DOI:
10.1039/C6SM00529B
(Paper)
Soft Matter, 2016,
12, 4221-4228
In situ modification of nanostructure configuration through the manipulation of hydrogen bonded amphiphile self-association†
Received
29th February 2016
, Accepted 31st March 2016
First published on 7th April 2016
Abstract
Herein, we report the synthesis of a novel amphiphilic salt containing a number of hydrogen bond donating (HBD) and accepting (HBA) functionalities. This amphiphile has been shown to self-associate via hydrogen bond formation in a DMSO solution, confirmed through a combination of NMR, UV-Vis and dynamic light scattering and supported by X-ray diffraction studies. The combination of different HBD and HBA functionalities within the amphiphile structure gives rise to a variety of competitive, self-associative hydrogen bonding modes that result in the formation of ‘frustrated’ hydrogen bonded nanostructures. These nanostructures can be altered through the addition of competitive HBD arrays and/or HBA anionic guests. The addition of these competitive species modifies the type of self-associative hydrogen bonding modes present between the amphiphilic molecules, triggering the in situ formation of novel hydrogen bonded nanostructures.
Introduction
In recent years a tremendous amount of interest has surrounded the incorporation of supramolecular chemistry into conventional amphiphile design. These systems utilise non-covalent interactions such as hydrogen bonding, π–π stacking, electrostatics and charge transfer to help drive self-association and nanostructure formation.1,2
Short chain peptide amphiphiles consisting of 8–30 amino acid residues are known to self-assemble stabilised by non-covalent supramolecular interactions to form aggregates showing significant promise in the development of drug delivery systems.3 The potential use of low molecular weight supramolecular-inspired amphiphiles for self-associated, hydrogen bonded drug/gene nanocarrier development has also been shown by Zhao and co-workers.4 These results further emphasise the need to understand the effects of hydrogen bond formation within these supramolecular-inspired systems at a fundamental level. Limited examples of this work include that from Faustino and co-workers,5 Oda and co-workers6 and Bong and co-workers.7 Effectively utilising knowledge of non-covalent interactions in amphiphile design will also inform the areas of surfactant, formulations and supramolecular material science, as recently illustrated by Steed and co-workers.8 This in turn has direct applications within the detergent and cosmetic industries, to name just two.
The work described here focuses on molecular level interactions and is designed to demonstrate how the in situ alteration of hydrogen bonding modes within amphiphile self-assembly may be used to influence nanostructure formation. These principles are already utilised in the field of supramolecular soft materials as illustrated in several recent reviews.9,10 Herein we have highlighted the complex area of hydrogen bond influenced self-association and hope to inspire further fundamental research into these types of systems. The use of hydrogen bonds in the formation of anion-receptor complexes11–14 and self-association,15–17 in particular those incorporating urea functionalities18,19 is well established, as is the self-assembly of amphiphilic compounds.20–22 Faustino and co-workers have completed extensive work, developing a range of supramolecular amphiphilic surfactants with a urea-spacer-anion structure.23–25 During these investigations, compounds from this family of surfactants were shown to exhibit critical micelle concentrations (CMC) competitive with other more traditional carboxylate surfactants such as sodium dodecanoate.26 This was attributed to the hydrogen bonding properties of the urea functionality27 also highlighted by Infante and co-workers.28
Here, we present the development of a novel tetrabutylammonium (TBA) sulfonate-urea (1) salt, Fig. 1, capable of producing hydrogen bonded nanostructures. Although similar aromatic, sulfonate-urea compounds have been previously synthesised (Muller and co-workers,29 Christensen and co-workers30–32) the self-association properties of this class of compounds remains unexplored.
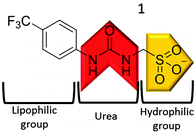 |
| | Fig. 1 Structure of compound 1. The structure of the TBA counter cation has been omitted for clarity. | |
Synthesis
Compound 1 (Fig. 1) contains a urea group capable of both donating and accepting hydrogen bonds (red), and a sulfonate group (yellow) acts only as a hydrogen bond acceptor. The presence of hydrogen bonding in the self-association of 1, through both of the urea NH groups, was confirmed by 1H NMR dilution studies in DMSO-d6/0.5% H2O. As shown in Fig. 2a, a down-field change in chemical shift for those resonances attributed to the NH groups of the urea functionality was observed with increasing concentration of 1. This proves intermolecular hydrogen bond formation which would be expected to lead to extended nanostructure. A deuterium exchange experiment confirmed that the hydrogen atoms of the urea functionality are readily able to exchange.
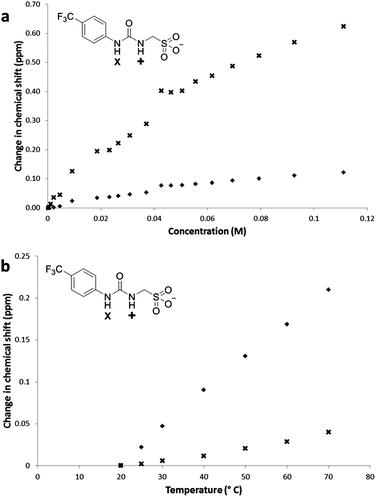 |
| | Fig. 2 (a) Graph illustrating the 1H NMR down-field change in chemical shift of urea NH resonances with changing concentration of 1 in DMSO-d6/0.5% H2O (298 K); (b) graph illustrating the 1H NMR up-field change in chemical shift of urea NH resonances with increasing temperature of 1 (55.63 mM) in a DMSO-d6. | |
The presence of intermolecular hydrogen bonding was further confirmed by variable temperature 1H NMR studies. Incrementally increasing the temperature of a DMSO-d6 solution containing compound 1 (55.63 mM) resulted in an up-field change in chemical shift for those resonances corresponding to the NH groups, as shown in Fig. 2b, illustrating breaking of the intermolecular hydrogen bonded network.
‘Frustrated’ hydrogen bonded self-association
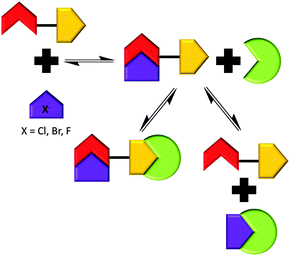
It is possible for compound 1 to adopt at least four different self-associative hydrogen bonding modes: syn- or anti-urea–urea, urea–anion stacking and urea–anion dimerisation (Scheme 1). These four binding modes cannot exist at the same time meaning that this system is ‘frustrated’. The type of self-association which prevails in these systems relies on the balance of these binding modes, which will in turn influence self-association and nanostructure formation. In order to investigate the strength of the possible self-associative interactions and likelihood of each different binding mode existing at any given time, model compounds 2 and 3 (TBA salt) were synthesised which contain the respective urea and TBA sulfonate functionalities. A stability constant of 63 M−1 was calculated by 1H NMR titration methods33 for the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hydrogen bonded complex, illustrated in Scheme 2, of 2 and 3 in a DMSO-d6/0.5% H2O solution. A 1:1 stoichiometry was confirmed by Job Plot analysis34 (Fig. S39, ESI†). This suggests the presence of the urea–anion binding mode in the self-association of 1 under similar solvent conditions. In this case the self-associative urea–urea interactions of 2 are out competed, allowing the formation of the urea(2)–anion(3) complex. A long range, through space 1H NOESY NMR experiment was also conducted with a DMSO-d6 solution of 1. The results of this experiment were compared with a through bond 1H NMR COSY (Fig. S19 and S20, ESI†) and found to be consistent with the formation of the urea–anion hydrogen bonded complex of 1 shown in Scheme 1. They are inconsistent in regard to the presence of the anti-urea–urea binding mode as there are no long range interactions identified between the urea or aromatic protons and those of the CH2 group. The presence of the syn-urea–urea binding mode is also likely disfavoured as it will result in the electrostatically unfavourable close contact of the negatively charged sulfonate groups.
:1 hydrogen bonded complex, illustrated in Scheme 2, of 2 and 3 in a DMSO-d6/0.5% H2O solution. A 1:1 stoichiometry was confirmed by Job Plot analysis34 (Fig. S39, ESI†). This suggests the presence of the urea–anion binding mode in the self-association of 1 under similar solvent conditions. In this case the self-associative urea–urea interactions of 2 are out competed, allowing the formation of the urea(2)–anion(3) complex. A long range, through space 1H NOESY NMR experiment was also conducted with a DMSO-d6 solution of 1. The results of this experiment were compared with a through bond 1H NMR COSY (Fig. S19 and S20, ESI†) and found to be consistent with the formation of the urea–anion hydrogen bonded complex of 1 shown in Scheme 1. They are inconsistent in regard to the presence of the anti-urea–urea binding mode as there are no long range interactions identified between the urea or aromatic protons and those of the CH2 group. The presence of the syn-urea–urea binding mode is also likely disfavoured as it will result in the electrostatically unfavourable close contact of the negatively charged sulfonate groups.
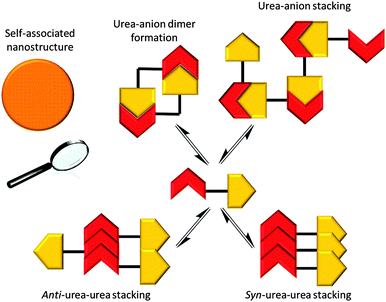 |
| | Scheme 1 The possible hydrogen bond mediated self-associated modes of 1. | |
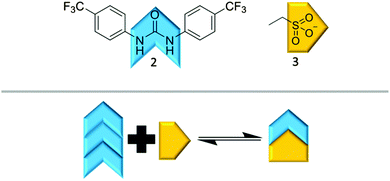 |
| | Scheme 2 The hydrogen bonded complex formation of 2 and 3 based on the NMR evidence presented herein. | |
A UV-Vis dilution study, Fig. 3, was also performed with a DMSO solution of 1 in an effort to establish the minimum concentration needed for self-association/nanostructure formation. At higher concentrations of 1, an absorbance maximum of 262 nm was observed and attributed to the self-associated nanostructures. The absorbance at 262 nm was found to decrease and exhibit a hypsochromic shift as the concentration of 1 was decreased. However, decreasing concentration of 1 also caused an increase in absorbance at 276 nm, the same absorbance maximum that is observed with solutions of 2 only. This absorbance at 276 nm is observed with concentrations of 1 ≤ 0.028 mM, which is an indication that the self-associated structures discussed in this manuscript are no longer present at this point.
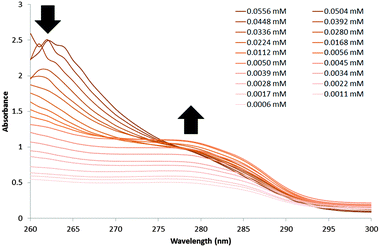 |
| | Fig. 3 UV-Vis spectra recorded for the serial dilution of 1 from 0.0556 mM to 0.0006 mM in DMSO. | |
Nanostructure formation
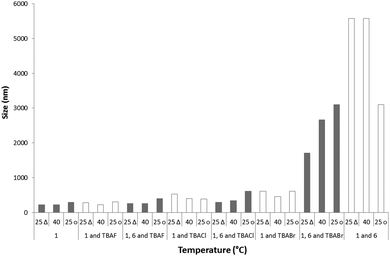
The sizes of the nanostructures formed by 1 in DMSO were identified by dynamic light scattering (DLS). Attempts were made to study the structures by TEM, however these results were ambiguous so are not presented here but are given for consideration within the ESI.† Nanostructure size was found to stabilise with the application of an annealing process in which solutions of 1 were heated from 25 °C to 40 °C, and cooled back to 25 °C, illustrated in Fig. 4. The nanostructures formed at 25 °C by direct solvation of 1 represent the initial kinetic products; raising the temperature to 40 °C destabilises the intermolecular interactions responsible for nanostructure formation, as illustrated in Fig. 2b. Cooling back to 25 °C then allows the intermolecular interactions to re-stabilise producing more thermodynamically stable nanostructures. This allows optimal self-association within the system and results in the increased uniformity of nanostructure size distribution, with maxima of 250–300 nm, despite changing concentration of 1 from 111.27 mM to 0.056 mM, as shown in Fig. 5. Analogous DLS studies conducted at 0.056 mM (Fig. S92, ESI†) and 0.0056 mM (Fig. S93, ESI†) shows evidence of nanostructure destabilisation/alteration. The count rate and raw correlation data also supports sample destabilisation at this point (see Fig. S42 and S57–S62, ESI†). The data shown in Fig. 5 was obtained by a continuous dilution process, as detailed in the experimental section of the ESI.† The sizes of the self-associated nanostructures are more susceptible to change during the annealing process as the concentration of the sample is decreased (5.56 mM and 0.56 mM). This correlates with the understanding that the hydrogen bonded network will be destabilised by decreasing concentration of 1 due to competitive association with the solvent molecules.
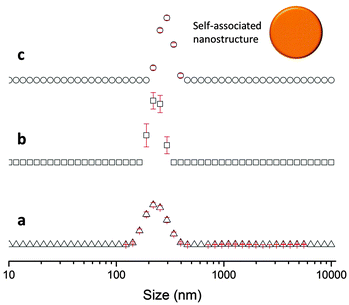 |
| | Fig. 4 Average intensity particle size distribution, calculated from 9 DLS runs, of superstructures formed by dissolving 1 (55.63 mM) in DMSO at (a) 25 °C, (b) heating to 40 °C and (c) cooling to 25 °C. Error given is the standard error of the mean. | |
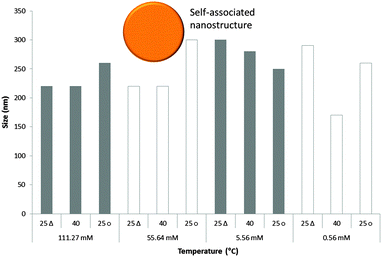 |
| | Fig. 5 Maxima observed from DLS average intensity size distributions (as shown in Fig. 4) for 1 at varying concentrations and temperatures. 25 △ – readings taken at 25 °C before heating to 40 °C; 25 ○ – readings taken at 25 °C after heating to 40 °C. | |
Solid state studies
X-ray diffraction studies conducted for analogous compounds 4 and 5, shown in Fig. 6 and 7 clearly illustrate two of the different self-associative hydrogen bonding modes that can be adopted by this family of sulfonate-urea based compounds in the solution state.‡
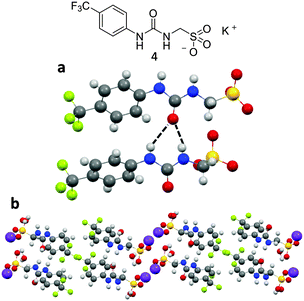 |
| | Fig. 6 Single crystal X-ray structure of compound 4 which was shows (a) hydrogen bonded self-association through syn-urea–urea stacking and (b) the formation of hydrophobic and hydrophilic regions. The potassium counter cation and associated water molecules have been omitted for clarity where necessary. Nitrogen atoms blue, hydrogen atoms white, carbon atoms grey, oxygen atoms red, potassium atoms purple, sulphur atoms yellow and fluoride atoms green. Crystals were obtained by slow evaporation of the pyridinium intermediate of 1 in water with approximately one equivalent of potassium hydroxide. | |
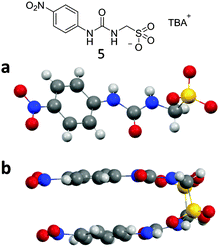 |
| | Fig. 7 Single crystal X-ray structure of (a) compound 5 which was shows (b) dimer formation through urea–anion hydrogen bond formation. The TBA counter cation and second sulfonate urea anion have been omitted for clarity where necessary. Nitrogen atoms blue, hydrogen atoms white, carbon atoms grey, oxygen atoms red, sulphur atoms yellow and fluoride atoms green. Crystals were obtained by slow evaporation of a chloroform solution containing 5. | |
Fig. 6a and b show the structure of compound 4 elucidated by single crystal X-ray diffraction. A syn-urea–urea hydrogen bonded network is observed in this instance resulting in the production of a synthetic bilayer in which the hydrophilic sulfonate and potassium ions are sandwiched between layers of lipophilic aromatic moieties. Each urea oxygen atom was found to form two intermolecular hydrogen bonds in the range N⋯O 2.764(3)–3.112(3) Å and bond angles N1–H⋯O5 140(18)°; N2–H⋯O5 145(17)°; N3–H⋯O1 153(17)°; N4–H⋯O1 153(18)°. Powder X-ray diffraction (PXRD) data was also obtained from the single crystal sample of 4, see Fig. S105 and S106 (ESI†). Good correlation was observed between the experimental and calculated diffraction pattern indicating the main bulk of the sample adopts the same phase illustrated in Fig. 6.
The structure of compound 5 elucidated by single crystal X-ray diffraction was not found to exhibit a urea–urea hydrogen bonded network but instead was shown to dimerise through hydrogen bonded urea–anion complex formation, as illustrated in Fig. 7a and b. Each sulfonate functionality was found to form two intermolecular hydrogen bonds with the urea group of a second molecule in the range N⋯O 2.860(3)–2.967(3) Å and bond angles N2–H⋯O12 154(17)°; N3–H⋯O10 162(16)°; N5–H⋯O4 165(15)°; N6–H⋯O6 167(13)°. Again a PXRD pattern was obtained from a sample of 5, see Fig. S107 (ESI†). The major differences between the experimental and calculated diffraction patterns indicate that the sample adopts a number of different phases. This is not unexpected due to the frustrated nature of these systems and their ability to adopt different self-associated modes as illustrated in Scheme 1.
In situ nanostructure modification
Addition of a competitive hydrogen bond donating array
The addition of the competitive hydrogen bond donating array (2) to a solution of 1 was shown to effect a change in the nanostructures observed with 1 only, through the formation of a hydrogen bonded complex as illustrated in Scheme 3, vide infra.
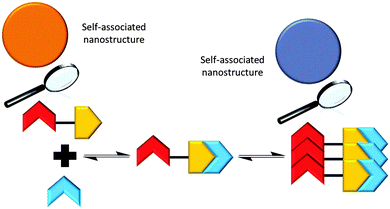 |
| | Scheme 3 Summary of the proposed hydrogen bonded complexes formed from solutions of 1 and solutions of 1 and 2 in DMSO, based on the NMR evidence presented herein. | |
A stability constant of 11 M−1 was calculated for the hydrogen bonded complex of 2 and 1, in a DMSO-d6/0.5% H2O solution by 1H NMR titration methods, with a 1:1 binding stoichiometry confirmed by Job Plot analysis (Fig. S38, ESI†). This weak stability constant is attributed to competition of the urea functionalities from both 1 and 2 towards the coordination of the sulfonate group (1), combined with the pre-existing nanostructure formation of 1 described in Fig. 4 and 5. An annealing process was not applied during the production of stability constant data which prevents the system from reaching a more thermodynamically stable state. However, an association between 1 and 2 is still observed. A comparative 1H NMR stack plot, Fig. 8, shows the effects for the addition of 1 to 2 in equimolar concentrations. A downfield change in chemical shift for the resonance corresponding to the NHs of 2, from 9.4 ppm to 9.7 ppm in the presence of 1 is accompanied by an up-field change in chemical shift for the NHs corresponding to 1. This indicates changes in hydrogen bond complex formation. Compound 1 in the presence of 2 now forms a hydrogen bonded complex which is either less favourable or utilises a less polar guest species, supporting the argument for the formation of self-associating urea–urea interactions. The reverse is true for 2 suggesting the formation of a urea–anion complex. This shift in self-associative hydrogen binding modes contributes to the in situ formation of a second, distinct type of nanostructure, incorporating both 1 and 2. A 1H NOESY NMR experiment (Fig. S23 and S24, ESI†) conducted with an equimolar DMSO-d6 solution of 1 and 2 was found to support the formation of the syn-urea–urea hydrogen bonded complex shown in Scheme 3. This binding mode would also allow 2 to form favourable π–π stacking interactions, further stabilising the resultant nanostructure, similar to that shown in Fig. 6.
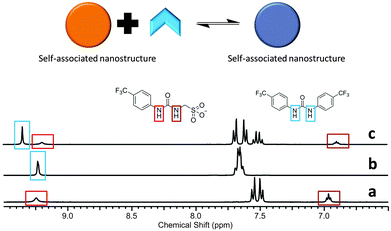 |
| | Fig. 8
1H NMR stack plot (a) compound 1 in DMSO-d6 (55.63 mM); (b) compound 2 in DMSO-d6 (55.63 mM); (c) compound 1 (55.63 mM) and compound 2 (55.63 mM) in DMSO-d6. | |
This in situ modification process of the original hydrogen bonded nanostructure, through the addition of 2, was confirmed by DLS. TEM studies were again found to give ambiguous results, which are discussed within the TEM section of the ESI.† DLS studies (Fig. 9) show that as previously observed, the annealing process (25 °C, 40 °C, 25 °C) produces an increased uniformity in nanostructure size distribution, with a maximum at 400 nm. This is 100 nm larger than the comparative solution of 1 only.
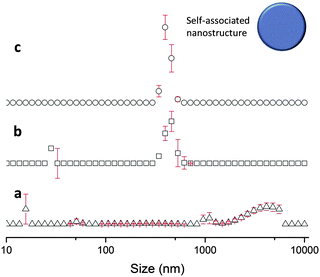 |
| | Fig. 9 Average intensity particle size distribution, calculated from 9 DLS runs, of superstructures formed by dissolving 1 (55.63 mM) and 2 (55.63 mM) in DMSO at (a) 25 °C, (b) heating to 40 °C and (c) cooling to 25 °C. | |
Reversible control of nanostructure formation through hydrogen bond manipulation
In order to further probe the effects of altering the self-associative, hydrogen bonding network on the nanostructure formation of 1 only, competitive anionic guests (fluoride, chloride and bromide) were added to solutions of 1 in DMSO as their tetrabutylammonium (TBA) salt. These competitive anionic guest species are expected to form a hydrogen bonded complex with the urea group of 1. Further to this the effects of anion sequestration on nanostructure formation were also explored through the addition of calix[4]pyrrole (6). As shown in Fig. 10, hydrogen bond donating compound 6 is capable of adopting a cone formation with all NH's available for participation in a 1:1 hydrogen bonded complex with an anionic guest species.35
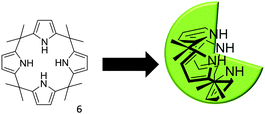 |
| | Fig. 10 The three dimensional conformation of calix[4]pyrrole, adopted for optimal anion coordination. | |
Preliminary investigations showed the addition of 6 to 1 in DMSO results in insoluble aggregate formation. Comparative 1H NMR studies conducted with solutions of 1, and a combination of 1 and 6 doped with acetone (0.002%) to act as an internal standard suggest that this precipitate consists of 6 only. The presence of these large nanostructures were also confirmed by TEM, see ESI.† The presence of the hydrogen bonded complex 1:6 suggested in Scheme 4 was confirmed by a 1H NOESY NMR experiment. Long range, through space interactions were identified as shown in Fig. 11. These interactions are most likely to be the result of the anti-urea–urea binding mode. Unlike the syn-urea–urea binding mode suggested for the self-association of the 1:2 complex, the lack of planar π-systems and the increased steric bulk of 6 compared to 2 would cause the 1:6 complex to favour self-association through the adoption of the anti-urea–urea binding mode.
 |
| | Scheme 4 Summary of the proposed hydrogen bonded complexes formed from solutions of 1 with combinations of 6 and competitive anionic guests in DMSO, based on the NMR evidence presented herein. | |
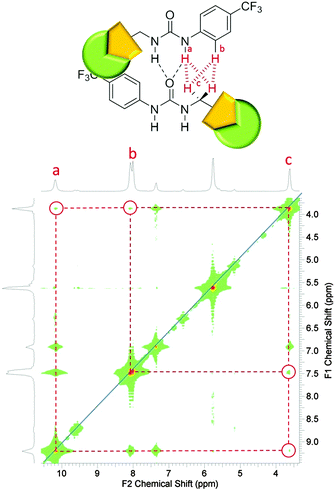 |
| | Fig. 11
1H NOESY of 1 (55.63 mM) and 6 (55.63 mM) in DMSO-d6. | |
The anion binding properties of 6 have been well established by Gale, Sessler and co-workers since 1996.35,36 Stability constants calculated for the formation of various 1:1 anionic complexes of 6 in CD2Cl2 show the following trend: fluoride (17170 M−1) > chloride (350 M−1) > bromide (10 M−1) > hydrogen sulfate (<10 M−1).35 The hydrogen bonding modes identified with different combinations of 1, 6 and various competitive anionic guests are outlined in Scheme 5. Proton NMR titration (Fig. S28–S33, ESI† conducted in a DMSO-d6/0.5% H2O mixture) and corresponding Job Plot studies (Fig. S40 and S41, ESI†) with 1 and TBAF, TBACl and TBABr showed that the anion:receptor complexes formed did adopt the expected 1:1 binding stoichiometry, but instead showed evidence of 2:1 halide:1 complex formation (TBAF:1, K1 = 680 M−1, K2 = 130 M−1; TBACl:1, K1 = 210 M−1, K2 = <10 M−1; TBABr:1, K1 = 40 M−1, K2 = <10 M−1). The stability constants are found to follow a similar trend to the 6:halide complex F > Cl > Br. Job Plot analysis was not possible for 1 and TBAF due to peak broadening and evidence of deprotonation. Zana and co-workers have shown that TBA counter cations can self-associate via hydrophobic interactions of the alkyl chains leading to the bridging of dodecyl sulfate micelles.37 This type of self-association in the presence of the nanostructures produced by 1 provides a plausible explanation of the unexpected 2:1 halide:1 complexes.
 |
| | Scheme 5 Summary of the proposed hydrogen bonded complexes formed from solutions of 1 with combinations of 6 and competitive anionic guests in DMSO, based on the NMR evidence presented herein. | |
Single point 1H NMR experiments conducted, Fig. 12, show the effects of competitive halide anion addition to a DMSO-d6 solution of 1 in equimolar concentration. These competitive anionic guests exhibit the following trend in basicity, a driving force for hydrogen bonded complex formation; fluoride > chloride > bromide. All three of these anions can be seen to interact with 1 through the formation of hydrogen bonds, as a downfield change in chemical shift/peak broadening of those resonances corresponding to the urea NHs of 1 is noted. The extent of these changes corresponds to the increasing basicity of the different anionic guests.
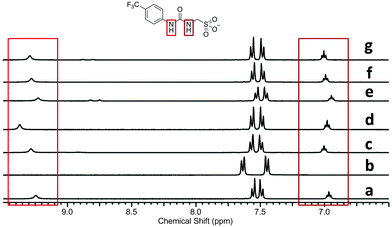 |
| | Fig. 12
1H NMR spectra in DMSO-d6 of (a) 1 (55.63 mM); (b) 1 (55.63 mM) and TBAF (55.63 mM); (c) 1 (55.63 mM), TBAF (55.63 mM) and 6 (55.63 mM); (d) 1 (55.63 mM) and TBACl (55.63 mM); (e) 1 (55.63 mM), TBACl (55.63 mM) and 6 (55.63 mM); (f) 1 (55.63 mM) and TBABr (55.63 mM); (g) 1 (55.63 mM), TBABr (55.63 mM) and 6 (55.63 mM). | |
Fig. 12, also shows the effects of adding 1 equivalent of 6 to DMSO-d6 solutions containing a combination of 1 and competitive anionic guest. This results in the regeneration of the urea NH resonances (1) in the presence of fluoride (comparative spectra b and c), as this anion is readily sequestered by 6. However, there is still a small downfield perturbation in chemical shift compared with a solution of 1 only (spectrum a), indicating that the presence of the fluoride ion is still producing an effect on hydrogen bonding mode. The addition of 6 to a solution of 1 and chloride (comparative spectra d and e) effectively sequesters the chloride resulting in an identical 1H NMR spectra to that of 1 only (spectrum a). This indicates that there is no longer a hydrogen bonded association between 1 and chloride.
The addition of 6 to a solution of 1 and bromide did not alter the position of those resonances corresponding to the urea NHs of 1 (comparative spectra f and g). In this instance 6 is ineffective in sequestering the bromide in order to regenerate free, un-complexed 1. Further evidence from both 1H NMR spectra (downfield change in chemical shift for the resonance corresponding to the NHs of 6 in the presence and absence of bromide) and DLS studies, Fig. 13, suggest that 6 in this case is free to form a hydrogen bonded complex with the sulfonate functionality of 1 or an insoluble precipitate of 6 only, which results in the formation of large aggregates (Scheme 4).
 |
| | Fig. 13 Major maxima observed from DLS average intensity size distributions for combinations of 1 (55.64 mM), 6 (55.64 mM), TBAF (55.64 mM), TBACl (55.64 mM) and TBABr (55.64 mM) in DMSO. 25 △ – readings taken at 25 °C before heating to 40 °C; 25 ○ – readings taken at 25 °C after heating to 40 °C. | |
Comparison of the major maxima observed by DLS for the average intensity size distribution of the nanostructures formed after the annealing process was found to increase with the introduction of a competitive anionic guest species. The general trend was found to be inversely proportional to halide ion basicity; 1 only 300 nm, 1 and fluoride 305 nm, 1 and chloride 390 nm, 1 and bromide 610 nm. The addition of 6 to these samples further increases the size of the major nanostructure formed in solution to 3100 nm, 400 nm, 610 nm and 1710 nm respectively. This size increase is inversely proportional to halide ion basicity and is therefore proportional to the quantity of 6 that is not involved in the formation of a 6:halide complex. This complexation process increases the solubility of 6 in the DMSO solution, preventing insoluble precipitate formation.
Conclusions
In conclusion, we have produced a study which highlights how hydrogen bonding may be used to influence the nanostructures formed from supramolecular-inspired amphiphiles at the molecular level. We have also illustrated how hydrogen bonding interactions may be targeted to affect an in situ change in nanostructure formation. This has been achieved through the synthesis of an anionic, hydrogen bond donating, TBA salt (1) which is capable of ‘frustrated’ nanostructure formation influenced by self-associative hydrogen bond formation in a DMSO solution. Addition of competitive hydrogen bond donating compounds (2/6) or anionic guest species was shown to alter the type of hydrogen bonding modes that influence nanostructure formation. This results in the transformation of nanostructure species in situ. Understanding and targeting hydrogen bonding in nanostructure formation is a promising novel way to develop systems for drug/gene delivery and triggerable sensing, encapsulation and remediation technologies. Research to this end is ongoing within our group as well as conducting further fundamental research to extend our knowledge of these types of complex systems at both the molecular and nanoscale levels.
Acknowledgements
J. R. H. would like to thank the University of Kent for the Caldin Fellowship, M. Howard (NMR), K. Howland (mass spectrometry), P. Saines (PXRD), I. Brown (TEM) and B. Goldmann (Nuffield student) for their assistance during the project. J. R. H. would also like to thank P. A. Gale for many years of help and support. B. W. and G. P. B. would like to thank the University of Kent for the funding of a summer placement.
Notes and references
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed.
- D. M. Leite, E. Barbu, G. J. Pilkington and A. Lalatsa, Curr. Top. Med. Chem., 2015, 15, 2277–2289 CrossRef CAS PubMed.
- X.-D. Xu, X. Li, H. Chen, Q. Qu, L. Zhao, H. Ågren and Y. Zhao, Small, 2015, 11, 5901–5906 CrossRef CAS PubMed.
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, J. Colloid Interface Sci., 2012, 367, 286–292 CrossRef CAS PubMed.
- C. Aime, R. Tamoto, T. Satoh, A. Grelard, E. J. Dufourc, T. Buffeteau, H. Ihara and R. Oda, Langmuir, 2009, 25, 8489–8496 CrossRef CAS PubMed.
- M. Ma, A. Paredes and D. Bong, J. Am. Chem. Soc., 2008, 130, 14456–14457 CrossRef CAS PubMed.
- A. E. Hooper, S. R. Kennedy, C. D. Jones and J. W. Steed, Chem. Commun., 2016, 52, 198–201 RSC.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- S. Wang, B. Wu, J. Duan, J. Fang and D. Chen, Prog. Chem., 2014, 26, 125–139 CAS.
- M. Yamanaka, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 33–48 CrossRef CAS.
- F. Lortie, S. Boileau and L. Bouteiller, Chem. – Eur. J., 2003, 9, 3008–3014 CrossRef CAS PubMed.
- F. Piana, M. Facciotti, G. Pileio, J. R. Hiscock, W. Van Rossom, R. C. D. Brown and P. A. Gale, RSC Adv., 2015, 5, 12287–12292 RSC.
- P. Foley, A. K. Pour, E. S. Beach and J. B. Zimmerman, Chem. Soc. Rev., 2012, 41, 1499–1518 RSC.
- J. P. Hill, L. K. Shrestha, S. Ishihara, Q. Ji and K. Ariga, Molecules, 2014, 19, 8589–8609 CrossRef PubMed.
- K. Jie, Y. Zhou, Y. Yao and F. Huang, Chem. Soc. Rev., 2015, 44, 3568–3587 RSC.
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, J. Phys. Chem. B, 2009, 113, 977–982 CrossRef CAS PubMed.
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, Biomacromolecules, 2009, 10, 2508–2514 CrossRef CAS PubMed.
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, J. Colloid Interface Sci., 2011, 359, 493–498 CrossRef CAS PubMed.
- E. Blanco, A. Gonzalez-Perez, J. M. Ruso, R. Pedrido, G. Prieto and F. Sarmiento, J. Colloid Interface Sci., 2005, 288, 247–260 CrossRef CAS PubMed.
- C. M. C. Faustino, A. R. T. Calado and L. Garcia-Rio, J. Colloid Interface Sci., 2010, 351, 472–477 CrossRef CAS PubMed.
- M. R. Infante, A. Pinazo and J. Seguer, Colloids Surf., A, 1997, 123, 49–70 CrossRef.
- G. W. Muller, J. C. Culberson, G. Roy, J. Ziegler, D. E. Walters, M. S. Kellogg, S. S. Schiffman and Z. S. Warwick, J. Med. Chem., 1992, 35, 1747–1751 CrossRef CAS PubMed.
- M. Pittelkow, J. B. Christensen and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3792–3799 CrossRef CAS.
- M. Pittelkow, C. B. Nielsen, A. C. Broeren, J. L. J. van Dongen, M. H. P. van Genderen, E. W. Meijer and J. B. Christensen, Chem. – Eur. J., 2005, 11, 5126–5135 CrossRef CAS PubMed.
- M. Pittelkow, C. B. Nielsen, A. Kadziola and J. B. Christensen, J. Inclusion Phenom. Macrocyclic Chem., 2009, 63, 257–266 CrossRef CAS.
- M. J. Hynes, Dalton Trans., 1993, 311–312 RSC.
- P. Job, Ann. Chim., 1928, 9, 113–203 CAS.
- P. A. Gale, J. L. Sessler, V. Kral and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- J. L. Sessler, D. E. Gross, W.-S. Cho, V. M. Lynch, F. P. Schmidtchen, G. W. Bates, M. E. Light and P. A. Gale, J. Am. Chem. Soc., 2006, 128, 12281–12288 CrossRef CAS PubMed.
- R. Zana, M. Benrraou and B. L. Bales, J. Phys. Chem. B, 2004, 108, 18195–18203 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: This includes experimental details and DLS, NMR, crystallography and UV-Vis data. See DOI: 10.1039/c6sm00529b |
| ‡ X-ray data were collected on a SuperNova, Dual, Cu at zero, AtlasS2 diffractometer. Crystal data for compound 4. CCDC 1453958, C18H20F6K2N4O10S2 (M = 708.70): monoclinic, space group P21/n, a = 9.229(18) Å, b = 7.2536(12) Å, c = 38.9707(9) Å, α = 90°, β = 95.758(19)°, γ = 90°, V = 2595.7(9) Å3, Z = 4, T = 100(10) K, μ(CuKα) = 5.716 mm−1, Dcalc = 1.813 g mm−3, 23171 reflections measured (13.032 ≤ 2Θ ≤ 136.502), 4737 unique (Rint = 0.0551, Rsigma = 0.0362) which were used in all calculations. The final R1 was 0.0418 (I > 2σ(I)) and wR2 was 0.1040 (all data). Crystal data for compound 5. CCDC 1453959, C24H44N4O6S (M = 516.69): monoclinic, space group P21/n, a = 16.9274(4) Å, b = 16.367(4) Å, c = 20.8867(6) Å, β = 103.701(3)°, V = 5621.2(3) Å3, Z = 8, T = 150(10) K, μ(CuKα) = 1.377 mm−1, Dcalc = 1.221 g mm−3, 13457 reflections measured (7.622 ≤ 2Θ ≤ 134.152), 13457 unique (Rsigma = 0.0302) which were used in all calculations. The final R1 was 0.0510 (I > 2σ(I)) and wR2 was 0.1429 (all data). |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Gianluca P.
Bustone
a,
Ben
Wilson
a,
Kate E.
Belsey
a and
Laura R.
Blackholly
b
*a,
Gianluca P.
Bustone
a,
Ben
Wilson
a,
Kate E.
Belsey
a and
Laura R.
Blackholly
b