the colloidal silica probe. Expectedly for a soft material, the indentation depth increased with the applied force. Such an indentation depth was not observed when the glass surface of the sample holder was indented at different forces with the same colloidal probe (ESI,† Fig. S1). We used these F–d curves to determine the average elastic modulus corresponding to a 9 μm2 area of the microbead rather than to nanometric areas that may not accurately represent its stiffness.34,35 Therefore, the mechanics of these micrometric areas reflects the stiffness of a single bead instead the mechanical properties of a single protein immobilized on that bead. Young's modulus was calculated by fitting the F–d curves with the Hertz model (eqn (1)) where E is the elastic modulus (Young's modulus), R is the radius of the colloidal probe, ν is Poisson's ratio and δ is the indentation depth.36,37
 | (1) |
Fig. 2 shows that the stiffness of agarose microbeads increases while sGFP is being covalently attached to the porous structure of Ag-G microbeads. The E values of the agarose microbeads increased from 300 to 1000 kPa during the immobilization process. Noteworthy, after 30 minutes E values reached a plateau although the immobilization of proteins on the beads continued. When we analyzed the immobilization kinetics of sGFP by monitoring its fluorescence at the supernatant under the same conditions as inside the AFM sample holder, we observed that the microbeads reached the maximum stiffness before the immobilization was complete. This saturation point for force spectroscopy seems to occur at protein loads of around 7 mg gbeads−1 (Fig. 2), while the protein immobilization continues to reach up to 90% of the theoretical yield (9 mg gbeads−1). This insight suggests that AFS finds a sensitivity limit owing to the fact that the indentation layer becomes saturated of proteins and the immobilization continues on deeper layers of the agarose microstructure.
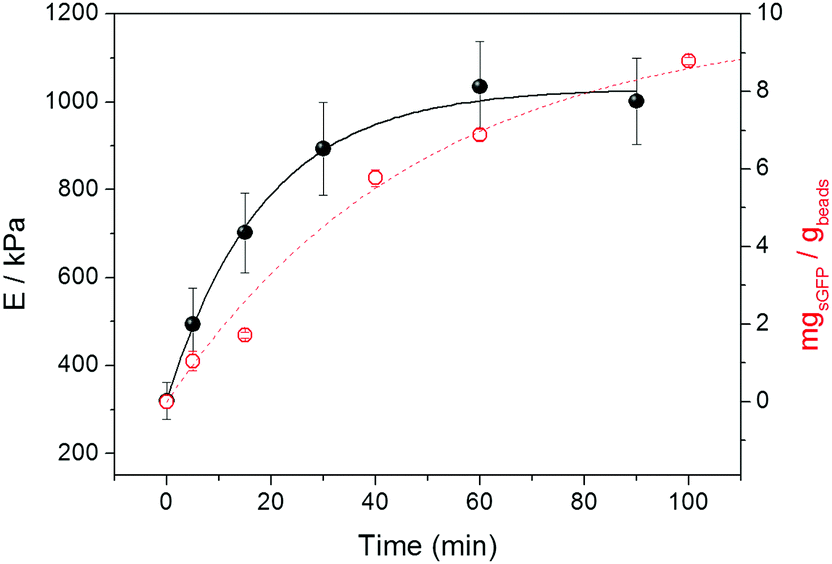 | ||
| Fig. 2 Real time covalent immobilization of sGFP onto Ag-G beads monitored by AFM. Young's modulus (black filled spheres) and immobilized proteins (red empty spheres) were monitored at different times. Immobilized proteins were indirectly calculated by measuring the remaining fluorescence at the supernatant. Protein immobilization was carried out at 25 °C in 100 mM of sodium carbonate buffer adjusted to pH 10. Young's modulus (E) is expressed as a mean value with its corresponding standard error. E values at time 0 correspond to the elasticity of the bare agarose microbeads, this value remains constant when the protein is not added to the solution. Loadings are expressed as the mean value of three experiments and their corresponding standard deviation. The solid and dashed lines are only guides to the eye. | ||
In contrast, when ethanolamine was attached to Ag-G under alkaline conditions, the resulting microbeads became 1.8-fold stiffer than the bare Ag-G beads, whereas Ag-G beads upon protein immobilization were 4.7-fold stiffer than the bare ones (Table S1, ESI†). The absolute E value is therefore 3 times higher when Ag-G immobilizes proteins than when tethers small molecules. This owes to the fact that ethanolamine can only establish one molecule–agarose bond, while several reactive residues in the protein surface drive to a multivalent interaction with the agarose fibers. In this regard, we suggest that the immobilization of sGFP on Ag-G promotes the protein decoration of the agarose fibers altering their elastic properties and significantly increasing the global stiffness of the microbead. We suggest that the proteins may act as a “biological varnish” that can coat and even cross-link the polymeric fibers through multivalent interactions, increasing their stiffness. Hence, these increasing E values along protein immobilization indicate that the formation of new protein–agarose bonds might affect some of the parameters that govern the microbead stiffness like the persistence length of the fibers.38 Similar results were obtained when a DNA-based hydrogel was functionalized with proteins resulting in stiffer hydrogel-like structures.39
Interestingly, the force map of a single Ag-G microbead covalently immobilizing sGFP was rather heterogeneous in terms of the spatial distribution of the E values (Fig. S2A, ESI†). The force map was consistent with the AFM image where patches of high protein density can be observed heterogeneously decorating the agarose fibers (Fig. S2B, ESI†). In fact, we expect this heterogeneity in the elastic properties of sub-μm regions since the agarose structure presents a wide fiber and pore size distribution across the microbead surface.8,40,41
Irreversible multivalency of protein–surface attachment affects the mechanics of agarose microbeads
We studied the effect of the number of covalent bonds between the sGFP molecules and the Ag-G surface on the E values of single microbeads. The attachment between sGFP and the glyoxyl groups on the Ag-G surface is multivalent and evolves with the incubation time under alkaline conditions.30To achieve a survey of immobilized preparations with different degrees of protein–surface interactions, we incubated sGFP with Ag-G microbeads at pH 10 for different times (1, 3 and 22 hours). The maximum immobilization yield (90%) was achieved in less than 1 h of incubation, thus all immobilized preparations were loaded with 9 mgsGFP gbeads−1 regardless of the incubation time. After incubation, the immobilized preparations were reduced with sodium borohydride to turn imine into amine bonds that make protein–surface interactions irreversible. Finally, we measured the E values of those different immobilized preparations under pH 7.4 (see Methods), finding higher E values for those microbeads incubated for longer times. This result suggests that longer incubation times under alkaline conditions allow establishing a higher number of rigid attachments between the proteins and the fibers, which increases the stiffness of the microbeads (Fig. 3). Pedroche et al. drew similar conclusions from the proteomic studies of trypsin immobilized on Ag-G at different incubation times. They quantified the number of protein–agarose bonds from different samples, observing that the enzyme immobilized for 1 and 24 hours under alkaline conditions established 3 and 7 protein–surface bonds, respectively.18 Noteworthily, when we studied the indentation events in single microbeads across a 9 μm2 area, we observed that the distribution of E values at 1 h of incubation was narrower – showing a sharp maximum at 427 kPa – than at 22 h incubation – two maxima at 461 and 698 kPa (Fig. 3). We suggest that the pore and fiber heterogeneity found in agarose besides the random distribution and the different reactivities of 20 lysines on the sGFP surface (Fig. S3, ESI†) provoke different interaction levels between sGFP and the agarose fibers at different times. At shorter times (1 h), the majority of sGFP molecules are likely immobilized through the most reactive lysines, those with lower apparent pKa values and higher accessibility to the media. In this case, the reactivity of these highly accessible lysines is negligibly affected by the steric hindrances underlying the agarose structure, thus most of the sGFP molecules should similarly interact with the agarose via few bonds, resulting in fibers with similar stiffness, driving to narrower histograms (Fig. 3). On the contrary, those less reactive lysines that are more hindered in the protein structure or present higher pKa values require longer times for the productive interaction with the agarose fibers, explaining the formation of more protein–agarose bonds that increase the stiffness of the microbeads upon 24 h immobilization. Moreover, the heterogenous structure of the agarose seems to dramatically affect the formation of these slower interactions, where less accessible lysine residues can never interact with the aldehydes located at those geometrically constrained areas of the fibers. However the same lysine residues of other sGFP molecules have the time to interact with the aldehydes located in more accessible areas of the agarose surface. These slow interactions affected by the geometrical constrains of some fibers create the wider distribution of Young's modulus histogram at longer incubation times (Fig. 3), conducting to protein populations with different levels of protein–agarose interactions, giving rise to fibers with different degrees of stiffness in the same micrometric area.
 | ||
| Fig. 3 Distribution of Young's modulus of sGFP immobilized on Ag-G microbeads for different incubation times. sGFP was immobilized at pH 10 on Ag-G for 1, 3 or 22 h and further reduced with sodium borohydride. Force spectroscopy measurements were performed in 10/150 mM HEPES/NaCl buffer at pH 7.4. The protein load of the three samples was 9 mgsGFP gbeads−1. Multi-modal distribution of E values is represented with the Kernel curve overlapping each histogram. The counts (Y-axis) represent the number of indentation events of the colloidal probe within the 3 × 3 μm3 area. | ||
Correlation between thermal stability and microbead stiffness
Functional and structural properties of immobilized proteins strongly rely on the immobilization chemistry through which the carrier attaches proteins.42 The reactive groups on the solid surface that enable protein immobilization thus determine the structural integrity and stability of different reactive groups. In addition to the irreversible and multivalent immobilization of sGFP on Ag-G, the fluorescent protein was also immobilized on agarose microbeads activated with cyanogen bromide groups (Ag-CB) that efficiently react with the protein N-terminus, creating a single irreversible bond under mild conditions. Finally, a sGFP tagged with 6 histidines at its N-terminus was immobilized on agarose microbeads activated with cobalt-chelates groups (Ag-Co2+) through one reversible and flexible attachment based on a coordination bond. The three immobilized preparations were analyzed by colloidal probe-AFS determining their absolute E values and normalizing them according to the absolute E values of their corresponding bare (without protein) agarose beads (Fig. 4A and Table S1, ESI†). Microbeads immobilizing sGFP through aldehyde chemistry presented a normalized Young's modulus 8-fold higher than agarose microbeads immobilizing the same protein through either cyanogen bromide or metal groups. These differences may rely on the multivalency of the protein–agarose interactions. To prove this fact, we modified Ag-G, Ag-CB and Ag-Co2+ with different small molecules under the specific immobilization conditions to mimic the univalent interaction between a single residue on the protein surface and one reactive group in the agarose fibers. When Ag-CB and Ag-Co2+ were modified with ethanolamine and imidazole, respectively, the normalized E values were similar to those measured for the corresponding microbeads upon protein immobilization (Fig. 4A). On the contrary, the normalized E value of Ag-G tethered with ethanolamine was similar to the ones for Ag-CB and Ag-Co2+ modified with small molecules, but 4.3 times lower than those for Ag-G microbeads upon sGFP immobilization (Table S1, ESI†). These data indicate that multivalent protein–agarose interactions (on Ag-G) significantly increase the microbead stiffness upon immobilization, whereas such an effect was negligible when the protein–agarose interactions are univalent (on both Ag-Co2+ and Ag-CB). On the other hand, Fig. 4B shows the dynamic thermal denaturation of the sGFP immobilized through different chemistries, and indicates that the immobilization chemistry that drove to stiffer microbeads, also made proteins more difficult to denature by temperature. sGFP immobilized on Ag-CB and Ag-Co2+ showed a similar behavior in thermal denaturation, linearly losing its fluorescence with increasing temperature until reaching an inflection point (T = 89–91 °C) where fluorescence dramatically decays to 0, likely due to protein unfolding (Fig. 4B and Fig. S4, ESI†).43 On the contrary, sGFP immobilized on Ag-G also followed the linear fluorescence reduction at higher temperatures but never reached the inflection point corresponding to protein unfolding. Therefore, the robust and multivalent nature of the aldehyde chemistry leads to more thermostable proteins than the flexible and univalent character of chemistries based on both cyanogen bromides and metal chelates. Similar insights have been found for a plethora of different proteins based on inactivation studies at high temperatures.44 More recently, our group has reported that the orientation and the intensity of the attachment of an enhanced fluorescent protein (structurally analogous to sGFP) immobilized through different chemistries promote different protein conformations with different dynamic and functional properties.45 Likewise, we herein observe that the nature of the protein–agarose bonding determines the mechanical properties of the beads as well as the thermal stability of the immobilized proteins (Fig. 4). Therefore, the immobilization chemistry determines both the protein orientation and the valence of the attachment; two factors that directly affect the conformation of the immobilized proteins resulting in more or less stable proteins, but also the mechanics of the materials upon the immobilization process. These results indicate that the immobilization chemistries that increase the stiffness of the agarose microbeads by creating rigid and multivalent protein–fiber interactions reciprocally rigidify the protein structures increasing their thermal stability. This experimental correlation between the thermal stability of the immobilized proteins and the global stiffness of the agarose microbeads upon immobilization opens a promising avenue to predict the experimental thermal stability of immobilized proteins by using force spectroscopy. We envision AFS as a label-free technique able to evaluate the capacity of different immobilization chemistries to promote protein stabilization, just screening them by colloidal probe-AFS. | ||
| Fig. 4 Normalized Young's modulus (A) and thermal denaturation (B) of sGFP immobilized on agarose microbeads through different reactive groups. sGFP was immobilized on agarose beads activated with aldehyde groups (Ag-G), cobalt chelates (Ag-Co2+) and cyanogen bromide groups (Ag-CB) for 24 hours under the corresponding conditions. Force spectroscopy measurements were performed in 10/150 mM HEPES/NaCl buffer at pH 7.4. Normalized Young's modulus values are calculated by subtracting the absolute Young's modulus value of the corresponding bare agarose beads from the absolute E values obtained for each type of agarose beads immobilizing either sGFP or small molecule (ethanolamine on Ag-G and Ag-CB, imidazol on Ag-Co2+) (Table S1, ESI†). Those values are expressed as a mean value with its corresponding standard error. The thermal denaturation of the immobilized sGFP was monitored by measuring the protein fluorescence at different temperatures (see Experimental methods). | ||
Force spectroscopy of single microbeads predicts the thermal stability of the resulting heterogeneous biocatalysts
The correlation between microbead stiffness and thermal stability of the immobilized proteins encouraged us to use force spectroscopy to characterize three oxidoreductases immobilized on agarose microbeads through different immobilization chemistries. Fig. 5 shows the normalized E values of three different enzymes; formate dehydrogenase (FDH), a homodimeric enzyme from Candida boidinii, glycerol dehydrogenase (GlyDH), a homoocatameric enzyme from Bacillus stearothermophilus, and NADH oxidase (NOX), a homodimeric enzyme from Thermus thermophilus, immobilized on either Ag-CB or Ag-G. The agarose microbeads immobilizing the different enzymes through the aldehyde chemistry were significantly stiffer than their counterparts immobilized on Ag-CB, likely owing to the higher number of bonds between the proteins and the Ag-G surface. In fact, enzymes attached through one bond (N-terminus) to Ag-CB slightly increased the microbead stiffness compared to bare Ag-CB microbeads (Table S1, ESI†). | ||
| Fig. 5 Young's modulus and thermal stability of different enzymes immobilized through different reactive groups. Each enzyme was immobilized on agarose beads activated either with aldehyde (Ag-G) or cyanogen bromide (Ag-CB) groups. The different insoluble preparations were inactivated at different temperatures for 1 h. * NOX was inactivated for 11 h. Residual activity means the percentage of the enzyme activity retained after incubation at high temperatures. Force spectroscopy measurements were performed in 10/150 mM HEPES/NaCl buffer at pH 7.4. Normalized Young's modulus values are calculated by subtracting the absolute Young's modulus value of the corresponding bare agarose beads to the absolute E values obtained for each type of agarose beads immobilizing each protein through different immobilization chemistries (Table S1, ESI†). Those values are expressed as a mean value with its corresponding standard error. | ||
On the other hand, immobilization of the enzymes on Ag-G gave rise to more stable heterogeneous biocatalysts than immobilization on Ag-CB. In light of both spectroscopic and observable data obtained for these three oxidoreductases, we confirm the results found for sGFP, which demonstrate that the multivalent protein–agarose interactions increase both the thermal stability of the immobilized proteins and the stiffness of the microbeads upon immobilization (Fig. 5). For example, GlyDH immobilized on Ag-G retains almost 5 times more activity at high temperatures and microbeads are 29 times stiffer than the same enzyme immobilized on Ag-CB. A similar trend was found for FDH and NOX, however the latter enzyme presented the lowest E value (495 ± 32 kPa) among the enzymes immobilized through the aldehyde chemistry, in spite of being the most thermostable heterogeneous biocatalyst tested herein.
We suggest that the low content in lysine residues of NOX (12 lysines, 6 per subunit) promotes less protein–agarose interactions when that enzyme is immobilized through the aldehyde chemistry, and thus the corresponding microbeads are significantly less stiff than their counterparts immobilizing either GlyDH (232 lysines, 29 per subunit) or FDH (64 lysines, 32 per subunit). Even though NOX is attached to the surface through few bonds, the immobilized enzyme is highly stable because of the intrinsic thermostability of the soluble protein due to its thermophilic origin. Therefore, the multivalency of the attachment ultimately depends on the lysine content of each protein.
These data substantiate that the mechanics of the microbeads can be used to indirectly elicit the thermal stability of different heterogeneous biocatalysts of the same enzyme with different levels of protein–agarose interactions, but cannot correlate thermal stability with microbead stiffness between two different enzymes immobilized through different, or even the same, chemistries. However, the stiffness of the agarose microbead immobilizing enzymes is a good indicator to evaluate the number of interactions between the enzymes and the solid surfaces. Hence, colloidal probe-AFS allows demonstrating that the immobilization chemistries which establish a high number of rigid bonds between the proteins and the fibers increase the stiffness of the microbeads upon immobilization and promote a significant thermal stabilization of the immobilized proteins. Therefore, microbead stiffness can be used as a measurable parameter to predict the optimal immobilization chemistry to achieve the highest thermal stability of one heterogeneous biocatalyst.
Conclusions
Nanoindentation by colloidal probe-AFS is revealed as a very informative technique to characterize proteins immobilized on porous microbeads. With this technique, we can investigate in real time the immobilization of proteins on a single microbead, monitoring the changes in Young's modulus of the agarose microbead during the immobilization process. In addition, we have discovered that microbead stiffness, as well as thermal stability, is affected by the immobilization chemistry that attaches the proteins to the solid material. This technique has been applied to forecast the stability of several immobilized enzymes with a high impact on industrial biotechnology, demonstrating that force spectroscopy is highly useful to indirectly unravel some functional properties of “ready-to-use” heterogeneous biocatalysts labeling neither the enzymes/proteins nor the solid materials where they are immobilized. Future research should focus on studying more complex systems formed by multi-enzyme consortiums as well as monitoring the changes in the microbead mechanics during the operational process with AFM. In this scenario, we may observe the real-time lixiviation of proteins from the solid surfaces under the reaction conditions, or protein unfolding that would reduce the rigidity of the coated fiber, decreasing the global stiffness of the microbeads.Acknowledgements
We acknowledge COST action CM103-System biocatalysis and IKERBASQUE for the funding to FLG. DG and SEM acknowledge the project MAT2013-48169-R from the Spanish Ministry of Economy (MINECO) and LV thanks financial support from CAM (project NANOAVANSENS, Ref. S2013/MIT-3029) and FIS2012-38866-C05-05 from MINECO. FLG also acknowledges the Spanish Ministry of Economy and Marie-Curie actions for funding the BIO2014-61838-EXP and NANOBIOENER, respectively. We finally acknowledge Mr Eneko San Sebastian for his support on image analysis and representation.References
- P. Tufvesson, W. Fu, J. S. Jensen and J. M. Woodley, Food Bioprod. Process., 2010, 88, 3 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, A. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, Chem. Soc. Rev., 2013, 42, 6290 RSC.
- J. M. Bolivar, I. Eisl and B. Nidetzky, Catal. Today, 2016, 259(part 1), 66 CrossRef CAS.
- Y. H. Tan, J. R. Schallom, N. V. Ganesh, K. Fujikawa, A. V. Demchenko and K. J. Stine, Nanoscale, 2011, 3, 3395 RSC.
- J. Ngunjiri, D. Stark, T. Tian, K. Briggman and J. Garno, Anal. Bioanal. Chem., 2013, 405, 1985 CrossRef CAS PubMed.
- N. Aissaoui, L. Bergaoui, S. Boujday, J.-F. Lambert, C. Méthivier and J. Landoulsi, Langmuir, 2014, 30, 4066 CrossRef CAS PubMed.
- S. A. Chizhik, Z. Huang, V. V. Gorbunov, N. K. Myshkin and V. V. Tsukruk, Langmuir, 1998, 14, 2606 CrossRef CAS.
- A. L. Weisenhorn, B. Drake, C. B. Prater, S. A. Gould, P. K. Hansma, F. Ohnesorge, M. Egger, S. P. Heyn and H. E. Gaub, Biophys. J., 1990, 58, 1251 CrossRef CAS PubMed.
- P. Y. Meadows, J. E. Bemis and G. C. Walker, Langmuir, 2003, 19, 9566 CrossRef CAS.
- M. Hartmann and X. Kostrov, Chem. Soc. Rev., 2013, 42, 6277 RSC.
- J. Chou, J. Wong, N. Christodoulides, P. Floriano, X. Sanchez and J. McDevitt, Sensors, 2012, 12, 15467 CrossRef PubMed.
- L. Arnal, D. O. Serra, N. Cattelan, M. F. Castez, L. Vázquez, R. C. Salvarezza, O. M. Yantorno and M. E. Vela, Langmuir, 2012, 28, 7461 CrossRef CAS PubMed.
- X. Chen, L. Mahadevan, A. Driks and O. Sahin, Nat. Nanotechnol., 2014, 9, 137 CrossRef CAS PubMed.
- V. Vadillo-Rodriguez and J. R. Dutcher, Soft Matter, 2011, 7, 4101 RSC.
- R. Hartmann, M. Weidenbach, M. Neubauer, A. Fery and W. J. Parak, Angew. Chem., Int. Ed., 2015, 54, 1365 CrossRef CAS PubMed.
- M. Rinaudo, Polym. Int., 2008, 57, 397 CrossRef CAS.
- C. Mateo, J. M. Bolivar, C. A. Godoy, J. Rocha-Martin, B. C. Pessela, J. A. Curiel, R. Muñoz, J. M. Guisan and G. Fernández-Lorente, Biomacromolecules, 2010, 11, 3112 CrossRef CAS PubMed.
- J. Pedroche, M. del Mar Yust, C. Mateo, R. Fernández-Lafuente, J. Girón-Calle, M. Alaiz, J. Vioque, J. M. Guisán and F. Millán, Enzyme Microb. Technol., 2007, 40, 1160 CrossRef CAS.
- C. A. Godoy, B. d. l. Rivas, V. Grazú, T. Montes, J. M. Guisán and F. López-Gallego, Biomacromolecules, 2011, 12, 1800 CrossRef CAS PubMed.
- J.-D. Pedelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79 CrossRef CAS PubMed.
- D. Gregurec, M. Olszyna, N. Politakos, L. Yate, L. Dahne and S. Moya, Colloid Polym. Sci., 2015, 293, 381 CAS.
- Y. Hu, E. P. Chan, J. J. Vlassak and Z. Suo, J. Appl. Phys., 2011, 110, 086103 CrossRef.
- J. Wiedemair, M. J. Serpe, J. Kim, J.-F. Masson, L. A. Lyon, B. Mizaikoff and C. Kranz, Langmuir, 2007, 23, 130 CrossRef CAS PubMed.
- J. P. Best, J. Cui, M. Müllner and F. Caruso, Langmuir, 2013, 29, 9824 CrossRef CAS PubMed.
- V. Normand, D. L. Lootens, E. Amici, K. P. Plucknett and P. Aymard, Biomacromolecules, 2000, 1, 730 CrossRef CAS PubMed.
- M. Ahearne, Y. Yang, A. J. El Haj, K. Y. Then and K.-K. Liu, J. R. Soc., Interface, 2005, 2, 455 CrossRef CAS PubMed.
- P.-E. Gustavsson, K. Mosbach, K. Nilsson and P.-O. Larsson, J. Chromatogr. A, 1997, 776, 197 CrossRef CAS.
- A. S. Medin, Doctoral dissertation, Uppsala University, 1995.
- J. Guisan, Enzyme Microb. Technol., 1988, 10, 375 CrossRef CAS.
- G. Fernandez-Lorente, F. Lopez-Gallego, J. M. Bolivar, J. Rocha-Martin, S. Moreno-Perez and J. M. Guisan, Curr. Org. Chem., 2015, 19, 1 CrossRef.
- M. E. McConney, S. Singamaneni and V. V. Tsukruk, Polym. Rev., 2010, 50, 235 CrossRef CAS.
- M. E. McConney, K. D. Anderson, L. L. Brott, R. R. Naik and V. V. Tsukruk, Adv. Funct. Mater., 2009, 19, 2527 CrossRef CAS.
- D. C. Lin, E. K. Dimitriadis and F. Horkay, Recent Research Developments in Biophysics Transworld Research Network, Kerala, 2006, vol. 5, p. 333 Search PubMed.
- W. F. Heinz and J. H. Hoh, Trends Biotechnol., 1999, 17, 143 CrossRef CAS PubMed.
- B. Cappella and G. Dietler, Surf. Sci. Rep., 1999, 34, 5 CrossRef.
- H. Hertz, J. Reine. Angew. Math., 1882, 1882, 156 Search PubMed.
- J. Domke and M. Radmacher, Langmuir, 1998, 14, 3320 CrossRef CAS.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651 CrossRef CAS PubMed.
- F. A. Aldaye, W. T. Senapedis, P. A. Silver and J. C. Way, J. Am. Chem. Soc., 2011, 132, 14727 CrossRef PubMed.
- N. Pernodet, M. Maaloum and B. Tinland, Electrophoresis, 1997, 18, 55 CrossRef CAS PubMed.
- K. Wadu-Mesthrige, N. A. Amro and G.-Y. Liu, Scanning, 2000, 22, 380 CrossRef CAS PubMed.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250 RSC.
- C. Kiss, J. Temirov, L. Chasteen, G. S. Waldo and A. R. M. Bradbury, Protein Eng., Des. Sel., 2009, 22, 313 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451 CrossRef CAS.
- A. H. Orrego, C. García, J. Mancheño, J. M. Guisán, M. P. Lillo and F. López-Gallego, J. Phys. Chem. B, 2016, 120, 485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting methods, immobilization parameters and absolute Young's modulus values for all the samples described in this work, topographical and force map of agarose beads and dF/dT plots representing the thermal denaturation of immobilized samples. See DOI: 10.1039/c6sm01435f |
| This journal is © The Royal Society of Chemistry 2016 |