
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoscale ordering in binary aqueous solvents induced by ion size asymmetry†
Monika
Witala
a,
Sebastian
Lages
b and
Kim
Nygård
*a
aDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-41296 Gothenburg, Sweden. E-mail: kim.nygard@chem.gu.se
bMAX IV Laboratory, Lund University, PO Box 118, SE-22100 Lund, Sweden
First published on 8th April 2016
Abstract
Surprising weak assembly behavior has lately been found in binary aqueous solvents containing antagonistic salt. The underlying mechanism is still under debate, particularly the role of ion size asymmetry. Here we use small-angle X-ray scattering to study the effect of ion size asymmetry on the mesoscale ordering in a binary solvent composed of water and 2,6-dimethylpyridine with added symmetrical quaternary ammonium salt. By systematically elongating the hydrocarbon side-chain lengths, and hence developing cation-to-anion size asymmetry, we provide the first experimental evidence of a gradual build-up of the solvent's mesoscale ordering. These results are in qualitative agreement with model-independent theoretical predictions.
1 Introduction
Antagonistic salts, which consist of hydrophilic cations and hydrophobic anions, have recently been found to induce mesoscale assembly in binary aqueous solvents.1,2 Already with a few mM of added salt mesoscale structures were observed, and increasing the salt concentration caused the appearance of an ordered lamella-like phase.1–3 These observations were initially assigned to the hydrophilic and hydrophobic nature of cations and anions, respectively, with the antagonistic salt behaving essentially as a surfactant.3 In contrast, no solvent ordering is observed upon adding simple inorganic salts. For a recent review on the topic, see ref. 4.The mechanism of the aforementioned assembled structures is still under debate. Some theoretical models emphasize the interpretation in terms of cation and anion preferential solubility, with the solvent ordering being caused by the unequal partitioning of hydrophilic and hydrophobic ions in water- and organic-rich phases.5,6 On the other hand, theoretical studies of antagonistic salts exhibiting no ion size asymmetry do not find mesoscale solvent ordering.7 Indeed, an alternative model-independent prediction indicates that such solvent structures can only be generated if the added salt exhibits significant asymmetry of ion size.8 Note that all previous experiments were conducted on a system containing antagonistic salt, which exhibits ion size asymmetry.
In this paper, we focus on the effect of ion size asymmetry on mesoscale ordering in binary aqueous solvents. For this purpose we have carried out a small-angle X-ray scattering (SAXS) experiment in a mixture of water and 2,6-dimethylpyridine (denoted as 2,6-DMP) by adding 10 mM of quaternary ammonium bromide. The SAXS data were collected for a salt series ranging from the symmetrical short-chained tetraethylammonium bromide to the longer-chained tetraheptylammonium bromide. Most importantly, our experimental results are the first to show that a systematic increase of ion size asymmetry leads to a gradual build-up of mesoscale order in binary aqueous solvents.
The reason for choosing a series containing symmetrical tetra-n-alkylammonium salts is dictated by their appealing properties. First, the cations become larger as the number of carbon atoms increases; since they are built in a symmetrical manner (four hydrocarbon chains), adding more carbons gives a bulky ion. Second, it is well known that their properties are linked with the structure and number of hydrocarbons.9 Therefore, we expected the data to depend on the length of alkyls. The choice of salts with the same anion results in a neat experiment where the effect of cation size can be studied systematically.
2 Materials
The studied sample was a binary solvent composed of water (Milli-Q) and 2,6-DMP (Sigma-Aldrich, purity ≥99%). We prepared six sample sets by adding 10 mM of quaternary ammonium salt – in a series from short-chained tetraethylammonium bromide to longer-chained tetraheptylammonium bromide (Sigma-Aldrich, purity ≥99%). Throughout this work we denote our samples containing different ammonium compounds as C2⋯C7 with respect to the carbon chain length in the alkyl group. Moreover, we collected data from our binary solvent containing the antagonistic sodium tetraphenylborate (Sigma-Aldrich, purity ≥99.5%) as a reference. All chemicals were used as received, without further purification.3 Results and discussion
The phase diagram of the neat mixture of water and 2,6-DMP is well known,10 exhibiting an immiscibility loop with a lower critical temperature TC ≈ 307 K at a volume fraction ϕ ≈ 0.3 of 2,6-DMP. Furthermore, the addition of ions usually alters the phase diagram. Hydrophilic inorganic salts enlarge the two-phase region and shift the critical point,11,12 in contrast to antagonistic salts which lead to the shrinking and eventual disappearance of the two-phase region as a function of the added salt concentration.13,14 The latter behavior is in line with the addition of ionic surfactants such as sodium dodecyl sulfate (SDS) and dioctyl sodium sulfosuccinate (often denoted as AOT).1,3Let us start by considering the effect of introducing quaternary ammonium salts into the water–2,6-DMP solvent. In Fig. 1 we present the experimental phase diagram as obtained following ref. 15, using a temperature step of 0.5 K and an equilibration time of 20 minutes. Strikingly, there is no effect of 10 mM tetraalkylammonium bromide on the lower critical point location of the water–2,6-DMP mixture, in stark contrast to the salts mentioned above. This finding suggests that the quaternary ammonium salts introduced here may be proven to be a highly useful model system to study salt-induced mesoscale ordering in binary aqueous solvents. In particular, the absence of a shift of the lower critical point facilitates our systematic study on the effect of ion-size asymmetry in salt-induced mesoscale solvent ordering. In the rest of this study, we will therefore focus on the critical composition ϕ = 0.3 and 10 mM added salt.
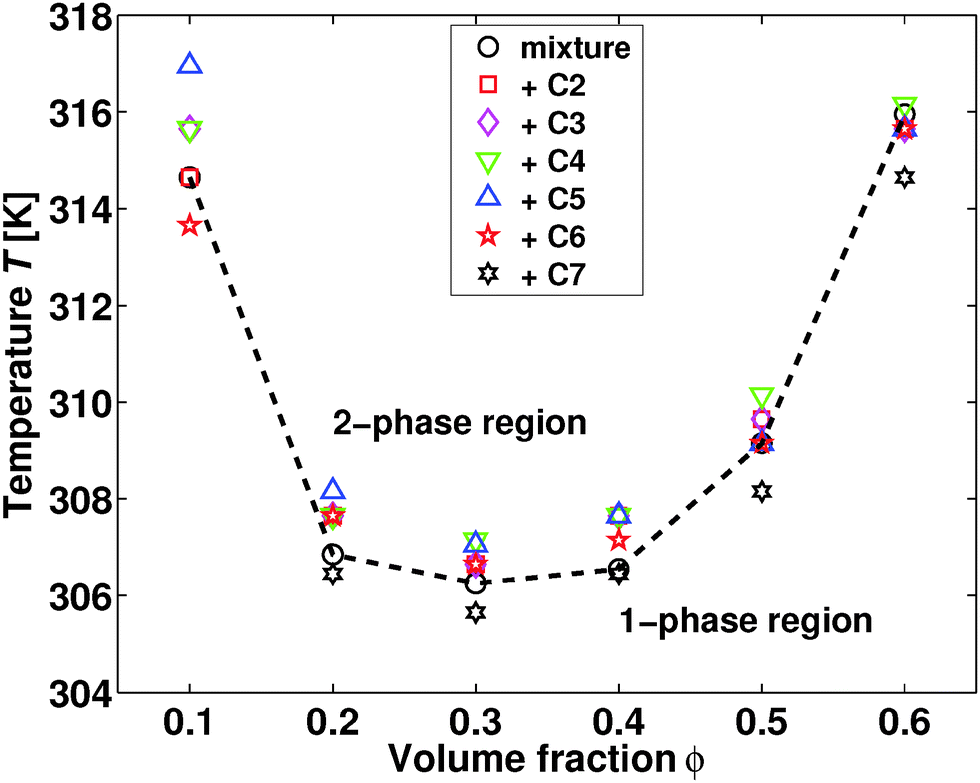 | ||
| Fig. 1 Coexistence points for the mixture of water and 2,6-dimethylpyridine. The data are presented as a function of 2,6-DMP volume fraction ϕ. The black circles represent data for the neat mixture, while the other symbols depict data obtained upon addition of 10 mM quaternary ammonium salts with increasing hydrocarbon chain length. The uncertainty in T is 0.5 K. | ||
The sample with 2,6-DMP content of ϕ = 0.1 and 10 mM of added tetraheptylammonium bromide (C7) was turbid already at room temperature, directly after mixing all components. This is in contrast to mixtures with ϕ ≥ 0.20, in which C7 dissolves completely. In order to verify this observation, the preparation was repeated with the same result. The limited solubility of C7 in the solvent provides a possible explanation for the turbidity of the sample. Adding 10 mM C7 to the mixture with a low fraction of 2,6-DMP could cause spontaneous emulsification, in line with the previous observations of oil-in-water emulsions containing symmetrical alkylammonium ions.16
Having verified that 10 mM of quaternary ammonium salts do not alter the phase diagram (except C7 at small ϕ), we next turn to the main objective of our study – solvent ordering induced by ion size asymmetry. For this purpose we have carried out a SAXS experiment at beamline I911-4 of the MAX-lab (Lund, Sweden), using an X-ray wavelength of λ = 0.91 Å. We kept the samples in thin-walled, 1.5 mm-diameter glass capillaries, and we recorded the scattered X-rays 1.9 m behind the sample using the two-dimensional pixel detector PILATUS 1 M. The X-ray beam focused onto the detector had a size of 0.22 × 0.3 mm at the sample position. We collected data at several temperatures below TC in the range of ΔT = TC − T ≈ 4⋯9 K and we used an exposure time of 300 s for each sample. For more specific beamline details, we refer the reader to ref. 17.
In order to exemplify the SAXS experiment, we compare in Fig. 2 data obtained upon adding either 10 mM hydrophilic tetraethylammmonium bromide18 or tetraheptylammmonium bromide. The data are presented for a temperature of 303 K in order to emphasize the characteristic feature of solvent ordering, and we have subtracted a temperature-independent background consisting of a linear combination of recorded water and 2,6-DMP data with a 2,6-DMP volume fraction ϕ = 0.3 (see the ESI† for details). The neat solvent can be described by the classical Ornstein–Zernike (OZ) model, I(q) = I(0)/(1 + q2ξ2), where I(q) is the scattered intensity as a function of scattering vector modulus q and ξ the bulk correlation length describing the concentration fluctuations.19 The latter is strongly temperature dependent and close to the critical point it follows the three-dimensional Ising universality class: ξ = ξ0(ΔT/TC)−ν, with the critical exponent ν ≈ 0.63, ΔT = TC − T, and ξ0 a sample-dependent constant. For the system composed of water and 2,6-DMP the constant has previously been found to be ξ0 = 0.20 ± 0.02 nm,10,20 and for the salt-containing mixtures studied here we observe ξ ≈ 1.7 nm at 298 K, in reasonable agreement with the neat case considering our accuracy in Fig. 1. First, we discuss the case of adding C2. A small amount of C2 does not induce ordering of the solvent, as is clear from the linear behavior in the OZ plot of Fig. 2(b). In this respect C2 behaves like simple inorganic salts.20 Second, if we change the salt to C7, it shows up as a concentration-dependent shoulder in the SAXS intensity of panel (a) and as a non-linearity in panel (c). This behavior is similar to that when adding the antagonistic sodium tetraphenylborate NaBPh4,3 and we thus expect this shoulder to be further enhanced when increasing the temperature towards the critical point.3,13 We emphasize that the C7 data of Fig. 2 can no longer be described by the OZ model.
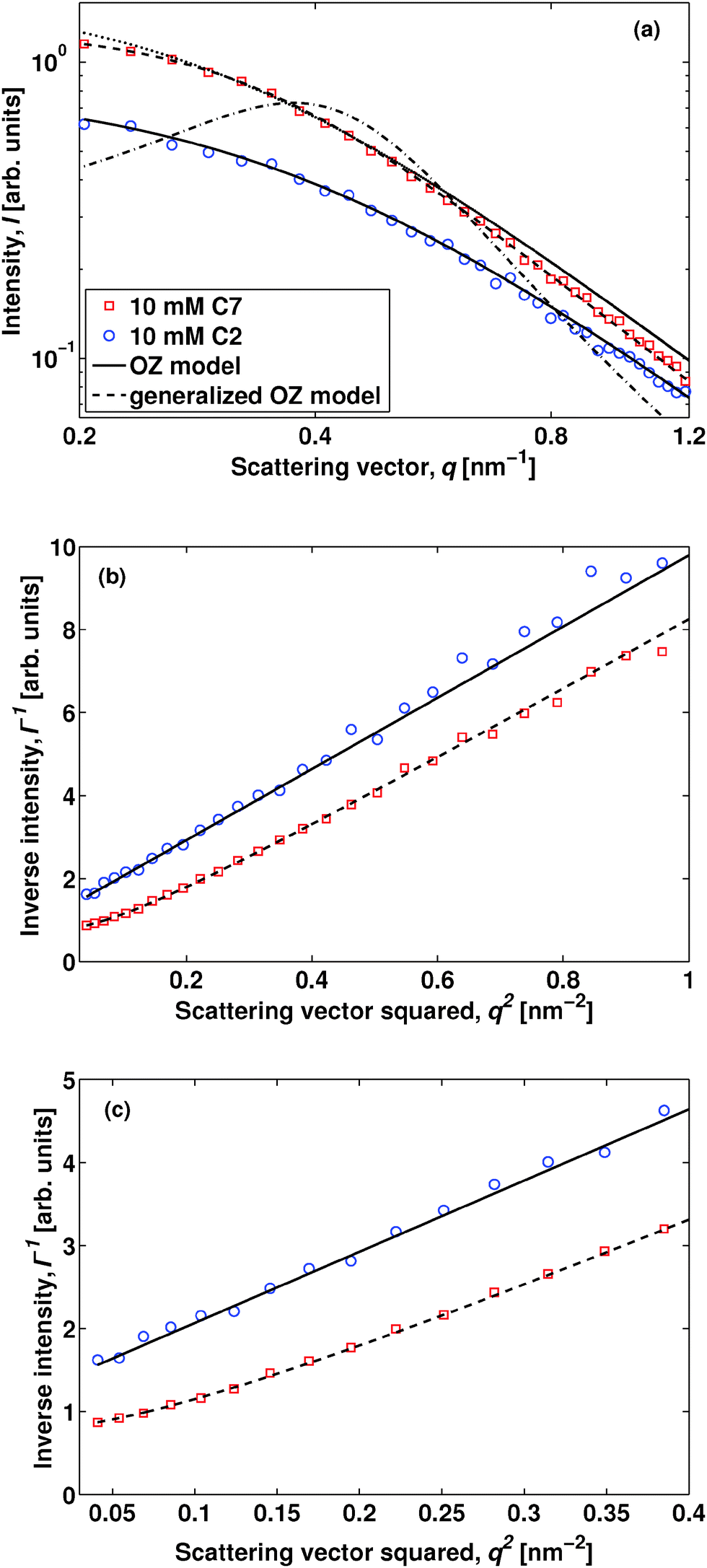 | ||
| Fig. 2 (a) Log–log plot of SAXS intensity as a function of scattering vector modulus, obtained from the critical mixture of water and 2,6-DMP with 10 mM added tetraethylammonium bromide (C2, blue circles) and tetraheptylammonium bromide (C7, red squares). The data were collected at a temperature of 303 K. The C2 data can be fitted using the classical OZ model (solid line). The C7 data have been fitted using a generalized OZ model (dashed line), and the dotted and dashed-dotted lines describe fits using a factor two smaller and larger interaction contrast |g|, respectively (see the text for details). (b) The same data set plotted as the inverse SAXS intensity vs. squared scattering vector. The C2 data follow the expected linear behavior of the OZ model, while the C7 data exhibit deviations from this trend. (c) As panel (b), except that the data are shown in a smaller q2 range, highlighting the non-linearity in the C7 data. | ||
Let us focus on the case of C7. In order to describe the experimental data properly and to capture the mesoscale structure, one needs to employ a generalized OZ model,5,6,8
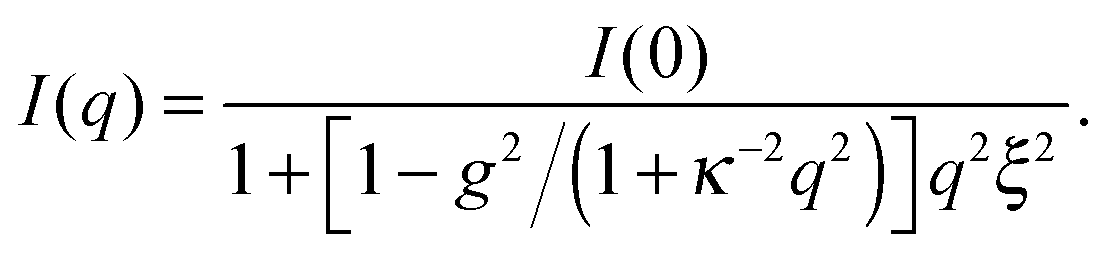 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 valency. Second, an interaction contrast g is introduced, i.e., a contrast between anion–solvent and cation–solvent interactions. We can relate g to the salt-induced mesoscale ordering in the solvent;5,6,8 with increasing g the solvent becomes more ordered, and for g > 1 a shoulder (or peak) apears in the structure factor. Note that in the limit of g = 0, and hence no interaction contrast, eqn (1) yields the classical OZ model.
:1 valency. Second, an interaction contrast g is introduced, i.e., a contrast between anion–solvent and cation–solvent interactions. We can relate g to the salt-induced mesoscale ordering in the solvent;5,6,8 with increasing g the solvent becomes more ordered, and for g > 1 a shoulder (or peak) apears in the structure factor. Note that in the limit of g = 0, and hence no interaction contrast, eqn (1) yields the classical OZ model.
It is worth noting that for low salt concentrations (as studied here) the different proposed theoretical models lead to the same functional form of the structure factor, although the underlying mechanisms differ; while some models5,6 assign the solvent ordering to the asymmetric solvation of ions, exhibiting surfactant-like behavior,6 the only model-independent prediction emphasizes asymmetric steric ion–solvent interactions.8
In Fig. 3 we show the modulus of the interaction contrast |g| versus the size of the cation, as obtained by fitting the generalized OZ model of eqn (1) to the SAXS data. In order to exemplify our sensitivity to the value of |g|, we also show in Fig. 2(a) theoretical SAXS data assuming interaction contrasts |g|/2 and 2|g|. Most importantly, we observe increasing |g| values upon elongating the hydrocarbon chains in the symmetrical tetraalkylammonium cations. We observe the same trend at larger temperatures, but observe the values of |g| to decrease as observed previously for the antagonistic salt.3,21 Whereas salt-induced solvent ordering has been observed before upon adding antagonistic salt consisting of small cations and large anions,3,21 the data of Fig. 3 provide the first direct evidence of a gradual build-up of solvent ordering by increasing the ion size asymmetry.
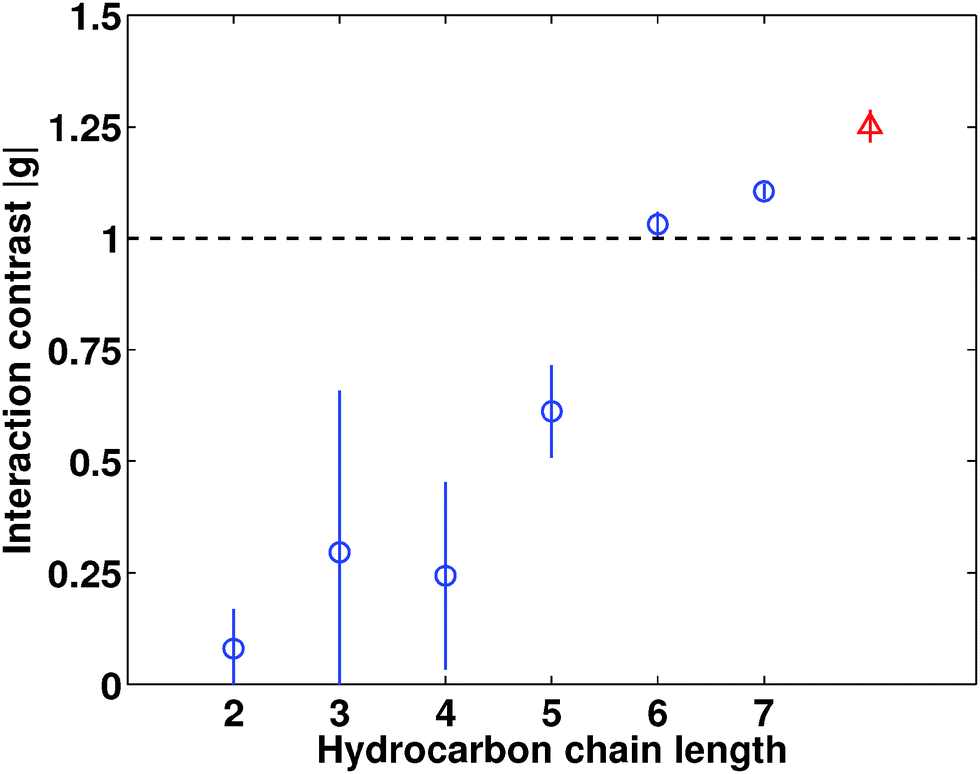 | ||
| Fig. 3 Interaction contrast |g| resulting from fitting the generalized OZ model to the SAXS data, obtained from the binary mixtures of water and 2,6-DMP with added 10 mM salt. Here we show the interaction contrast |g| as a function of elongated hydrocarbon chain length for quaternary ammonium salts (blue circles) and sodium tetraphenylborate (red triangle) as a reference. All data were collected at room temperature (298 K). | ||
How can we understand the ion-size-induced gradual mesoscale ordering of the solvent, as seen in Fig. 3? It is not observed upon adding simple inorganic salts,7 where the ions are of comparable size, and it was recently suggested by Bier and Harnau to be induced by steric ion–solvent interactions.8 Our data of Fig. 3 can be readily reconciled with this prediction; by gradually increasing the hydrocarbon chain length we increase the difference between steric cation–solvent and anion–solvent interactions, thereby increasing |g| and inducing mesoscale ordering of the solvent. We further note that tetraheptylammonium bromide exhibits an ion size asymmetry comparable to that of sodium tetraphenylborate.9,22 If steric ion–solvent interactions are the dominant mechanism of solvent ordering, we would thus expect comparable interaction contrast |g| for C7 and NaBPh4. This is indeed the case as can be seen from Fig. 3.
An alternative explanation for the gradual build-up of solvent ordering is given by preferential ion solvation.5 With increasing size one expects the ions to become hydrophobic,23 and tetraalkylammonium homologues are indeed known to exhibit a carbon chain length-dependent preferential solubility in non-aqueous liquids.9 Although we are not aware of tabulated free energies of transfer for the present salts and solvent components, other than C2 preferring water over pyridine,18 we can expect the quaternary ammonium ions to acquire a preference for 2,6-DMP over water with increasing size, and this will contribute to |g|.5,6 This preference can be identified through the surface activity of the salt series, akin to NaBPh4 which lowers the surface tension γ of water.24 The tendency of lowering the surface tension of water has been reported for C2–C5, and the longer the hydrocarbon chains the more prominent the effect.25 We have verified these results for 10 mM aqueous electrolytes (using the ring method; Sigma 70, Crelab instruments); while C2–C4 have a negligible effect on the surface tension, further elongation of the hydrocarbon chains gradually decreases γ. More specifically, 10 mM of C5 and C6 decreases the surface tension of water by ≈10 and 25 mN m−1, respectively. However, in order to estimate |g| using such an approach, numerical values for the free energies of transfer for C2–C7 between water and 2,6-DMP are needed.
4 Conclusions
In summary, we have investigated salt-induced weak assembly of binary aqueous solvents. Previous ordering of the solvent has been observed upon adding either antagonistic salts or ionic surfactants, but here we report similar structures induced by quaternary ammonium salts. Our results show a gradual build-up of mesoscale order with increasing cation-to-anion size asymmetry, in qualitative agreement with model-independent theoretical predictions. These results are of high importance in the fields of soft matter and colloidal science, where similar ordering in the so-called pre-Ouzo region is applied for designing stable surfactant-free microemulsions26,27 and for mesoscale solubilization using hydrotropes instead of surfactants.28Finally we note that more pronounced mesoscale solvent ordering has been observed upon increasing the concentration of antagonistic salts.29 Here we have focused on a low salt concentration, where no salt-induced shift of the lower critical point occurs, thus facilitating our systematic study as a function of ion-size asymmetry. At this low salt concentration we observe only the initial build-up of mesoscale ordering. In the future we foresee extending these studies to larger salt concentrations and different solvent compositions, where more pronounced mesoscale solvent ordering is expected.
Acknowledgements
We acknowledge the MAX-lab for providing beamtime. KN thanks the Swedish Research Council (Grant No. 621-2012-3897) and the Stenbäck foundation for financial support.References
- K. Sadakane, A. Onuki, K. Nishida, S. Koizumi and H. Seto, Phys. Rev. Lett., 2009, 103, 167803 CrossRef PubMed.
- K. Sadakane, M. Nagao, H. Endo and H. Seto, J. Chem. Phys., 2013, 139, 234905 CrossRef PubMed.
- K. Sadakane, N. Iguchi, M. Nagao, H. Endo, Y. B. Melnichenko and H. Seto, Soft Matter, 2011, 7, 1334 RSC.
- A. Onuki, S. Yabunaka, T. Araki and R. Okamoto, Curr. Opin. Colloid Interface Sci., 2016, 22, 59 CrossRef CAS.
- A. Onuki and H. Kitamura, J. Chem. Phys., 2004, 121, 3143 CrossRef CAS PubMed.
- F. Pousaneh and A. Ciach, Soft Matter, 2014, 10, 8188 RSC.
- M. Bier, A. Gambassi and S. Dietrich, J. Chem. Phys., 2012, 137, 034504 CrossRef PubMed.
- M. Bier and L. Harnau, Z. Phys. Chem., 2012, 226, 807 CrossRef CAS.
- Y. Marcus, J. Solution Chem., 2008, 37, 1071 CrossRef CAS.
- E. Gülari, A. F. Collings, R. L. Schmidt and C. J. Pings, J. Chem. Phys., 1972, 56, 6169 CrossRef.
- C. Y. Seah, C. A. Grattoni and R. A. Dawe, Fluid Phase Equilib., 1993, 89, 345 CrossRef CAS.
- V. Balevicius and H. Fuess, Phys. Chem. Chem. Phys., 1999, 1, 1507 RSC.
- K. Sadakane, H. Endo, K. Nishida and H. Seto, J. Solution Chem., 2014, 43, 1722 CrossRef CAS.
- A. Onuki, T. Araki and R. Okamoto, J. Phys.: Condens. Matter, 2011, 23, 284113 CrossRef PubMed.
- M. Witala, R. Nervo, O. Konovalov and K. Nygård, Soft Matter, 2015, 11, 5883 RSC.
- K. Aoki, M. Li, J. Chen and T. Nishiumi, Electrochem. Commun., 2009, 11, 239 CrossRef CAS.
- A. Labrador, Y. Cerenius, C. Svensson, K. Theodor and T. Plivelic, J. Phys.: Conf. Ser., 2012, 425, 072019 CrossRef.
- H. D. Inerowicz, W. Li and I. Persson, J. Chem. Soc., Faraday Trans., 1994, 90, 2223 RSC.
- J. P. Hansen and I. R. McDonald, Theory of Simple Liquids, Academic Press, Amsterdam, 3rd edn, 2006 Search PubMed.
- U. Nellen, J. Dietrich, L. Helden, S. Chodankar, K. Nygård, J. F. van der Veen and C. Bechinger, Soft Matter, 2011, 7, 5360 RSC.
- J. Leys, D. Subramanian, E. Rodezno, B. Hammouda and M. A. Anisimov, Soft Matter, 2013, 9, 9326 RSC.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995 RSC.
- D. Chandler, Nature, 2005, 437, 640 CrossRef CAS PubMed.
- D. Michler, N. Shahidzadeh, M. Westbroek, R. van Roij and D. Bonn, Langmuir, 2015, 31, 906 CrossRef CAS PubMed.
- K. Tamaki, Bull. Chem. Soc. Jpn., 1974, 47, 2764 CrossRef CAS.
- J. Marcus, D. Touraud, S. Prevost, O. Diat, T. Zemb and W. Kunz, Phys. Chem. Chem. Phys., 2015, 17, 32528–32538 RSC.
- V. Fischer, J. Marcus, D. Touraud, O. Diat and W. Kunz, J. Colloid Interface Sci., 2015, 453, 186–193 CrossRef CAS PubMed.
- A. E. Robertson, D. H. Phan, J. E. Macaluso, V. N. Kuryakov, E. V. Jouravleva, C. E. Bertrand, I. K. Yudin and M. A. Anisimov, Fluid Phase Equilib., 2016, 407, 243–254 CrossRef CAS.
- K. Sadakane, H. Seto, H. Endo and M. Shibayama, J. Phys. Soc. Jpn., 2007, 76, 113602 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details on SAXS data reduction. See DOI: 10.1039/c6sm00580b |
| This journal is © The Royal Society of Chemistry 2016 |