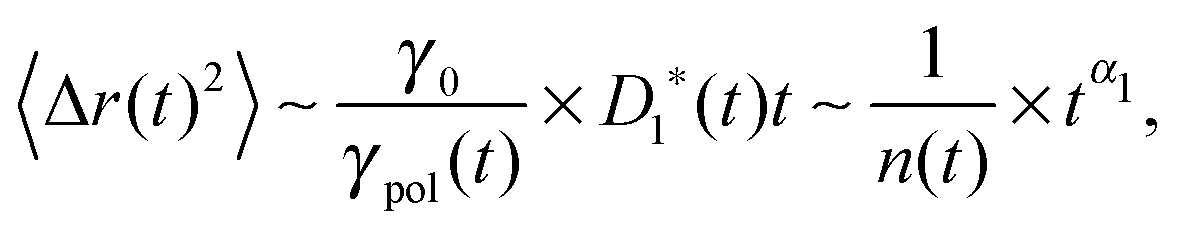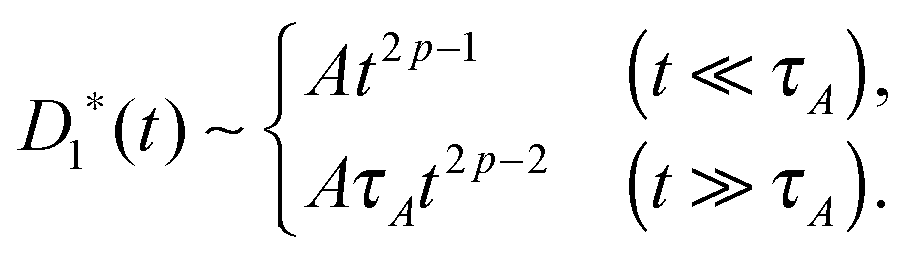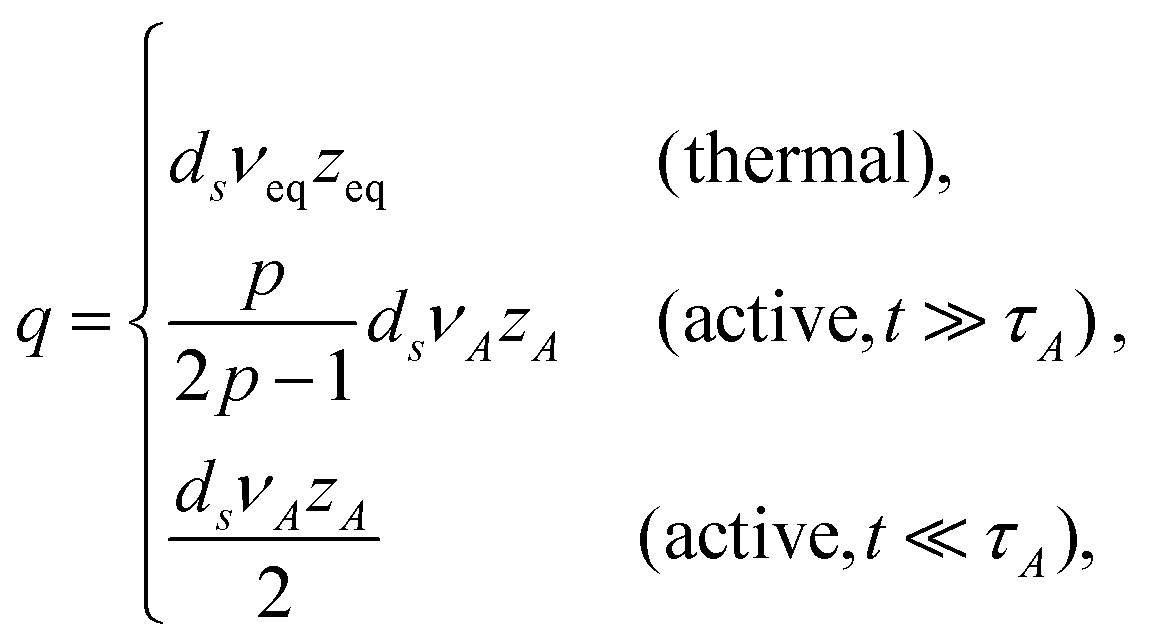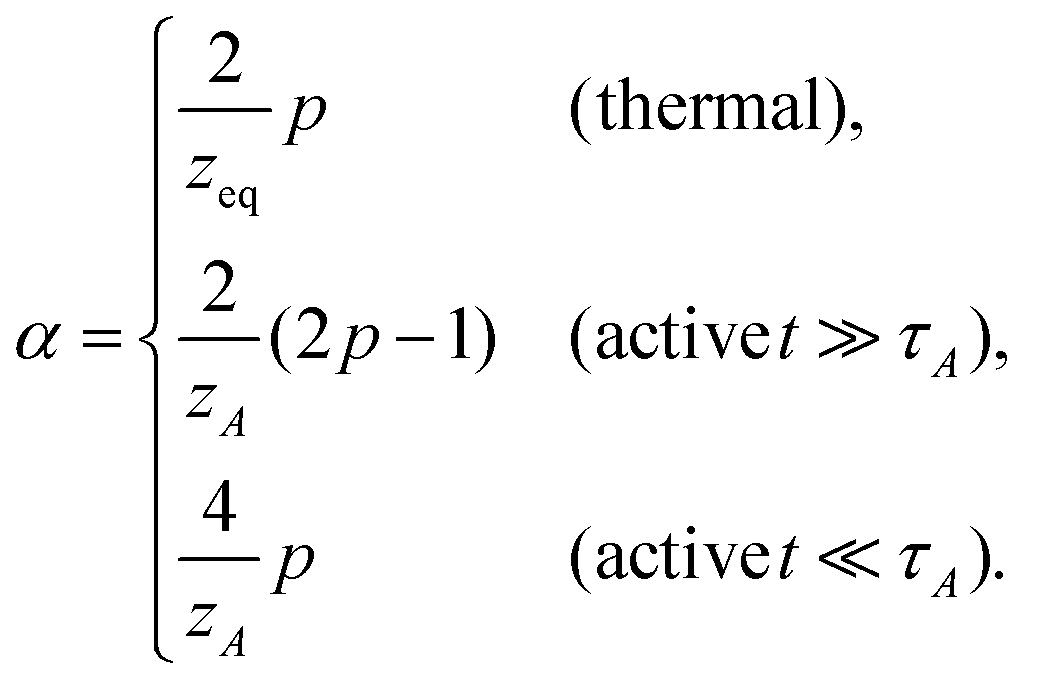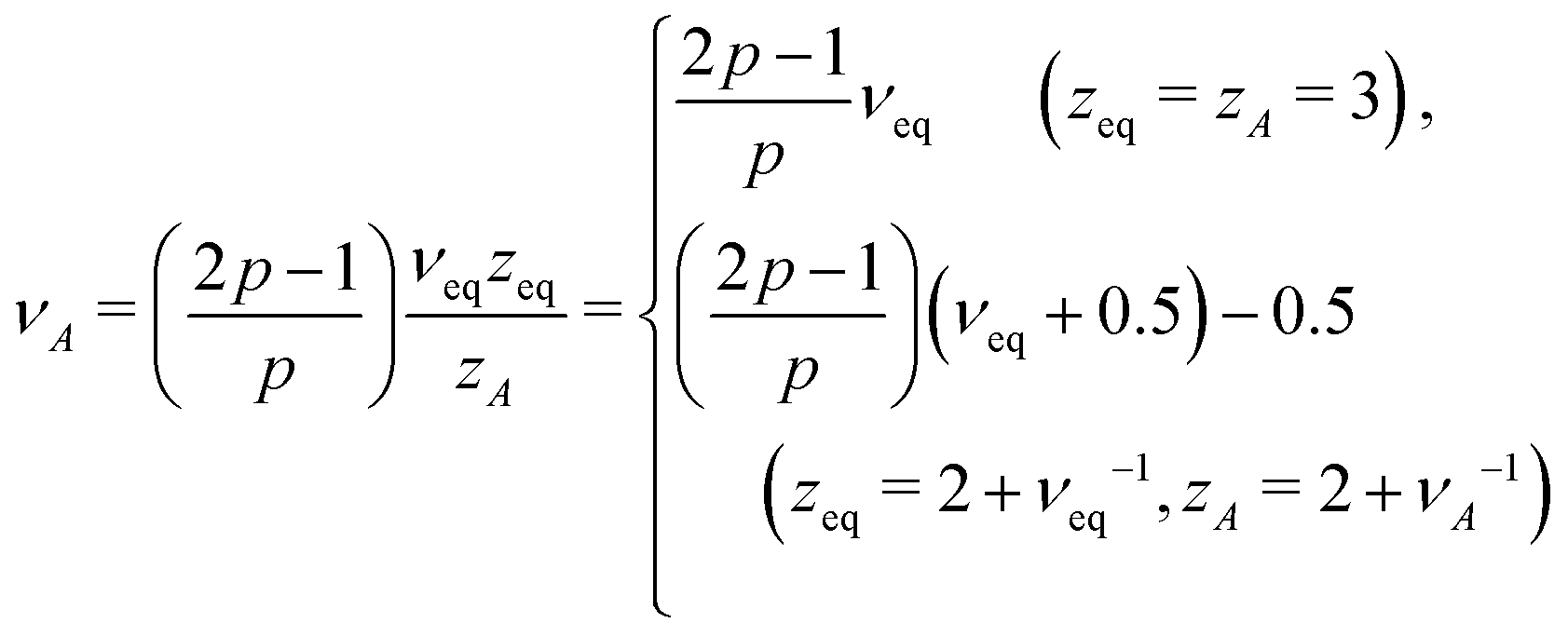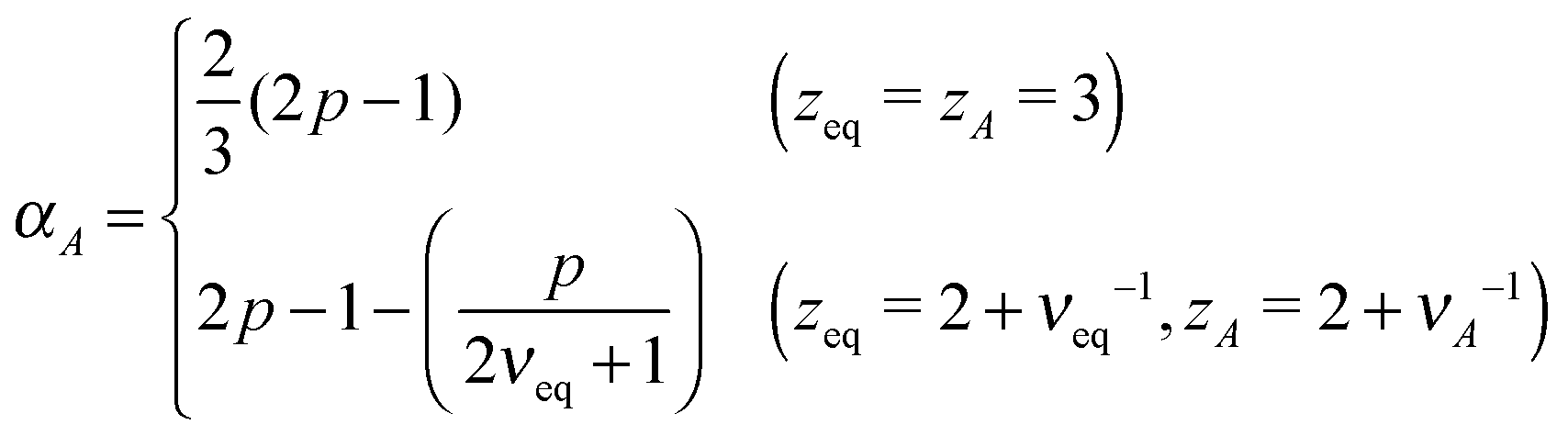DOI:
10.1039/C6SM00775A
(Communication)
Soft Matter, 2017,
13, 81-87
Active diffusion of model chromosomal loci driven by athermal noise
Received
31st March 2016
, Accepted 25th May 2016
First published on 25th May 2016
Abstract
Active diffusion, i.e., fluctuating dynamics driven by athermal noise, is found in various out-of-equilibrium systems. Here we discuss the nature of the active diffusion of tagged monomers in a flexible polymer. A scaling argument based on the notion of tension propagation clarifies how the polymeric effect is reflected in the anomalous diffusion exponent, which may be of relevance to the dynamics of chromosomal loci in living cells.
I Introduction
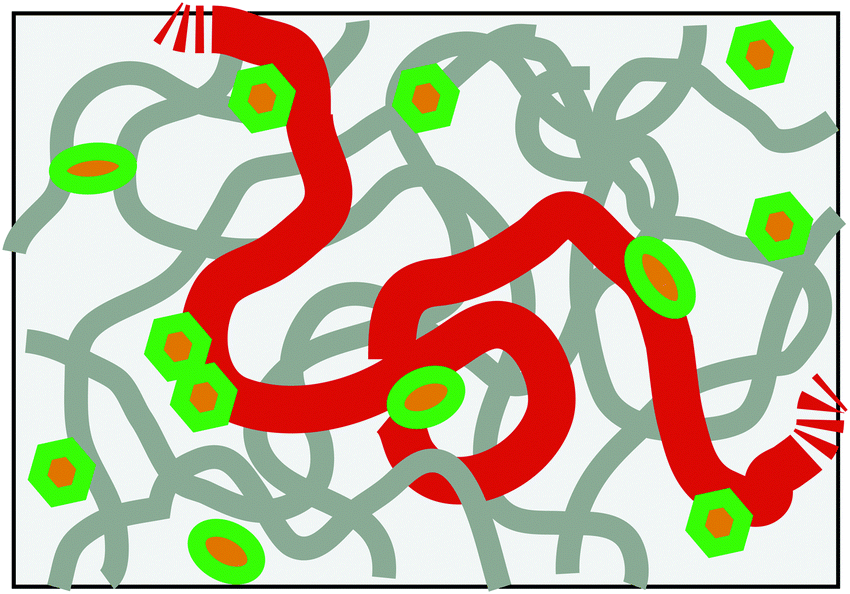
It is now well acknowledged that chromosomes in living cells are highly dynamic entities.1,2 Clarifying their dynamic structure in various length and timescales is one of the major challenges in the current biological science. In this respect, it is remarkable that recent experiments are starting to reveal the rich fluctuating dynamics of chromosomal loci in prokaryotic cells and eukaryotic nuclei.3–5 As a rule, the motion of loci looks apparently random, but there are indications from the measurement of the mean square displacement (MSD) 〈Δr(t)2〉 ∼ tα that its nature is different from ordinary Brownian motion, i.e., α ≠ 1.6 Several reasons can be conceived: (i) chromosomes exist in a highly crowded environment in cells, which may endow the viscoelastic memory effect for the motion. (ii) The metabolic activity in living cells drives the system out-of-equilibrium. Several experiments evidenced that the in vivo motion of loci is ATP-dependent, thus non-thermal.2,3,5,7 (iii) Being long DNA molecules complexed with structure proteins, chromosomes behave as flexible polymers in length scale larger than ca. 30 nm. Any chromosomal locus is, thus, a part of a long polymer chain (Fig. 1).
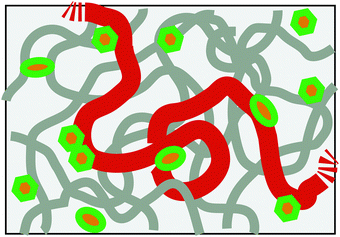 |
| | Fig. 1 Schematic of the model chromosome chain in a nuclear-like crowded, active environment. The boldest curve shows only a part of the chain with the remaining part of the same chain and other chains being drawn in thinner curves. Other crowding agents are drawn as small objects, some of which act as the active noise generators consuming chemical fuels (ATP). | |
The motion of chromosomal loci in cells is, in a sense, similar to the sub-diffusional motion, i.e., 〈Δr1(t)2〉 ∼ tα1 with α1 < 1, of micron-sized non-polymeric, say colloidal particles in complex medium (here the subscript “1” is introduced to mark non-polymeric particles, which will be referred to as simple probes hereafter).8–12 However, the measured exponent α for the loci, though dependent on various factors (species, cell cycle, the drug treatment, etc.), is often substantially smaller than that for simple probes, i.e., α < α1.4,13 Given the point (iii) in the preceding paragraph, one asks if it is a general consequence of the polymeric nature of chromosomes.
In ref. 13, the Rouse model supplemented with a cytoplasmic viscoelasticity has been suggested to account for the sub-diffusional motion of loci in bacterial cells, where the motion of an n-th monomer (n ∈ (0,N)) reads in the continuum limit
| | 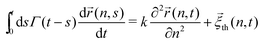 | (1) |
where
Γ(
t) = (
γ0/
τ0)(2 −
p)(1 −
p)|
t/
τ0|
−p (0 <
p < 1) is a memory kernel. The spring constant
k = 3
kBT/
a2 (
kBT: the thermal energy,
a: the size of monomers) for the chain connectivity and the monomeric friction coefficient
γ0 define the microscopic timescale
τ0 =
γ0/
k. In addition,
![[small xi, Greek, vector]](https://www.rsc.org/images/entities/i_char_e1cd.gif) th
th(
n,
t) is the thermal noise with zero mean whose second moment obeys the fluctuation-dissipation theorem (FDT) 〈
th(
n,
t)
th(
m,
s)〉 =
kBTΓ(
t −
s)
δ(
n −
m)
I with
I being 3 × 3 identity matrix. The main outcome from the model is that, while the sub-diffusion of the simple probes subjected to the same kernel
Γ(
t) is characterized by the MSD exponent
α1 =
p, the exponent of tagged monomers,
i.e., chromosomal loci, halves
α =
p/2, see
eqn (5) below. More recently, the model has been extended to include the effect of the activity in living cells, in which the noise term in
eqn (1) is modified as
th(
t) →
(
t) =
th(
t) +
A(
t).
14 The newly added active noise
A(
t) with zero mean was supposed to have the correlation of the form
| | | 〈A(n,t)A(m,s)〉 = Ae−|t−s|/τAδ(n − m)I | (2) |
where
A and
τA characterize the strength and the persistence time of the active noise. The analysis of the model indicates a much richer scenario including a super-diffusion behavior in a certain time interval, which is connected to the out-of-equilibrium nature of the system manifested by the fact that the noise
(
t) does not obey FDT.
Our purpose here is to try to extract a key physics in the behaviors of polymers in the viscoelastic active medium from a simple scaling argument based on the notion of tension propagation. This is particularly interesting due to its potential relevance to the problem of chromosomal loci in cells. As a main result, we pinpoint how the motion of chromosomal loci, modeled as tagged monomers in a long polymer, differs from that of simple probes in various situations (viscous, viscoelastic fluids with or without active noise). We also discuss the stationary conformation of polymers in active media; while the large scale behavior is characterized by an exponent, which may (or may not) be different from conventional Flory's, there is a certain subtlety in the model regarding the short scale behavior. Another merit of our argument lies in its flexibility for various generalizations, for instance, to include the effect of the self-avoidance, hydrodynamic interactions, non-linear molecular architecture, and possibly the convective flow which may be present in the medium.
II Tagged-monomer diffusion viewed from tension propagation dynamics
Even without entanglements, the dynamics seen in polymeric systems differs in many aspects from those in other non-polymeric counterparts. The anomalies in the former often trace back to the viscoelastic response associated with the dynamics of tension propagation. This notion of tension propagation is getting increasingly popular in recent studies on non-equilibrium dynamics of single polymers,15–18 in particular, in the context of polymer translocation.19–23 The term “non-equilibrium” here indicates the transient process, in which the large driving force induces the conformational distortion. Not only in the transient process, however, the tension propagation is equally important as a decisive factor in the dynamics and rheology of polymers in the stationary state. In fact, the dynamics of tagged monomers in a long polymer chain can be most easily described in terms of the time-dependent friction effected by the tension propagation. We first illustrate it using the Rouse polymer in viscous media, and then proceed to look into the viscoelasticity and/or active noise effects. In Section 3, we discuss various generalizations.
In viscous media
A sub-diffusion of tagged monomers is a generic property in long polymer molecules, which can be found already in the Rouse model, the simplest model for polymer dynamics.†24 Here we provide two arguments for it. Eqn (1) reduces to a classical Rouse model by replacing the left-hand side with γ0d![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) (n,t)/dt (or equivalently by taking the limit p → 1). One can see that the deterministic part of the Rouse equation of motion takes a form of the diffusion equation with the diffusion coefficient k/γ0. This implies that a disturbance at particular sites inside the chain will diffusively propagate along the chain, i.e., the number n(t) of monomers that can adjust themselves to the disturbance grows as (t/τ0)1/2, which defines a terminal timescale τN ≃ τ0N2 at which the tension propagates up to the chain end. In the first argument (I), we appreciate that n(t) corresponds to the number of monomers that move coherently, leading to the time-dependent friction γ*(t) = γ0n(t) ≃ γ0(t/τ0)1/2 for the motion of the tagged monomer. We thus find the MSD scaling
(n,t)/dt (or equivalently by taking the limit p → 1). One can see that the deterministic part of the Rouse equation of motion takes a form of the diffusion equation with the diffusion coefficient k/γ0. This implies that a disturbance at particular sites inside the chain will diffusively propagate along the chain, i.e., the number n(t) of monomers that can adjust themselves to the disturbance grows as (t/τ0)1/2, which defines a terminal timescale τN ≃ τ0N2 at which the tension propagates up to the chain end. In the first argument (I), we appreciate that n(t) corresponds to the number of monomers that move coherently, leading to the time-dependent friction γ*(t) = γ0n(t) ≃ γ0(t/τ0)1/2 for the motion of the tagged monomer. We thus find the MSD scaling| | | 〈Δr(t)2〉 ≃ D*(t)t ≃ a2(t/τ0)1/2 | (3) |
where D*(t) = kBT/γ*(t) is the effective diffusion coefficient according to the Einstein relation. The second argument (II) is based on the observation that the section of polymers with n monomers can equilibrate itself during the time interval τn ≃ τ0n2. This implies that the amplitude of the tagged-monomers' positional fluctuation in the time interval coincides with the equilibrium size an1/2 of the section. Assuming the sub-diffusion scaling 〈Δr(t)2〉 ∼ tα for t < τN, we thus write 〈Δr(τn)2〉 ≃ n2α ≃ n1, from which we again find α = 1/2. One may first wonder whether these two arguments are redundant. However, a closer inspection reveals that while the argument (II) invokes the equilibrium conformational properties of the polymer, the argument (I) does not. The equivalent result is reached, however, through the Einstein relation (FDT). This point manifests later, when we discuss the dynamics in the active media. Finally, we note that the exponent α = 1/2 can be compared to the normal diffusion α1 = 1 for simple probes in viscous fluids.
In viscoelastic media
A power-low memory kernel Γ(t) ∼ |t|−p implies the time-dependent friction γ1*(t) ∼ t1−p for the motion of simple probes, hence, the MSD 〈Δr1(t)2〉 ≃ D1*(t)t with D1*(t) = kBT/γ1*(t), i.e., a sub-diffusion with exponent α1 = p. To see how the medium viscoelasticity affects the motion of tagged monomers, we point out that the dynamics of tension propagation is governed by the following fractional diffusion equation;| | 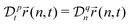 | (4) |
where the operator 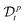 represents the fractional derivative of order p with respect to time.‡ For the “spatial” derivative along the chain (n-coordinate), the order is q = 2 for Rouse polymers.
represents the fractional derivative of order p with respect to time.‡ For the “spatial” derivative along the chain (n-coordinate), the order is q = 2 for Rouse polymers.
Eqn (4) indicates that the tension propagates along the chain as n(t) ∼ (t/τ0)p/2. Then, the effective friction grows as γ*(t) ∼ γ1*(t)n(t) ∼ t1−(p/2), where we have taken into account the time dependence of the effective monomeric friction γ1*(t) due to medium viscoelasticity. This leads to the MSD 〈Δr(t)2〉 ≃ D*(t)t with D*(t) = kBT/γ*(t), hence
Perhaps more illuminating is to express the tagged monomer MSD as
| | 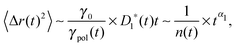 | (6) |
where
γpol/
γ0 represents the friction enhancement due to the polymeric effect, which coincides with the number
n(
t) of associated monomers in the Rouse model. This form explicitly indicates the contribution of memory from the tension dynamics to otherwise sub-diffusing simple probes with the exponent
α1.
In the above, we have followed the line of argument (I). One can easily re-derive the same result from the argument (II), by noting the terminal time scaling τN ≃ τ0N2/p from the tension propagation dynamics.
In viscoelastic active media
Here again, one can follow the same step: first identify the behaviors of simple probes, then, superimpose the effect of tension propagation. One should note, however, that the steady-state conformation of the polymers may no longer be a simple random walk. Indeed, the additive nature of the active and thermal noises and their mutual statistical independence implywhere Rn is the stationary spatial size of polymers with the number n of constituting monomers, and b is related to the length scale (effective segment size) introduced by the active dynamics. Below we focus on the situation, in which the active noise dominates over the thermal noise Rn ∼ nνA, and determine how the exponent νA is related to the dynamics.
Assuming that the medium viscoelasticity is still described by power-law kernel with the exponent p,§ the MSD of simple probes is calculated as 〈Δr1(t)2〉 ≃ D1*(t)t, where the effective diffusion coefficient depends on the timescale
| | 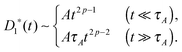 | (8) |
The MSD exponent is thus
α1,A = 2
p at
t ≪
τA and
α1,A = 2
p − 1 at
t ≫
τA. Such behaviors arise from the combined effects of medium caused time-dependent friction
γ1*(
t) ∼
t1−p and the persistent (random) action of the active noise in the short (long) timescale, and have been observed in the dynamics of engulfed microspheres in a living eukaryotic cell.
25 Notice that unlike passive systems driven by the thermal noise, the active fluctuation is free from the Einstein relation.
We now turn our attention to the dynamics of tagged monomers in the timescale t ≫ τA. The dynamics of tension propagation is governed by eqn (4), thus, n(t) ≃ (t/τ0)p/2. Applying the argument (I), this leads to the MSD of tagged monomers 〈Δr(t)2〉 ∼ (1/n(t)) × D1*(t)t ∼ (1/n(t)) × tα1,A, thus
| | | αA = α1,A − (p/2) = (3p/2) − 1 (t ≫ τA). | (9) |
The stationary size of a polymer can be found from the condition imposed from the argument (II); 〈Δ
r(
t)
2〉 ∼
tαA ∼
n(
t)
2νA, hence
Combining it with
eqn (9), we find
| | | νA = (3/2) − p−1 (t ≫ τA), | (11) |
in agreement with rigorous calculations in
ref. 14.
In shorter timescale t ≪ τA, the dynamics of simple probes is different (eqn (8)), which implies α, thus, νA as well may be different from those at t ≫ τA. The simple probe behavior (eqn (8) upper line) combined with tension propagation dynamics suggests
again in agreement with
ref. 14. There is, however, a certain subtlety in the short scale properties of the model. To see it, let us define the characteristic number of monomers
nA ≃ (
τA/
τ0)
p/2. For a smaller section of chain with
n <
nA monomers, the action of the active noise may be too strong to over-swell it. Indeed, applying the argument (II) in the timescale
t <
τA, one again expects the scaling relation of
eqn (10), which combined with
eqn (12) leads to
νA = 3/2 > 1.
¶ A related issue is associated with the bond stretching. In Rouse and many other flexible polymer models, the connectivity is ensured by the entropic spring constant
k ≃
kBT/
a2, and its ratio to the monomeric friction coefficient determines the microscopic timescale
τ0 =
γ0/
k. In active media, the strength of active noise affects the entropic nature of the spring. In particular, in the timescale
t ≫
τA, the active noise enters the “equi-partition” relation
kAaA2/
kBT = 3[1 + (
TA/
T)], where we introduce the effective temperature
TA =
AτA/(
kBγ0),
14 and thus, the spring constant
kA and/or the bond length
aA, and the monomeric timescale
τ0A as well may be altered. Two extreme cases are the followings: (i) the fixed spring constant
kA =
k implies the bond stretching
aA ≃
a(
TA/
T)
1/2 and
τ0A =
τ0; (ii) fixed bond length
aA =
a, which results in the stiffening
kA ≃
kTA/
T and the reduction in the microscopic timescale
τ0A ≃
τ0T/
TA ≃ (
γ0a)
2/(
AτA).
III Generalization
There are situations where the monomers' self-avoidance and/or the solvent mediated hydrodynamic interactions become apparent. One may be also interested in behaviours of objects with a fractal connectivity, i.e., polymeric fractals, for which we need to distinguish the total number N of monomers from the number M of monomers in a given direction along the chemical structure. To deal with these aspects, we introduce several exponents ν, z and ds. The first relates the equilibrium (stationary) spatial size Rn of the polymer with the number n of constituting monomers Rn ≃ anνeq; in active media, this relation is replaced by Rn ≃ anνeq + bnνA, see eqn (7). Hereafter, we write Rn ∼ nν with ν = νeq or νA in thermal or active media (TA ≫ T), respectively; if necessary, we shall adopt the same notation for other exponents as well. The second exponent specifies the ratio of the friction coefficient of the polymer to that of monomers as γpol/γ0 = (Rn/a)z−2, which is thus related to the dissipation mechanism. For free-draining polymers z = 2 + ν−1, the ratio is equal to the number of monomers γpol/γ0 = n irrespective of the conformation (often called Rouse dynamics). In dilute solutions, however, the hydrodynamic interactions between monomers are usually relevant to alter the exponent to z = 3; then the ratio is proportional to the spatial size of the polymer γpol/γ0 = (Rn/a), reminiscent to the Stokes formula (often called Zimm dynamics).26,28 The last one is used for the relation N ≃ Mds, where the spectral dimension ds characterizes the topology, or the chemical formula of the object (describing linear sequences and branch points), but is independent of the spatial conformation of the object.27
Crucial to our discussion is the dynamics of tension propagation described by eqn (4). Now the self-avoidance and/or hydrodynamic interactions introduce long-range spatial memories, making the order q non-integer in general cases. Since the tension propagates along the path of connectivity as m(t) ≃ (t/τ0)p/q, the number of monomers which contributes to the effective friction grows as n(t) ≃ (t/τ0)pds/q. The terminal time is thus τ ≃ τ0Mq/p = τ0Nq/(dsp). We now apply the argument (I) to calculate the MSD of tagged monomers 〈Δr(t)2〉 ≃ (a/Rn(t))z−2D1*(t)t, where the time-dependent effective diffusion coefficient D1*(t) for simple probes is already given (before eqn (4) for thermal media, and eqn (8) for active media, respectively). With Rn(t) ∼ n(t)ν ∼ m(t)dsν ∼ tpdsν/q, this leads to the MSD exponent
| | | α = α1 − pdsν(z − 2)q−1 | (13) |
On the other hand, the argument (II) suggests the relation 〈Δ
r(
t)
2 〉 ∼
tα ∼
n(
t)
2ν, from which we obtain the generalized version of
eqn (10)|| Consistency between two arguments requires
α1 =
pdsνz/
q, thus
| | 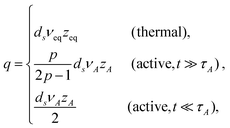 | (15) |
and
| | 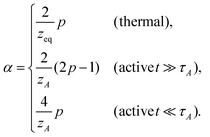 | (16) |
These results deserve some appreciation.
In thermal media
The expression of qeq is seen to be the generalization of the form suggested from the Gaussian approximation, where the equation of motion is linearized by means of pre-averaging with the equilibrium monomer distribution function.18,28 Since νeq is known as an equilibrium property, once the dissipation mechanism (zeq) is specified, the MSD exponent can be determined.
In active media (t ≫ τA)
In this long timescale, the active noise acts as non-correlated random kick reminiscent of the white noise. This suggests νA → νeq in the case of viscous monomer dynamics p → 1, since in this limit, the noise and the friction terms satisfy the proportionality relation, analogous to the FDT, albeit with the effective temperature TA. Inserting p = 1 in eqn (15: middle), we thus find qA = dsνeqzeq(=qeq), which allows us to obtain| | 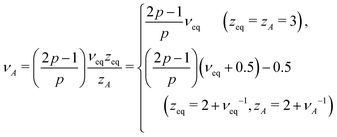 | (17) |
and, from eqn (16: middle),| | 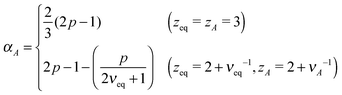 | (18) |
For Rouse polymers, their defining properties q = 2 and z = 2 + ν−1 make it possible to rewrite the middles of eqn (15) and (16) as νA = (2p − 1)/(pds) − 0.5 and αA = 2p − 1 − (pds)/2, which reduce to eqn (11) and (9), respectively, already obtained for linear polymers ds = 1.
In active media (t ≪ τA)
In this scale, the spectrum of active noise does not match that of friction kernel for any exponent p, so a strong out-of-equilibrium feature is expected. To get some insight into the short scale behaviors, let us again find the exponents for Rouse polymers; with q = 2 and z = 2 + ν−1, the bottoms of eqn (15) and (16) lead to νA = (2/ds) − 0.5 and αA = (4 − ds)p/2, respectively, which reduce to the results already obtained for linear polymers ds = 1. The pathological over-swelling behaviour νA > 1 for Rouse polymers with ds = 1 may be avoided by the non-local spatial interactions. From eqn (15: bottom), we have νA = (qA/ds) − 0.5 for free draining polymers. Thus, for linear polymers ds = 1, the upper limit νA = 1, i.e., elongation, can be realized by qA = 3/2. This modifies the MSD exponent at a short time from eqn (12) to αA = 4p/3. Another way to look at the effect is the following. Polymers synthesized from the mixture of bi- and tri-functional units develop tree-like structures with random branching, which may be characterized by the fractional spectral dimension ds ≃ 4/3. The fractal dimension of such objects without self-avoidance is known to be 4 in thermal equilibrium.24 This is consistent with νeq = 1/4 derived from eqn (15: top) with Rouse polymer characteristics qeq = 2 and zeq = 2 + νeq−1. Such over-collapsing behavior (νeq−1 > space dimension) indicates the significant effect of the self-avoidance. In the active media, however, the active noise inhibits the shrinkage, and leads to the stretching in the short length scale. For the branched polymers with a quenched connectivity, the upper limit of elongation is νA = 1/ds. This again implies the necessity of the non-local correlation with qA = (2 + ds)/2 ≃ 5/3. Numerical simulations of model polymers with bond length restriction, e.g. finitely-extensible bonds, may be interesting to resolve the issue. Notice that the analogue of the length scale nA introduced below eqn (12) has been observed in a recent simulation of a semiflexible filament in the active media, where the undulation modes from bending energy control the tension dynamics.29
IV Discussion
To what extent is the model analyzed relevant to real chromosomal dynamics? While we are confident in revealing fundamental polymeric aspects of active dynamics, we feel that more elaboration is needed to answer the above question. The following remarks and questions may be pertinent to the further development of the model.
Medium viscoelasticity
The present model assumes the viscoelasticity of the medium in a phenomenological way through the non-trivial memory kernel exponent p ≠ 1 for the monomer motion. This has been motivated in ref. 13 from the observation that the fluorescently labeled RNA molecules (with size ∼100 nm supposed to be simple probes in the present terminology) in live E.coli cells exhibit sub-diffusion with α1 ≃ 0.7.30 By setting p = α1, the authors have claimed that eqn (1) describes the observed sub-diffusion of chromosomal loci with α ≃ 0.4 reasonably well; cf.eqn (5). So the question to be addressed is the molecular origin of the memory kernel.
Such power-law memory kernels are ubiquitously found in complex fluids, which characterize their mechanical properties in the scale larger than the mesh size.31 For instance, in a standard microrheology set-up, one monitors the motion of simple probes in dense polymer solution. Here, the motion of the probe, whose size is larger than the mesh size of the polymer solution becomes sub-diffusive due to the viscoelastic response of the polymers.10 The medium where chromosomes are packed in cells is highly crowded, so certain viscoelastic properties would be conceivable. Microrheological measurements have reported a sort of hopping dynamics of probe particles with size 100 nm.32 The motion of larger probes with size 1 μm is caged over long time, but the external force larger than ∼102 pN enables the probes to hop between cages.33 These studies provide an estimate for the intra-nuclear mesh size 200–300 nm, which would be larger than the effective monomer size of a chromosomal polymer. This might suggest that the chromosomal monomers feel the local (interstitial) viscosity, and if so, the limit p → 1 in eqn (1) and (4), hence the usual viscous dynamics would be appropriate for the dynamics in a monomeric scale, which results in αeq = αA,t≫τA = 2νeq/(2νeq + 1). The possible alternative mechanism for the non-Rouse exponent α ≠ 1/2 could be related to the topological constraints, which may either restrict the motion of monomers on fractal support (random walk trajectory) or may induce a non-trivial fractal conformation;34 see the following remarks.
Topological constraints
The motion of tagged monomers in entangled polymer solutions is a simple example of (anomalous) diffusion on fractal. There is an intermediate time region with the MSD exponent of tagged monomers α ≃ 1/4 as a consequence of the Rouse dynamics eqn (3) inside the entanglement tube which makes random walk spatial configuration.24,28 This has indeed been suggested as a possible mechanism to explain the sub-diffusion of telomeres with α ≃ 0.3.4 The apparently same result is obtained with p = 1/2 in thermal media (eqn (5)), but the underlying physics leading to the result is different.
Note that the entanglement in the linear polymers invoked above is a matter of timescale. Growing evidence indicates that the conformation of long chromosomes in vivo is distinct from the entangled Gaussian conformation characteristic of linear polymers in their equilibrium melt;34 see ref. 35 for the estimates for the reptation time of chromosomal chains in various species in comparison to the characteristic time of cellular processes. The loci dynamics in the non-trivial chromosome conformation would be different from that in the conventional reptation picture in the entangled melt.** A numerical simulation on the tagged monomer diffusion in crumple globule, i.e., a knot-free self-similar compact structure with ν = 1/3,36 has reported the exponent α ≃ 0.38.37 In our frame in Section 3, this corresponds to the passive dynamics with ds = 1 and zeq = 2 + νeq−1 = 5, thus, eqn (16: top) yields αeq = 0.4. In addition, qeq = 5/3(<2) from eqn (15: top) effects the long-range spatial correlation along the chain, which effectively ensures the compact conformation compared to the ideal conformation with q = 2. A similar scaling argument for α exponent can be found in ref. 18 and 37 as well. Finally, due to the reason of topological constraints mentioned above, the melt of unlinked and unknotted rings has attracted considerable interest as a model system of the interphase chromosome,38 for which a recent theory predicts the exponent α = 2/7.39
Active noise
Inside cells and nuclei, there are various ATP-driven active processes taking place along chromosomes.40 Examples include nucleosome repositioning by chromatin remodeling enzymes, transcription by RNA polymerases and local condensation/decondensation controlled by various histone modifications. Recent experiments have evidenced that the effect of metabolic activity is at work on the chromosomal dynamics in living cells.2,3,5,7,33 For instance, ref. 5 reported that ATP depletion leads to the reduction in the apparent diffusion coefficient of loci, while the sub-diffusive exponent α ≃ 0.4 remains unchanged.†† There are also reports on the ATP-dependent jump motion3 and the super-diffusion of loci.41
In the present paper, the effect of metabolic activity is incorporated via the active noise with the property (2). This is one of the simplest ways, and indeed seems to explain the observed active dynamics of simple probes in cells,25 but there may be other possibilities to model the active effect on the chromosome dynamics42,43,44 such as the convective flow induced by the conformational change of molecular machines in the medium or the chromatin itself. We stress, however, that the present argument is rather generic; representative formula (6) is a consequence of the collective dynamics of the polymer determined by its mode spectra,18 and does not depend on the details of the noise. Hence, the idea should be applicable to the semiflexible filaments, too.29,45 It would be also interesting to look at the dynamics of the polymer driven by self-propelled particles from the present viewpoint.‡‡46,47 Furthermore, aside from the situation, where the active noise dominates over the thermal noise TA ≫ T, a further study should clarify how the competition between these two noises leads to various crossover dynamical behaviors. We hope that its simplicity helps us understand the complex dynamics of chromosomal loci, not only the anomalous fluctuation driven by noise but also the directional motion driven by external bias,18e.g. chromosome segregation, and other related macromolecular dynamics including, for instance, the translocation across a nanopore19–23 and the rotational dynamics48 in- and out-of-equilibrium situations.
Acknowledgements
The present analysis was motivated by the discussion with C. Vanderzande, H. Vandebroek and E. Carlon, which was done when one of us (T. Sakaue) stayed at KU Leuven. We thank T. Sugawara, S. Shinkai and A. Mikhailov for discussion on chromosome and active dynamics in living cells. This work is supported by KAKENHI (No. 16H00804, “Fluctuation and Structure”) from MEXT, Japan.
References
- S. M. Gasser, Science, 2002, 296, 1412–1416 CrossRef CAS PubMed.
- A. Zidovska, D. Weitz and T. Mitchison, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15555–15560 CrossRef CAS PubMed.
- V. Levi, Q. Ruan, M. Plutz, A. S. Belmont and E. Gratton, Biophys. J., 2005, 89, 4275–4285 CrossRef CAS PubMed.
- I. Bronstein, Y. Israel, E. Kepten, S. Mai, Y. Shav-Tal, E. Barkai and Y. Garini, Phys. Rev. Lett., 2009, 103, 018102 CrossRef CAS PubMed.
- S. C. Weber, A. J. Spakowitz and J. A. Theriot, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7338–7343 CrossRef CAS PubMed.
- E. Barkai, Y. Garini and R. Metzler, Phys. Today, 2012, 65(8), 29–35 CrossRef CAS.
- F. MacKintosh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7138–7139 CrossRef CAS PubMed.
- F. Amblard, A. C. Maggs, B. Yurke, A. N. Pargellis and S. Leibler, Phys. Rev. Lett., 1996, 77, 4470–4473 CrossRef CAS PubMed.
- I. Y. Wong, M. L. Gardel, D. R. Reichman, E. R. Weeks, M. T. Valentine, A. R. Bausch and D. A. Weitz, Phys. Rev. Lett., 2004, 92, 178101 CrossRef CAS PubMed.
- L.-H. Cai, S. Panyukov and M. Rubinstein, Macromolecules, 2011, 44, 7853–7863 CrossRef CAS PubMed.
- C. Xue, X. Zheng, K. Chen, Y. Tian and G. Hu, J. Phys. Chem. Lett., 2016, 7, 514–519 CrossRef CAS PubMed.
- A. Godec, M. Bauer and R. Metzler, New J. Phys., 2014, 16, 092002 CrossRef.
- S. C. Weber, A. J. Spakowitz and J. A. Theriot, Phys. Rev. Lett., 2010, 104, 238102 CrossRef PubMed.
- H. Vandebroek and C. Vanderzande, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 060601 CrossRef PubMed.
- T. Sakaue, T. Saito and H. Wada, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 011804 CrossRef PubMed.
- P. Rowghanian and A. Y. Grosberg, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 011803 CrossRef PubMed.
- R. Rrederickx, T. in't Veld and E. Carlon, Phys. Rev. Lett., 2014, 112, 198102 CrossRef PubMed.
- T. Saito and T. Sakaue, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 012601 CrossRef PubMed.
- T. Sakaue, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 021803 CrossRef PubMed.
- T. Sakaue, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 041808 CrossRef PubMed.
- T. Ikonen, A. Bhattacharya, T. Ala-Nissila and W. Sung, J. Chem. Phys., 2012, 137, 085101 CrossRef CAS PubMed.
- T. Saito and T. Sakaue, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 135 CrossRef CAS PubMed; T. Saito and T. Sakaue, Eur. Phys. J. E: Soft Matter Biol. Phys., 2012, 35, 125 CrossRef.
- V. Palyulin, T. Ala-Nissila and R. Metzler, Soft Matter, 2014, 10, 9016–9037 RSC.
-
M. Rubinstein and R. Colby, Polymer Physics, Oxford University Press, Oxford, 2003 Search PubMed.
- A. Caspi, R. Granek and M. Elbaum, Phys. Rev. Lett., 2000, 85, 5655–5658 CrossRef CAS PubMed.
-
P.-G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, Ithaca, 1979 Search PubMed.
-
D. Stauffer and A. Aharony, Introduction to Percolation Theory, Taylor and Francis, London, 1994 Search PubMed.
-
M. Doi and S. Edwards, The Theory of Polymer Dynamics, Clarendon Press, Oxford, 1986 Search PubMed.
- A. Ghosh and N. Gov, Biophys. J., 2014, 107, 1065–1073 CrossRef CAS PubMed.
- I. Golding and E. Cox, Phys. Rev. Lett., 2006, 96, 098102 CrossRef PubMed.
- T. Mason and D. Weitz, Phys. Rev. Lett., 1995, 74, 1250–1253 CrossRef CAS PubMed.
- Y. Tseng, J. Lee, T. Kole, I. Jiang and D. Wirtz, J. Cell Sci., 2004, 117, 2159–2167 CrossRef CAS PubMed.
- F. Hameed, M. Rao and G. Shivashankar, PLoS One, 2012, 7, e45843 CAS.
- A. Bancaud, C. Lavelle, S. Huet and J. Ellenberg, Nucleic Acids Res., 2012, 40, 8783–8792 CrossRef CAS PubMed.
- A. Rosa and R. Everaers, PLoS Comput. Biol., 2008, 4, e1000153 Search PubMed.
- A. Grosberg, S. Nechaev and E. Shakhnovich, J. Phys., 1988, 49, 2095–2100 CAS.
- M. Tamm, L. Nazarov, A. Gavrilov and A. Chertovich, Phys. Rev. Lett., 2015, 114, 178102 CrossRef CAS PubMed.
- J. Halverson, J. Smrek, K. Kremer and A. Grosberg, Rep. Prog. Phys., 2014, 77, 022601 CrossRef PubMed.
- T. Ge, S. Panyukov and M. Rubinstein, Macromolecules, 2016, 49, 708–722 CrossRef CAS PubMed.
-
B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Gerland Science, New York, 2002 Search PubMed.
- A. Javer, N. Kuwada, Z. Long, V. Benza, K. Dorfman, P. Wiggin, P. Cicuta and M. Lagomarsino, Nat. Commun., 2014, 5, 4854 CrossRef PubMed.
- R. Bruinsma, A. Grosberg, Y. Rabin and Y. Zidovska, Biophys. J., 2014, 106, 1871–1881 CrossRef CAS PubMed.
- A. S. Mikhailov and R. Kapral, Proc. Natl. Acad. Sci. U. S. A., 2015, 29, E3639–E3644 CrossRef PubMed.
- J. M. Yeomans, D. O. Pushkin and H. Shum, Eur. Phys. J. Spec. Top., 2014, 223, 1771–1785 CrossRef.
- C. Brangwynne, G. Koenderink, F. MacKintosh and D. Weitz, Phys. Rev. Lett., 2008, 100, 118104 CrossRef PubMed.
- A. Kaiser and H. Löwen, J. Chem. Phys., 2014, 141, 044903 CrossRef CAS PubMed.
- J. Shin, A. Cherstvy, W. Kim and R. Metzler, New J. Phys., 2015, 17, 113008 CrossRef.
- M. Laleman, M. Baiesi, B. P. Belotserkovskii, T. Sakaue, J.-C. Walter and E. Carlon, Macromolecules, 2016, 49, 405–414 CrossRef CAS.
- R. Metzler, J. Jeon, A. Cherstvy and E. Barkai, Phys. Chem. Chem. Phys., 2014, 16, 24128–24164 RSC.
- D. Panja, J. Stat. Mech.: Theory Exp., 2010, P06011 Search PubMed.
- H. Hajjoul, J. Mathon, H. Ranchon, I. Goiffon, J. Mozziconacci, B. Albert, P. Carrivain, J. Victor, O. Gadal, K. Bystricky and A. Bancaud, Genome Res., 2013, 23, 1829–1838 CrossRef CAS PubMed.
Footnotes |
| † Among various classes of stochastic processes for anomalous diffusion,49 the tagged monomer dynamics in free space discussed here corresponds to the fractional Brownian motion. See ref. 18 and 50 for microscopic derivation of the generalized Langevin equation with power-law memory kernel. |
| ‡ Eqn (4) with q = 2 is the deterministic part of eqn (1). One can easily check that the left-hand sides of these equations are equivalent in a scaling sense. A concise explanation can be found in the ESI of ref. 14. |
| § Note that the active noise may disturb the media so that the exponent p may be altered from the thermal system. |
| ¶ This analysis suggests, unlike the large scale behavior, the short scale conformation has no p dependence. The calculation of Rouse modes in viscous solution under the condition n ≪ nA ⇔ t ≪ τA shows 〈Rn2〉 ≃ (kBT/kA)n + AkA−2n3, where kA is the spring constant of the polymer in active media (see the main text). |
| || Notice the correspondence to the problem of random walk on fractal objects, in which the MSD of random walker is 〈Δr(t)2〉 ∼ tds/df.27 The random walk (thus, p = 1 and q = 2) along the path and the walker's displacement in real space correspond to the tension dynamics (described by eqn (4)) and the amplitude of the monomer's positional fluctuation, respectively, in our problem. |
| ** There is a recent report that the MSD of loci in budding yeast follows a scaling consistent with the Rouse model, i.e., α = 1/2.51 This is presumably because the entanglement effect would be very weak in budding yeast as estimated from the length and density of yeast chromosomes in nucleus38. |
| †† Within the viscoelastic Rouse model (p ≠ 1, q = 2), the lack of variation of α on ATP was interpreted as a sign that the ATP-driven active noise has a frequency dependence similar to that of thermal noise in viscoelastic media.5,7 Our analysis indicates an alternative possible scenario p = 1, q ≠ 2 with the active noise which acts as random kick at t ≫ τA; compare eqn (16: top) and (18). |
| ‡‡ Indeed, the large scale exponent νA = νeq, cf.eqn (17) with p = 1, and the non-trivial expansion and stiffening observed in the simulation of short polymers46 are in line with our predictions. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
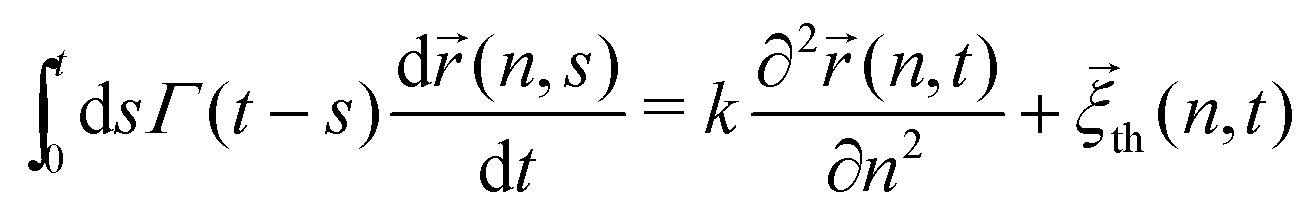
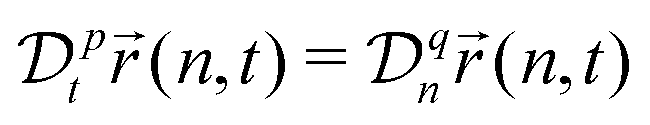
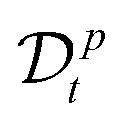 represents the fractional derivative of order p with respect to time.‡ For the “spatial” derivative along the chain (n-coordinate), the order is q = 2 for Rouse polymers.
represents the fractional derivative of order p with respect to time.‡ For the “spatial” derivative along the chain (n-coordinate), the order is q = 2 for Rouse polymers.