
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of conductive hydrogel composites of pH-responsive microgels and carbon nanotubes†
Zhengxing
Cui
a,
Mi
Zhou
b,
Paula J.
Greensmith
a,
Wenkai
Wang
a,
Judith A.
Hoyland
bc,
Ian A.
Kinloch
 a,
Tony
Freemont
bc and
Brian R.
Saunders
*a
a,
Tony
Freemont
bc and
Brian R.
Saunders
*a
aSchool of Materials, The University of Manchester, MSS Tower, Manchester, M13 9PL, UK. E-mail: Brian.Saunders@manthester.ac.uk
bCentre for Tissue Injury and Repair, Institute for Inflammation and Repair, Faculty of Medical and Human Sciences, University of Manchester, Oxford Road, Manchester, M13 9PT, UK
cNIHR Manchester Musculoskeletal Biomedical Research Unit, Manchester Academic Health Science Centre, Manchester, UK
First published on 6th April 2016
Abstract
Conductive gel composites are attracting considerable attention because of their interesting electrical and mechanical properties. Here, we report conductive gel composites constructed using only colloidal particles as building blocks. The composites were prepared from mixed dispersions of vinyl-functionalised pH-responsive microgel particles (MGs) and multi-walled carbon nanotubes (CNTs). MGs are crosslinked pH-responsive polymer colloid particles that swell when the pH approaches the pKa of the particles. Two MG systems were used which contained ethyl acrylate (EA) or methyl acrylate (MA) and around 30 mol% of methacrylic acid (MAA). The MA-based MG is a new pH-responsive system. The mixed MG/CNT dispersions formed thixotropic physical gels. Those gels were transformed into covalent interlinked electrically conducting doubly crosslinked microgel/CNT composites (DX MG/CNT) by free-radical reaction. The MGs provided the dual roles of dispersant for the CNTs and macro-crosslinker for the composite. TEM data showed evidence for strong attraction between the MG and the CNTs which facilitated CNT dispersion. An SEM study confirmed CNT dispersion throughout the composites. The mechanical properties of the composites were studied using dynamic rheology and uniaxial compression measurements. Surprisingly, both the ductility and the modulus of the gel composites increased with increasing CNT concentration used for their preparation. Human adipose-derived mesenchymal stem cells (AD-MSCs) exposed to DX MG/CNT maintained over 99% viability with metabolic activity retained over 7 days, which indicated non-cytotoxicity. The results of this study suggest that our approach could be used to prepare other DX MG/CNT gel composites and that these materials may lead to future injectable gels for advanced soft-tissue repair.
Introduction
The interest in hydrogels has been growing rapidly,1,2 due to remarkable improvements in gel modulus,3 ductility,4 swelling ratios5 and toughness6 which have great importance for soft matter science, tissue engineering and regenerative medicine.7 Moreover, the range of properties available increases greatly for hydrogel composites.8 However, the overwhelming majority of hydrogel-based research has used small molecules (monomers and crosslinkers) for construction of the gel component which contrasts to the approach used in the present study which involved microgel (MG) particles. Most hydrogels are intrinsically brittle. To overcome this limitation a number of toughened hydrogels have been established which include double network hydrogels,9 polyampholyte gels10 and hybrid gels of polyacrylamide and ionically crosslinked alginate.11,12 In each case a network of sacrificial crosslinks provided energy dissipation which opposed crack propagation. Indeed, the development of toughened hydrogels has enabled a key problem that plagued hydrogel composites to be addressed; that is, the tendency of high modulus particulates to cut through soft hydrogel networks when strained.13 Toughened hydrogels have also enabled new fabric-reinforced hydrogel composites to be prepared that have potential for use as prosthetics.14 Here, we construct a new family of gel composites that have improved mechanical properties when carbon nanotubes are present without the use of a special toughening methodology.MG particles are crosslinked polymer particles that swell when the pH approaches the pKa of the particles.15 Here, these MGs are termed singly crosslinked microgels (SX MGs) because they are internally crosslinked. Vinyl-functionalised SX MGs can be subsequently covalently interlinked16 to form macroscopic hydrogels, termed doubly crosslinked microgels (DX MGs). Our unique hydrogel construction approach enables structural control at the colloidal length scale (∼10–1000 nm) and does not involve small monomer building blocks. The use of monomers can limit potential for gel construction in the presence of cells. The latter concern restricts potential for minimally-invasive studies (e.g., by injection) because it is difficult to prevent small molecule building blocks from rapidly migrating away from the gel construction site. In an effort to overcome these drawbacks we have used colloidal MG particles to prepare new hydrogels16 that can be constructed in the presence of cells.17 In this study hydrogel composites are prepared from MGs and multi-walled carbon nanotubes (CNTs) for the first time. We hypothesised that the MGs could provide the dual roles of dispersant for the CNTs and macro-crosslinker for DX MG/CNT construction. The CNTs also had dual roles, which were provision of electrical conductivity and modulus enhancement. The motivation for this study was to establish an injectable conductive gel (based on colloidal building blocks) with mechanical and electrical properties that may enable future biomaterials applications, e.g., intervertebral disc (IVD) repair17 or electronic skin.18
CNTs have attracted much interest since Iijima's 1991 paper19 due to their excellent combination of mechanical and electrical properties. CNTs also have a very high Young's modulus (∼1000 GPa) and tensile strength (∼100 GPa).20,21 They have been investigated in a wide variety of contexts which include biosensing,22 drug delivery23 and tissue engineering.24,25 CNTs aggregate in water due to their very large van der Waals attractive interactions,26 low entropy and high hydrophobicity which has required the use of surfactants, linear polymers or bespoke polymers for their aqueous dispersion.27–29 The requirement for dispersion necessarily limits the range of gel composites that can be prepared. Shi et al. studied PVA/CNT gels prepared by the freeze–thaw method.30 UV-photocrosslinking was used to prepare gelatin methacrylate and polyethylene glycol methacrylate/CNT mixtures gels.31,32 Non-covalent polymer/CNT gels have been prepared by ionic crosslinking of alginate.33 Bottom up approaches using free-radical polymerisation of monomers such as N-isopropylacrylamide have also been used to construct these composites.34 However, gel composites prepared from MGs and CNTs have not been reported. Here, we show for the first time that CNTs can be dispersed using MGs and that this process enables preparation of covalently inter-linked MG/CNT gel composites.
In addition to the systems mentioned above a number of other gel composites have been investigated which include nanocomposite gels involving LAPONITE®,35 or silica nanoparticles,36 graphene oxide37 or CNTs.38–40 All of these gel composites have either been prepared from small molecule or linear polymer building blocks. By contrast our approach for constructing gel composites the building blocks uses pre-formed MGs which were also responsible for dispersing the additive prior to macrogel formation. Because the DX MG phase was not specially toughened (e.g., by double network formation41) one might expect that the mechanical properties of the DX MG/CNTs to be inferior to those of the parent DX MGs. However, the mechanical properties of the gel composites are shown to be improved and a mechanism is proposed for this behaviour (later).
Recently, we investigated DX MG/graphene oxide (DX MG/GO) gel composites42 which were prepared using blended dispersions of MGs and GO. The MGs and GO were both negatively charged, electrostatically repulsive and did not associate in mixed dispersions. The role of the MGs in that work was solely to act as macro-crosslinkers which enabled formation of a DX MG network which contained dispersed GO. The DX MG/GO gels were not electrically conductive which rules them out of future studies that require conductive gels. To improve the versatility of DX MG gel composites we sought to replace GO with conductive nanoparticles. Very recently we showed that pH-responsive MGs could stabilise oil-in-water emulsions.43 Because CNTs are hydrophobic we postulated that MGs could also be used to disperse CNTs and enable preparation of DX MG/CNT gel composites. In contrast to the DX MG/GO composites42 we show in the present study that attractive MG-to-CNT interaction are essential for hydrogel composite preparation. Whilst the previously studied DX MG/GO modulus values increased linearly with GO concentration the ductility was constant.42 At variance with the latter data the ductility of the DX MG/CNT composites studied will be shown to increase with CNT concentration.
The first MG system investigated here (abbreviated as MG1) consisted of pH-responsive poly(ethyl acrylate-co-methacrylic acid-co-1,4-butanediol diacrylate) (poly(EA/MAA/BDDA)) particles that had been vinyl-functionalised with glycidyl methacrylate (GMA) – see Scheme 1. A second MG system based on poly(MA/MAA/BDDA) (MA is methyl acrylate), abbreviated as MG2, was used to demonstrate versatility. MG2 is a new pH-responsive microgel and has not been previously reported. Formation of the gel composites involved using concentrated MG dispersions and pH-triggered particle swelling (Scheme 1). The MG particles were in their collapsed (latex) form at pH values less than their respective pKa values. Concentrated dispersions formed viscous fluids when the pH reached the pKa. A fluid-to-gel transformation occurred when the pH increased to above the pKa that produced a physical (SX MG/CNT) gel composite. This process occurred at constant total volume which means that the free volume (i.e., the water-filled space between the microgel particles) decreased. The vinyl groups of the swollen MG particles came into close contact and the presence of radical fragments from the initiator enabled conversion of the physical gels into covalently inter-linked gel composites (DX MG/CNT). Uniquely, the building blocks used to construct the hydrogel/CNT composites (MGs and CNTs) were colloidal.
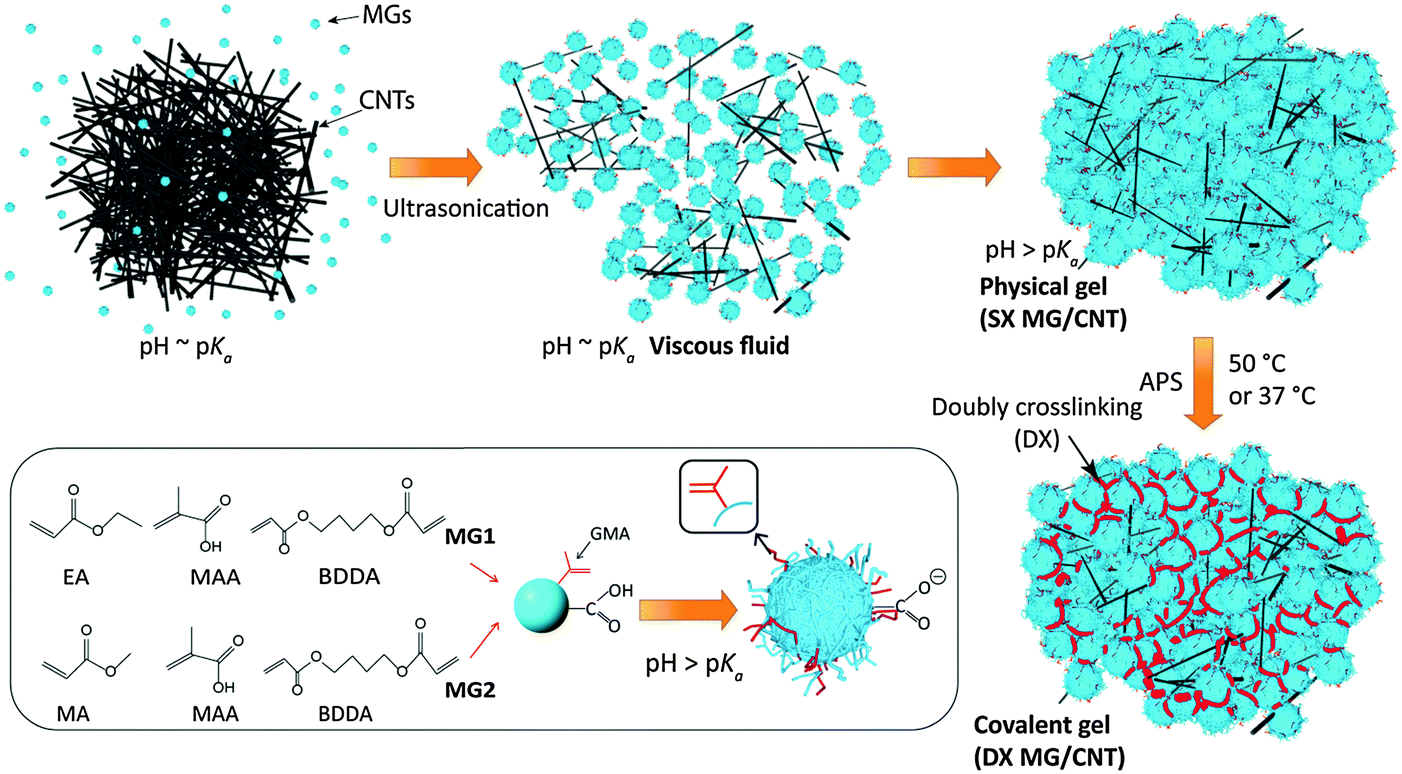 | ||
| Scheme 1 Dispersion of CNTs using microgels and DX MG/CNT formation. The CNTs were initially dispersed in the presence of a viscous fluid of partially swollen vinyl-functionalised MG particles. The pH was increased to above the pKa, triggering further MG swelling, and the formation of a physical gel (SX MG/CNT). In the presence of ammonium persulphate (APS) covalent interlinking (shown in red) occurred and the DX MG/CNT gel composite formed. | ||
In this study we first characterise the MG and CNT colloidal building blocks used for gel composite construction. Then, the interaction between the MGs and CNTs was investigated followed by a study of the electrical and pH-responsive properties of the DX MG/CNT gel composites. The morphology and mechanical properties of the composites gels were also investigated. We show that both the modulus and ductility increased with increasing CNT concentration and a mechanism for this behaviour is proposed. Whilst this study focuses on fundamental properties, the favourable electrical and mechanical properties for our new injectable electrically conductive, pH-responsive gel composites may lend themselves to future applications involving soft tissue repair and electronic skin.44
Experimental details
Reagents
CNTs (multi-walled, outer-diameter × length 6–13 nm × 2.5–20 μm, Sigma), ethyl acrylate (EA, 99%), methyl acrylate (MA, 99%), methacrylic acid (MAA, 99%), 1,4-butanediol diacrylate (BDDA, 90%), glycidyl methacrylate (GMA, 97%), sodium hydroxide (NaOH, 97%), ammonium persulphate (APS, 98%), sodium dodecyl sulfate (SDS, ≥92.5%), dipotassium phosphate (K2HPO4, 97%), phosphate buffered saline (PBS, pH = 7.4 bioreagent), α-MEM medium (minimum essential medium eagle, α modification), foetal bovine serum (FBS), L-ascorbic acid-2 phosphate were all purchased from Sigma-Aldrich and used as received. All water was distilled and deionised.Microgel preparation
The synthesis of poly(EA/MAA/BDDA) microgel (MG1) used seed-feed emulsion polymerisation method and followed earlier work.16 Briefly, a mixed monomer solution (250 g) of EA (62.9 mol%), MAA (36.6 mol%), BDDA (0.5 mol%) was prepared. Seed formation was conducted using a portion of the monomer mixture (31.5 g) after addition of water (517.5 g) containing SDS (1.8 g), K2HPO4 (3.15 g of 7 wt% solution) and APS (10 g of 2 wt% solution). The seed was formed at 80 °C with mechanical stirring under a nitrogen atmosphere. The remaining monomer solution was added uniformly with a rate of 2.4 g min−1. After feed completion the temperature was maintained for a further 2.5 h. The product was extensively dialysed against water. MG2 (poly(MA/MAA/BDDA)) was prepared using the same method as described for MG1 and the same monomer mole ratios. The method for functionalising the MG particles with GMA was identical to that reported elsewhere.17 Composition and property data for MG1 and MG2 are given in Table S1 (ESI†).Preparation of mixed microgel/carbon nanotube dispersions
The MG content for all gels used in this study was 10 wt% unless otherwise stated. To prepare SX MG1/CNTy viscous fluids (y is the wt% of CNTs) the pH of a concentrated mixed dispersion containing 10 wt% MG1 was adjusted to 6.8 using NaOH solution (0.5 M) and then the appropriate mass of CNTs added. Mechanical stirring (700 rpm) was used until a mixed dispersion was formed. The mixture was subsequently placed into an ice bath and an ultrasonic probe used (ultrasonic processor, 750 W) for 60 min combined with mechanical stirring. Ultrasonication used sequential power step cycles of “on” (30 s) and “off” (30 s). The total sonication time was 30 min and the energy used was ∼96 kJ. A similar method was used to prepare the SX MG2/CNT physical gels. However, in that case the pH of MG2 before addition of the CNTs was ∼6.4.Preparation of doubly crosslinked microgel/carbon nanotube gel composites
The DX MG/CNT gel composites were prepared at 37 or 50 °C. The following example describes the preparation of DX MG1/CNT1.0. The latter was prepared by adding NaOH (4 M, 40 μl) and also APS solution (3.4 wt%, 230 μl) to the mixed MG1/CNT1.0 viscous dispersion (6 g) described above. The final pH was adjusted to 7.4. The crosslinking process was initiated by heating at 50 °C for 8 h within sealed molds. Identical methods were used for the preparation of the other DX MG/CNTy gel composites. DX MG/CNT gel composites were also prepared at physiological temperature (37 °C). In these cases APS solution (3.4 wt%, 230 μl) and TEMED (0.08 M, 200 μl) were added to the parent viscous mixed dispersions. The crosslinking process was initiated by heating at 37 °C for 20 min. Unless otherwise stated the gels investigated were prepared at 50 °C. Some gel composites were prepared at 37 °C and these are identified.Physical measurements
Potentiometric titration measurements were performed using a Mettler Toledo DL 15 titrator in the presence of a supporting electrolyte (0.1 M NaCl). Photon correlation spectroscopy (PCS) measurements were performed using a BI-9000 Brookhaven light scattering apparatus equipped with a 20 mW HeNe laser and the detector was set at a scattering angle of 90°. SEM images were obtained using a Philips FEGSEM instrument. Samples were dried at room temperature (MG particles) or by freeze-drying (gels). TEM measurements were obtained using a JEOL JEM-2011F operating at an accelerating voltage of 200 kV and the samples were supported by 200 mesh copper TEM grids. TGA measurements were performed in a N2 atmosphere using a TA instrument Q500 with a heating rate of 10 °C min−1. The electrical conductivity measurements were performed on gel composite samples in the hydrated state using a Jandel multi-height four-point probe station with cylindrical tungsten carbide four probe head (spacing 1.00 mm between each probe head). The measurement results were recorded by Keithley 2440 multimeter. The samples were prepared inside O-rings with a constant diameter of 20.8 mm and thickness of 2.43 mm. Swelling experiments for the DX microgels were performed by placing samples in buffer solutions for 7 days. The buffers were refreshed every day. To measure the weight and the volume change, a sample was removed from buffer, excess solution from the surface carefully removed using absorbent tissue and the sample weighed and then returned to the buffer solution. The volume swelling ratio of the gel composites (QDX) was calculated from the difference of the sample weight before and after swelling and the data were corrected for polymer and CNT densities. Dynamic and static rheology measurements were conducted using a TA Instruments AR-G2 temperature-controlled rheometer equipped with an environmental chamber. A parallel plate geometry (20 mm) was used. For the frequency-sweep data a strain of 1% was used; whilst for the strain-sweep data a frequency of 1 Hz was used. The uniaxial compression tests were conducted using an Instron series 5569 load frame equipped with a 100 N compression testing head. Nominal stress and strain values are reported. A cylindrical geometry was used for the samples (diameter of 12 mm and height of 12.5 mm).Cytotoxicity studies
Adipose derived human mesenchymal stem cells (AD-MSCs) were grown in α-MEM medium supplemented with 10% v/v FBS, L-ascorbic acid-2 phosphate (10 μM), 1× Glutamax (Life Invitrogen, UK) and an antibiotic mixture of penicillin (100 units per ml), streptomycin (100 μg ml−1) and amphotericin (0.25 μg ml−1) were used for cytotoxicity studies of the gel composites. Cells were regularly passaged at 80% confluence and cells at passage 3 were used in the cytotoxicity assessments. The gel composites were fabricated into round disc-like shape with a diameter of 12 mm. Prior to assessments the hydrogels were pre-soaked in PBS for 24 h. AD-MSCs were trypsinized and cultured in a density of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2 into 6-well plates. The cells were allowed to adhere to the bottom of the well for 24 h before exposure to the hydrogels. The hydrogels in tissue culture inserts were then introduced into the wells and was maintained for a further 7 days. Cell viability and proliferation was measured respectively at 1, 4, and 7 days post hydrogel introduction. At each time point, samples were incubated in a live/dead solution containing calcein AM and ethidium homodimer-1 (Life Invitrogen, UK), and the cells were imaged using an Olympus BX51 fluorescence microscope fixed with a Qimaging Retiga-SRV camera. Other samples were incubated at the same time in normal medium containing Alamar Blue solution (Life Invitrogen, UK) for 2 h and fluorescence changes of the medium supernatant were measured at a 560 nm EX (excitation)/590 nm EM (emission) setting using a BioTek FLx800 fluorescence plate reader combined with a Gen5 data analysis software.
000 cells per cm2 into 6-well plates. The cells were allowed to adhere to the bottom of the well for 24 h before exposure to the hydrogels. The hydrogels in tissue culture inserts were then introduced into the wells and was maintained for a further 7 days. Cell viability and proliferation was measured respectively at 1, 4, and 7 days post hydrogel introduction. At each time point, samples were incubated in a live/dead solution containing calcein AM and ethidium homodimer-1 (Life Invitrogen, UK), and the cells were imaged using an Olympus BX51 fluorescence microscope fixed with a Qimaging Retiga-SRV camera. Other samples were incubated at the same time in normal medium containing Alamar Blue solution (Life Invitrogen, UK) for 2 h and fluorescence changes of the medium supernatant were measured at a 560 nm EX (excitation)/590 nm EM (emission) setting using a BioTek FLx800 fluorescence plate reader combined with a Gen5 data analysis software.
Results
Microgel and carbon nanotube characterisation
Two MG systems were used to prepare the DX MG/CNT gel composites studied in this work (Scheme 1). MG1 was used for most of the study. To demonstrate generality for our approach we also prepared MG2, which is a new MA-based microgel. Whilst MG1 and MG2 contained 38.2 and 27.4 mol% MAA respectively (Table S1, ESI†) they contained mostly the primary (or structural) monomer (EA or MA) and can be considered to be different pH-responsive MG systems. The extent of GMA functionalisation of the MGs determined from the titration data (Fig. S1, ESI†) was about 8.0 mol% for both MG1 and MG2 (Table S1, ESI†). SEM data (Fig. 1a) showed that the MG1 particles had a number-average diameter (dSEM) of 74 nm. The MG2 particles (Fig. 1b) were significantly larger with a dSEM value of 111 nm which indicates that a relatively small number of MG2 particles formed prior to stabilisation during emulsion polymerisation. For comparison a representative SEM image for the CNTs (Fig. 1c) shows they were anisotropic and had a tendency to form bundles. The Raman spectrum for the CNTs (Fig. S2a, ESI†) showed characteristic D and G bands and is similar to that reported elsewhere.45 The D and G bands correspond to disorder induced and tangential Raman modes, respectively.46 The ratio of the D to G band intensities gives a measure of the defect concentration.47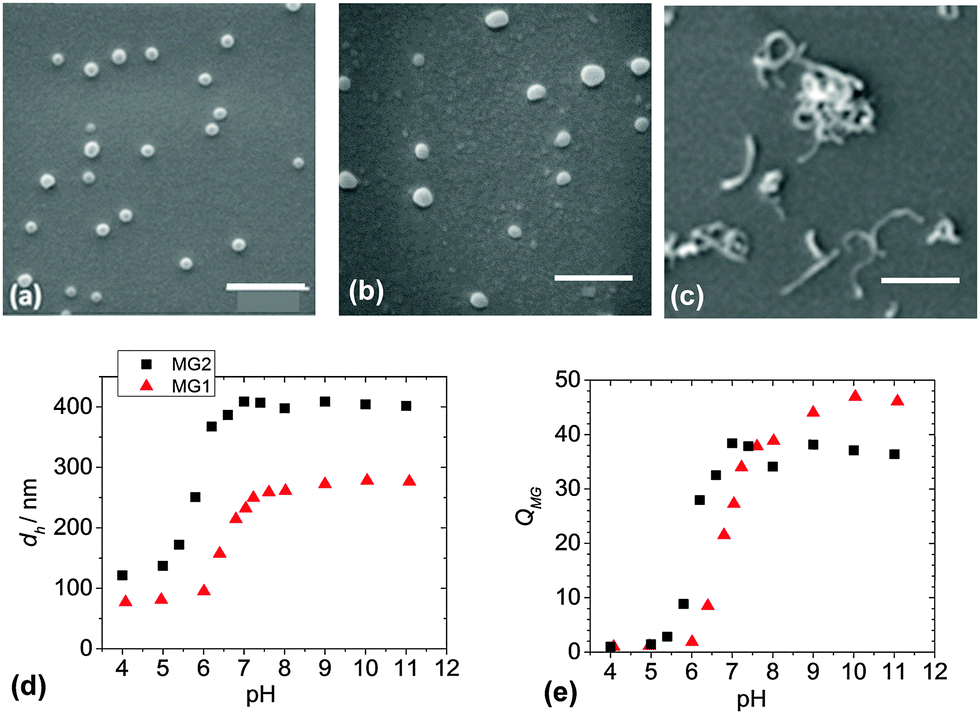 | ||
| Fig. 1 Microgel and carbon nanotube characterisation. SEM images are shown for (a) MG1 (b) MG2 and (c) CNTs. The scale bars are 500 nm. (d) and (e) Show the variations of the hydrodynamic diameters (dh) and volume swelling ratios (QMG) with pH for MG1 and MG2. Values for QMG were calculated using QMG = (dh/dh(4))3, where dh(4) was measured at pH 4.0. | ||
The apparent pKa values for MG1 and MG2 were 6.3 and 4.9, respectively, as determined from potentiometric titration (Fig. S1, ESI†). This difference shows that the primary monomer (EA or MA) has a strong effect on the relative ease for –COOH neutralisation. The MG1 and MG2 particles were pH-responsive (Fig. 1d) and their hydrodynamic diameter values (dh) increased for pH values greater than their respective pKa values. MG particle swelling occurred because neutralisation of the –COOH groups produced –COO− which electrostatically repelled neighbouring groups resulting in chain expansion. The MG volume swelling ratios, QMG, were estimated from the PCS data and were 47 and 37, respectively, for MG1 and MG2 at pH = 10 (Fig. 1e). The higher QMG value for MG1 is due to the higher MAA content for that system (Table S1, ESI†). It can be concluded that both MG systems showed pronounced pH-triggered swelling which was governed by their respective pKa values and this, in turn was determined by the nature of the primary monomer. Strong pH-triggered particle swelling is a requirement for the formation of DX MGs because this favours inter-segment contact for neighbouring MGs and efficient covalent inter-linking16 (Scheme 1).
CNT dispersion using pH-responsive microgels
A commonly used approach for dispersing CNTs involves the use of small molecule surfactants or linear polymers.27,28,48 Here, a study was conducted of the ability of MGs to disperse CNTs. There were several features of the MG particles that were expected to favour CNT dispersion. Firstly, the MG particles contained high concentrations of –COOH groups and these groups are known to facilitate CNT dispersion.49 Secondly, MG1 particles have been shown to stabilise oil-in-water emulsions, which demonstrates their affinity for hydrophobic surfaces.43 In our initial attempts at dispersing CNTs with MG1 dispersions we used pH values below the pKa. Interestingly, the CNTs formed macroscopic aggregates (Fig. S3, ESI†). This observation confirmed that strong, attractive, MG–CNT interactions were present (which is a new result for MG research) and contrasts to our earlier work involving mixed MG/GO dispersions where the two components were repulsive.42We established that by increasing the pH to 6.8, where partial MG1 particle swelling occurred (Fig. 1e), the CNTs could be dispersed within viscous mixed MG1/CNT dispersions (Fig. 2a). The CNTs could be subsequently kinetically trapped in the dispersed state by further increasing the pH to 7.4 in order to trigger the fluid-to-physical gel transition (Fig. 2b). The MG1 particles were found to be more effective than SDS at dispersing CNTs. SDS caused the CNTs to aggregate strongly (Fig. 2c). The optical micrographs showed that some spherical aggregates were present for the mixed MG1/CNT dispersions (Fig. 2a and b), which were most likely CNT bundles. (CNT bundles were also present within pure CNT dispersions – see Fig. 1c.) The physical gels exhibited thixotropic behaviour as evidenced by shear stress vs. shear rate data (Fig. 2d) where the gradient (instantaneous viscosity) and shear stress decreased after the maximum shear rate was reached. The gels could be injected (Fig. 2b(i)) and retained their shape once shear ceased which allowed them to be molded into various shapes (Fig. 2b(ii)).
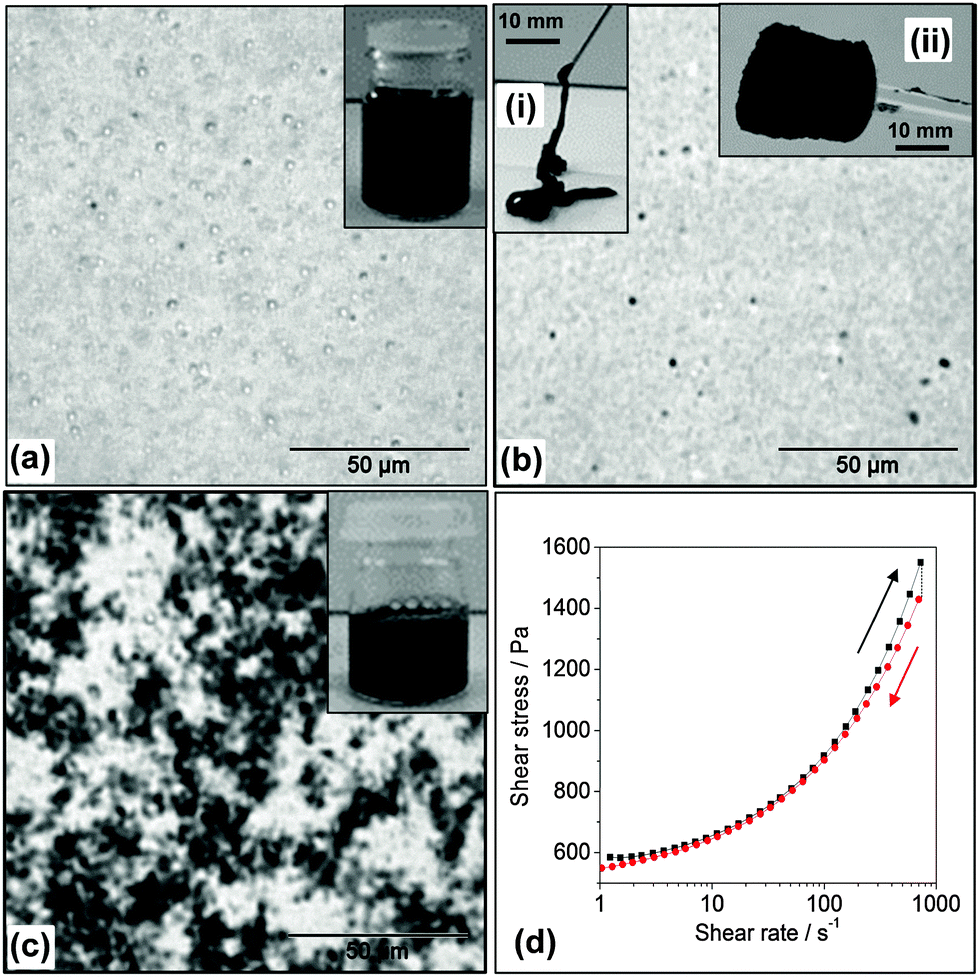 | ||
| Fig. 2 Investigation of CNT dispersion using microgels or a surfactant. Images and optical micrographs for CNTs (1 wt%) (a) dispersed in MG1 (10 wt%, pH = 6.8) as a viscous fluid, (b) dispersed in the physical gel state, SX MG1/CNT (10 wt%, pH = 7.4) or (c) dispersed in aqueous SDS (2.5 wt%) solution. The insets show the tubes (a and c) or physical gel (b). (d) Shear stress vs. shear rate data for the MG1/CNT dispersion. The thixotropic physical gels were injectable (b(i)) and moldable (b(ii)). | ||
An important question concerns what the mechanism is that enabled MGs to facilitate CNT dispersion. We propose that the MG particles enabled CNT dispersion through a combination of attractive MG–CNT interactions (likely involving hydrophobic association) that favoured CNT separation during shear and steric repulsion between MG particles that opposed CNT movement and aggregation in the physical gel state. The swollen MG particles would also have prevented the dispersed CNTs from agglomerating due to the large excluded volume of the MGs. According to this mechanism the SX MG1/CNT physical gel contained CNTs located between the MG particles. Our MG-assisted dispersion/kinetic trapping method enabled CNTs to be dispersed at CCNT values up to 1 wt%. The latter value is relatively high compared to CCNT values reported for CNTs dispersed by conventional surfactants.50,51
It is well known that CNT dispersion processes (especially those involving covalent attachment) can disrupt the CNT structure.27 To probe for changes in conjugation Raman spectroscopy was used to examine the MG1/CNT dispersion (Fig. S2b, ESI†). The relative height of the D and G bands was identical to that of the as-supplied CNTs (Fig. S2a, ESI†). Therefore, the physical CNT dispersion process established here occurred without a detectable increase of defect density or shortening of the nanotubes. Dispersion processes that do not shorten the nanotubes, or decrease their conjugation, should maximise CNT conductivity and enable maximum charge transport.
Additional evidence for an attractive MG–CNT interaction was found from TEM images of a diluted SX MG1/CNT1.0 mixture (see Fig. 3). The CNT walls are clearly evident and the images are similar to those reported for CNTs elsewhere.27 Entangled CNTs were evident where one CNT had threaded another looped CNT (Fig. 3(i)). This type of morphology should have been present within the DX MG/CNT composites and is potentially important for energy dissipation (discussed later). Importantly, MGs were adsorbed directly onto the CNTs (Fig. 3(ii)), which confirms an attractive MG–CNT interaction. It can be seen that the MGs were able to bridge neighbouring CNTs (Fig. 3(iii), arrow) which is likely to be a key interaction for stress transfer within the gel composites. We discuss stress distribution and dissipation within DX MG/CNT later.
 | ||
| Fig. 3 Evidence for an attractive MG–CNT interaction from TEM. The TEM images are of dried SX MG1/CNT1.0 after dilution. The pH of the mixed MG1/CNT dispersion before dilution was 6.8. The insets show (i) entangled CNTs with one CNT threading another looped CNT, (ii) MGs adsorbed on a CNT and (iii) a MG particle bridging two CNTs. | ||
Conductive DX MG/CNT morphology and pH-triggered swelling
The thixotropic SX MG1/CNT physical gels (Fig. 2b) were covalently inter-linked to form robust DX MG1/CNT gel composites (Fig. 4a). The covalent inter-linking between the MG particles was confirmed by negligible sol fractions from swelling experiments (later). DX MG1/CNT morphology was probed using SEM (Fig. 4b and c). The microporous morphology of the freeze-dried samples (Fig. 4b) is typical of that reported for hydrogels.52 The large voids are attributed to ice formation. Closer examination of the surface shows MG particles and also rod-like features (Fig. 4b((i) and (ii)) and also 4c, yellow arrows). The MG particles could be seen at the surface (Fig. 4c, white arrow) and were inter-linked. A similar morphology was also observed from SEM images for DX MG2/CNT1.0 (Fig. S4, ESI†). Scrutiny of the rod-like features shown in Fig. 4c reveals that they have much larger diameters that those apparent for the CNTs (Fig. 1c). The rod-like features (Fig. 4c) are ascribed to strain-induced damage of the DX MG matrix caused by the CNTs as a result of modulus mismatch between the CNTs and the DX MG. These rod-like features can be thought of as deformed impressions of a CNT. A gel composite sample was fractured and then freeze-dried and the fracture surface examined (Fig. 4d). Close examination of a fracture surface shows many protruding CNTs (Fig. 4d(i) to (iii)). These images provide evidence of CNTs that had been pulled out of the matrix during the fracture process. | ||
| Fig. 4 DX MG1/CNT morphology. (a) DX MG1/CNT1.0 cylinder. (b) and (c) Show SEM images of freeze-dried DX MG1/CNT0.5. The insets for (b) show rod-like features ((i) and (ii)). (c) Shows a higher magnification image from (b). The white and yellow arrows show, respectively, MG particles and deformation patterns caused by CNTs (see text). (d) A sample was fractured and then freeze-dried and the image shows a fracture surface. The insets ((i)–(iii)) show CNTs protruding from the fracture surface (identified with arrows). (e) Shows SEM images for DX MG1/CNT0.5 gel after heating at 850 °C in N2. The insets ((i) and (ii)) show increasing magnifications of the entangled CNT mesh. | ||
CNTs are known to have thermal stability at 900 °C in an inert atmosphere.53 We exploited this property to probe the distribution of CNTs within the gel composites. A freeze-dried DX MG1/CNT1.0 sample was heated at 850 °C in N2. This process removed more than 80% of the MG1 phase. (The TGA profile for the experiment is shown in Fig. S5, ESI†). The remaining matrix had a nanoporous morphology with CNTs distributed throughout pore walls (Fig. 4e). The higher magnification images (Fig. 4e(i) and (ii)) show that the CNTs formed an entangled 3D mesh. It follows that the as-made DX MG/CNTs contained a network of entangled CNTs. Indeed entangled CNTs were clearly common as evidenced by Fig. 3(i). However, inter-linked MG particles were also present. We therefore propose that these gel composites contained interpenetrating networks of inter-linked MGs and entangled CNTs.
In order to probe CNT–CNT contacts within the gel composites electrical conductivity measurements were performed (Fig. 5a). The conductivity values (σ) of DX MG1/CNT for CCNT > 0.25% was comparable to that reported by Hur et al.44 for their hydrogel composites that were used for bio-electrodes. Indeed, the latter workers reported a σ of 0.35 S cm−1 which is larger the values shown in Fig. 5. However, the maximum σ we measured (0.031 S cm−1 for DX MG1/CNT1.0) is much larger than the values of 7.6 × 10−4, 5 × 10−3 and 10−4 S cm−1 reported for polyacrylamide/CNT,54 self-assembled graphene hydrogels55 and polyaniline/chitosan blend hydrogels,56 respectively. Taken as a whole, the σ values for our DX MG1/CNTy compared favourably to related materials.
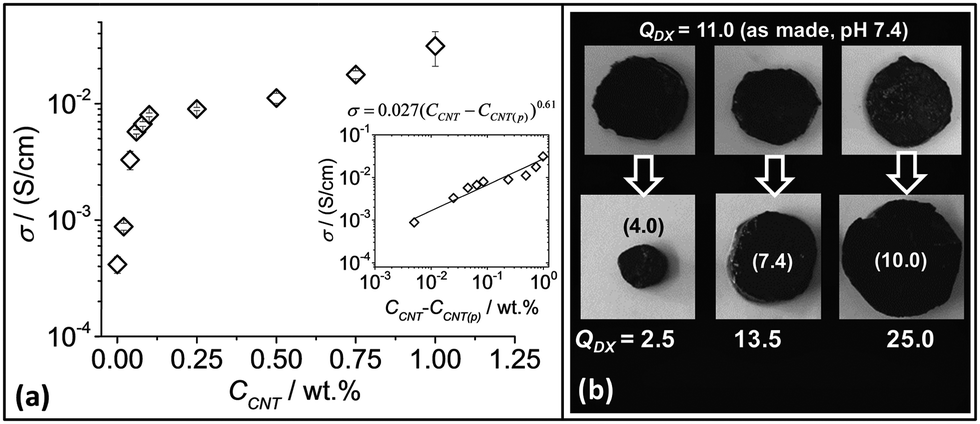 | ||
| Fig. 5 Conductive gel composite properties. (a) Conductivity variation with CNT concentration for DX MG1/CNTy. The inset shows a fit for the data to the percolation model (eqn (1)). At least three samples were tested for each data point and the error bars include the minimum and maximum values. (b) pH-Responsive swelling of DX MG1/CNT1.0 after three samples prepared at pH 7.4 (top row) were placed in buffer solution. The pH values for the latter are shown in brackets for the bottom row. QDX is the equilibrium gel composite volume swelling ratio. | ||
The percolation model57 was applied to the data.
| σ = σo(CCNT − CCNT(p))s | (1) |
Because the DX MG/CNT composites contained high MG particle concentrations (10 wt%) their ability to undergo pH responsive swelling was studied. Three DX MG/CNT1.0 samples (initial pH 7.4) were placed in buffer solutions with pH values of 4.0, 7.4 and 10.0 (Fig. 5b). The equilibrium volume-swelling ratio (QDX) for the as-prepared gel was 11.0. These pH-responsive gel composites collapsed at pH 4.0 and swelled at pH 7.4 and 10.0. Moreover, they did not fragment and had negligible sol fractions which demonstrated effective inter-MG linking. By contrast SX MG1/CNT physical gels re-dispersed when placed in buffer solutions (not shown). When compared to the QMG values for the parent MG1 particles (Fig. 1e) the QDX values (Fig. 5b) were larger at pH 4 (cf. QMG1 = 1.0) and smaller at pH values of 7.4 (cf. QMG1 ∼ 35) and 10 (cf. QMG1 = 47). These differences are attributed to the CNT and inter-linked MG networks within the hydrogel composites which opposed deswelling and swelling at low and high pH values, respectively.
Mechanical properties of SX MG/CNT and DX MG/CNT hydrogel composites
The mechanical properties of both the physical gels (SX MG1/CNT) and covalent gels (DX MG1/CNT) were investigated using dynamic rheology. The frequency-sweep and strain-sweep dynamic rheology data are shown in Fig. S6 (ESI†). Whilst there was little difference in the frequency dependences of the storage modulus (G′) for DX MG1/CNT and SX MG1/CNT, the gradients for tan δ (=G′′/G′, where G′′ is the loss modulus) were much lower for DX MG1/CNT due to covalent MG interlinking. The strain-sweep data for DX MG1/CNT and SX MG1/CNT generally showed linear viscoelastic behaviour when the strain was less than 10%.The variation of G′ with CCNT is shown in Fig. 6a. The data show pronounced differences for DX MG/CNT and SX MG/CNT. Clearly, double crosslinking increased the modulus and the rate of G′ increase with CCNT. For a given CNT concentration the ratio of the G′ value for DX MG/CNT to that for the respective SX MG/CNT gives a measure of the relative ratio of elastically-effective chains. The fact that this ratio increased from ∼3.0 (for CCNT = 0%) to 11.0 (for CCNT = 1.0%) shows that double crosslinking strongly increased the ability of the gel composites to distribute stress. Because G′ is a measure of network connectivity it follows that double crosslinking strongly increased the ability of the CNTs to participate in and extend the elastic networks present.
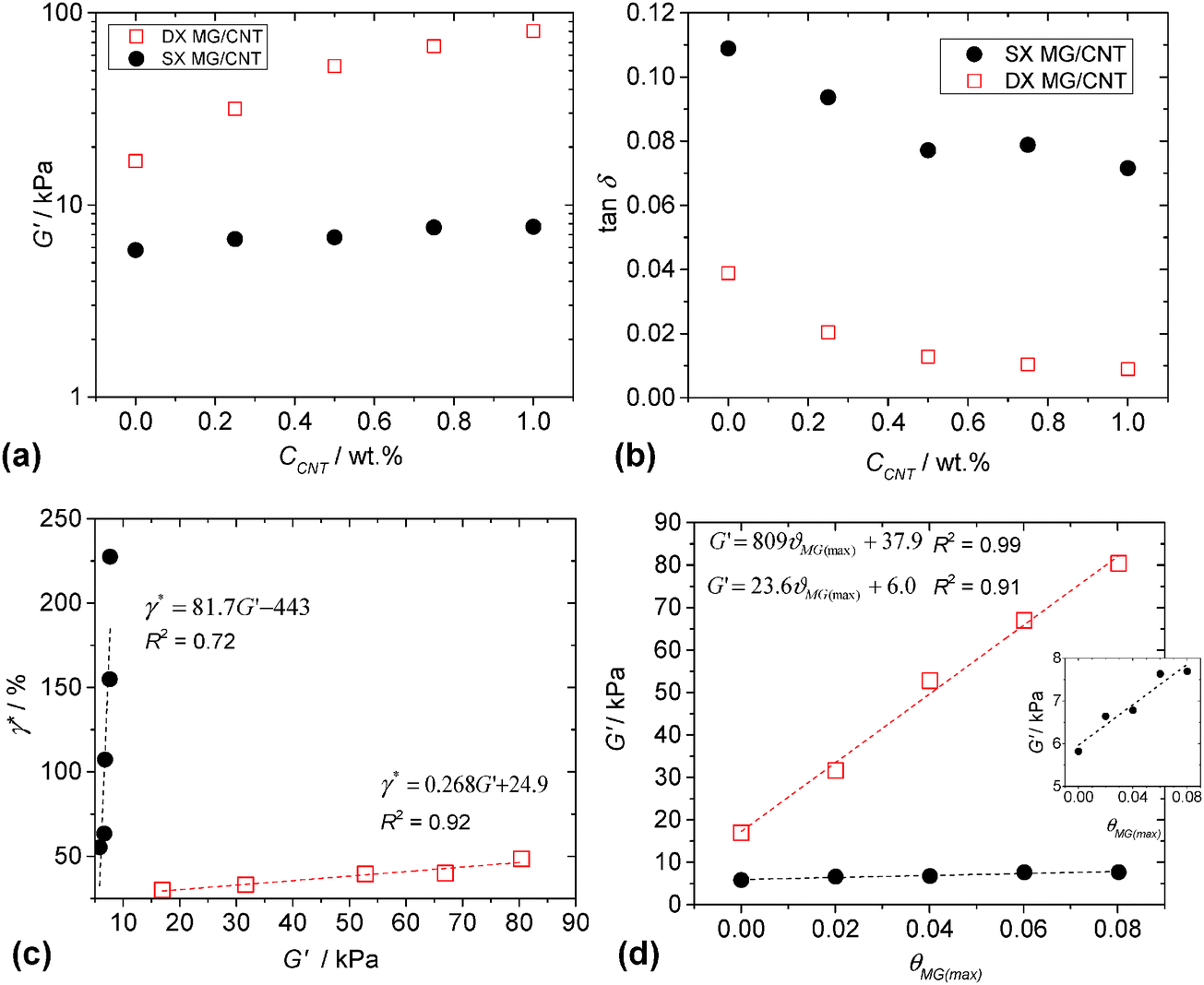 | ||
| Fig. 6 Rheological properties for SX MG1/CNT and DX MG1/CNT. Variation of G′ (a) and (b) tan δ with CNT concentration. These values were measured at a frequency of 1 Hz. (c) Shows the critical strain as a function of G′. (d) Variation of G′ with maximum average fractional coverage of the MG particles with CNT (see text). The legends for (a) and (b) also apply to (c) and (d). The inset for (d) shows an expanded view of the SX MG1/CNT data. | ||
The value of tan δ is a measure of the proportion of energy due to strain that is stored. For a conventional network tan δ values that approach zero indicate efficient energy storage and that the network is free of elastically ineffective linkages, such as dangling chains and loops. The values of tan δ for both the composite systems decreased with increasing CCNT (Fig. 6b). We propose that the CNTs forced the MG particles to more effectively participate in energy storage during deformation. Presumably, this occurred through attractive MG–CNT interactions (discussed above). The effect was amplified for the DX MG/CNT system (which had lower tan δ values) because of covalent MG inter-linking which restricted MG movement and also the increased contribution from the highly elastic CNTs.
It is usually the case that gels with higher modulus values are increasingly brittle, i.e., the modulus is inversely proportional to yield strain.60 This trend is expected because the modulus is proportional to the number density of elastically effective chains (νeff) and the strain at break is an increasing function of the molecular weight of the elastically effective chains (Meff). These two preparameters are inversely related, i.e., νeff ∼ 1/Meff. A widely used rheological measure of gel ductility is the critical strain (γ*). The value for γ* is the strain at which G′ = G′′ and was obtained from strain-sweep data (Fig. S6, ESI†). Fig. 6c shows that the value for γ* was proportional to G′ for both SX MG1/CNT and DX MG1/CNT, which is a remarkable trend. Because the gradient for the data was lowest for DX MG1/CNT it follows that the relationship between ductility and modulus could be tuned by varying the extent of inter-MG crosslinking, implying good potential for convenient mechanical property modification. Fig. 6d shows the variation of G′ with fractional maximum average fractional coverage (θMG(max), eqn (2)) for the MG particles by the CNTs. For both the SX MG1/CNT and DX MG1/CNT data G′ was proportional to θMG(max) and it follows that MG–CNT interfacial interactions were important for stress distribution (see later discussion).
Our previous work demonstrated that DX MGs have the long term potential to restore the mechanical properties of degenerated intervertebral discs (IVDs).17 An interesting question was whether composites prepared at 37 °C had similar mechanical properties to those discussed above (which were prepared at 50 °C). Therefore, DX MG1/CNT1.0 were prepared at 37 °C using TEMED as a redox accelerator and the mechanical properties for DX MG1/CNT1.0 assessed using rheology (see Fig. S7, ESI†). The frequency-sweep and strain-sweep data for the composites were nearly superimposable showing that the mechanical properties were very similar. It is concluded that these new gel composites can be prepared with similar covalent inter-linking extents using conventional or redox-assisted free-radical coupling. All systems discussed hereafter were prepared at 37 °C.
Static uniaxial compression measurements were performed for DX MG1/CNT (Fig. 7a) and also DX MG2/CNT gel composites (Fig. 7b) to further probe composite gel mechanical properties. All systems showed strain hardening (Fig. 7a). Importantly, the modulus (Fig. 7c) and strain at break (Fig. 7d) values both increased with increasing CCNT. These data confirm the trends apparent from the rheology data (Fig. 6c). Moreover, the data for DX MG2/CNT1.0 (Fig. 7b–e) also showed the same trend of increasing ductility (and toughness) upon inclusion of CNT. (The fracture toughness values were determined from the area under the stress–strain curves.61) The toughness for DX MG1/CNT showed a linear relationship with CCNT (Fig. 7e). These results, which were obtained using two different microgels, demonstrate that the ability of CNTs to increase modulus, ductility and toughness of DX MG/CNT composites is general. There may be potential for IVD repair in the future for these composites because the modulus values are comparable to those reported for DX MGs that provided load support for model degenerated IVDs.17
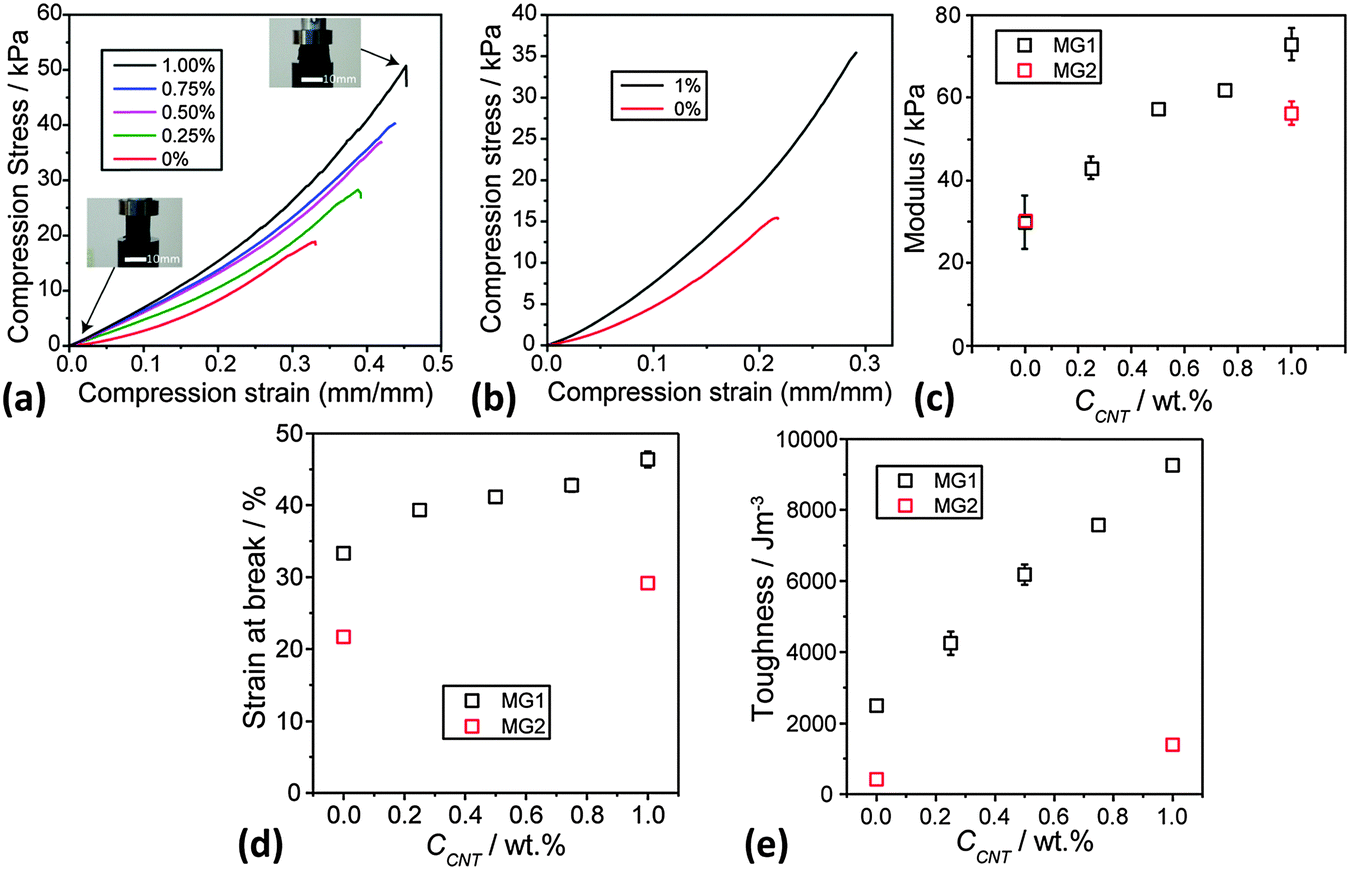 | ||
| Fig. 7 Uniaxial compression data for DX MG/CNT gel composites. (a) and (b) Compression stress–strain data for DX MG1/CNT and DX MG2/CNT gels, respectively. The legends show the CNT concentration (CCNT) used. (c), (d) and (e) Show the variations of compression modulus, strain at break and toughness with CCNT, respectively. Some of the data points in (c), (d) and (e) were larger than the error bars. The insets of (a) show the uncompressed (left) and compressed (right) samples of DX MG1/CNT1.0. At least three samples were tested for each data point shown in (c), (d) and (e) and the error bars include the minimum and maximum values. | ||
It can be seen from the toughness values (Fig. 7e) that this parameter was much lower for the DX MG2 and DX MG2/CNT systems compared to the respective MG1-based systems. The gels containing MG2 were less ductile than those containing MG1. From Fig. 1e it can be seen that the MG1 particles were able to swell more extensively than the MG2 particles. Whilst it is understood that those differences were driven by a higher MAA content for MG1 the data do demonstrate greater ability for deformation of the MG1 particles. It is tentatively proposed that the increased ductility for the MG1-containing DX systems discussed above originates from greater intra-particle ductility. More work is required to unravel the relationships between MG swelling and DX MG ductility and is currently underway.
Cytoxicity studies for DX MG1/CNT1.0
The cytotoxicity of the gel composites was studied using live/dead and Alamar blue assays. Human adipose-derived human mesenchymal stem cells (AD-MSCs) were used because they have potential for regenerative medicine/tissue engineering applications which includes IVD repair.62,63 Here, cell viability was studied in the presence of the gel composites using an insert (See Experimental details). Live/dead assay images (Fig. 8a and b) showed that over 99% of AD-MSCs remained alive after 7 days' exposure to DX MG1/CNT1.0. Furthermore, cell metabolism data at 1, 4, 7 day time points were similar to cells not exposed to the gel composites (Fig. 8c). Therefore exposure of adhered AD-MSC cells to DX MG1/CNT maintained normal cell viability and also metabolic activity. We conclude that DX MG1/CNT was not cytotoxic under the conditions used.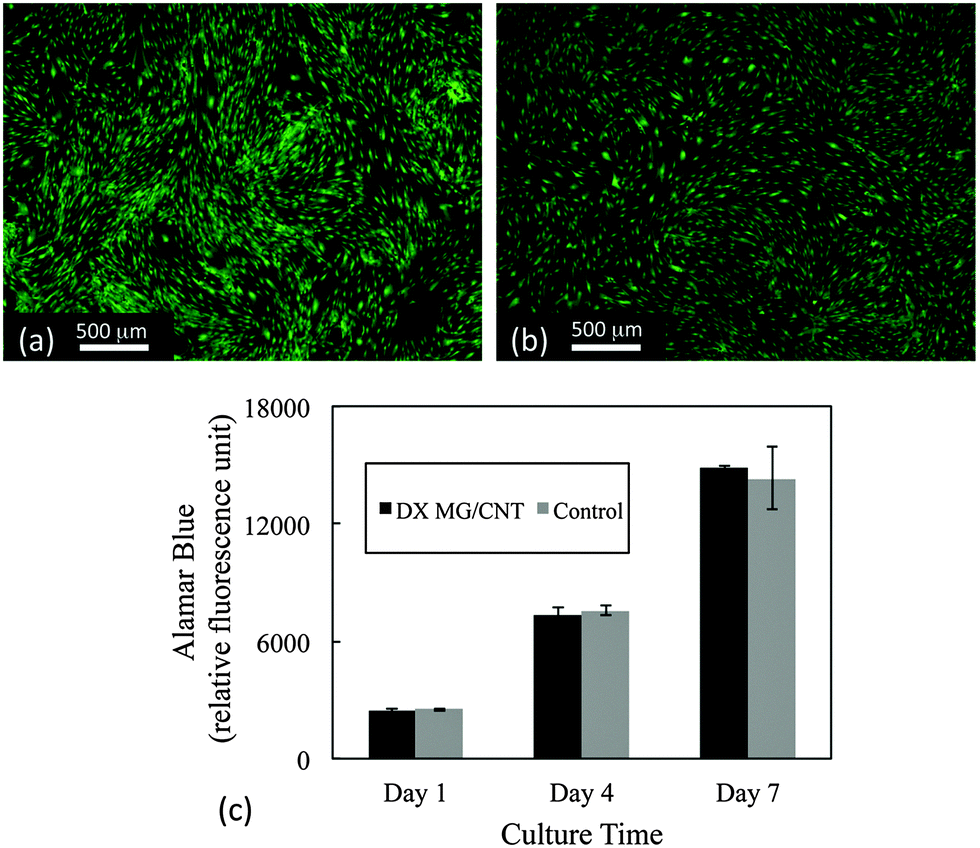 | ||
| Fig. 8 Cytotoxicity study for DX MG1/CNT1.0. Live–dead staining showing (a) AD-MSCs exposed to DX MG1/CNT1.0 gel at day 7 and (b) cells on glass without exposure to the composite at day 7. (c) Metabolic activity of AD-MSCs after exposure to the gels for different time periods. | ||
Discussion
In the following we discuss the mechanisms governing modulus and ductility for the DX MG/CNT hydrogel composites. Firstly, we consider the mechanism by which stress is transferred between the CNT and MG components within SX MG/CNTs and DX MG/CNTs. The evidence discussed above (Fig. S3, ESI† and Fig. 3) indicated significant attractive MG–CNT interfacial interactions. To explore the potential importance of the MG–CNT interface the maximum average fractional coverage (θMG(max)) for the MG particles (present at a total polymer volume fraction of ϕMG(T)) by the CNTs (present at a total volume fraction of ϕCNT(T)) within a perfectly mixed composite was estimated using eqn (2).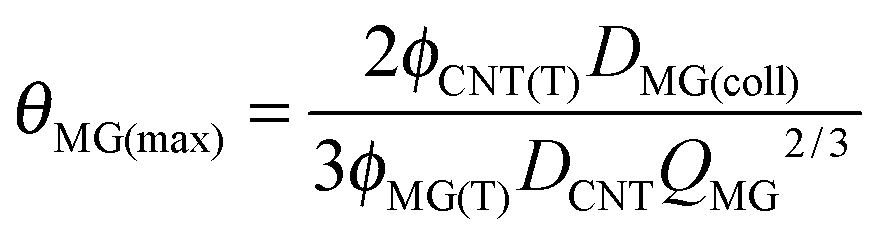 | (2) |
Arguably, the most intriguing question for these DX MG/CNT gel composites concerns what the mechanism is for the increase of ductility observed for Fig. 6c and d. There are three key mechanisms envisaged. This first mechanism (suggested by a referee) envisaged the DX MG/CNT gel composites as a double network with strong CNT and soft DX MG phases. As deformation occurred the CNT phase was able to fracture, dissipating energy, without causing catastrophic failure of the DX MG network. The second mechanism is associated with the CNTs and involves their “pull out” (or debonding) from the DX MG matrix, or from entanglements during strain, both of which would dissipate energy and oppose crack propagation. A pull out mechanism has been postulated to be responsible for the improved toughness of CNT elastomer composites;64 however, that mechanism is new for CNT hydrogel composites. A third mechanism is decreased inter-MG covalent linking due to co-location of CNTs and vinyl groups from the MGs at the MG interface. The latter would be an excluded volume effect. Because the values for θMG(max) were at most 8% (Fig. 6d) and the SX MG/CNT systems also showed a ductility increase with CCNT (Fig. 6c) we do not consider the third mechanism to have been significant.
The SEM images of the DX MG/CNT fracture surface (Fig. 4d(i)–(iii)) showed clear evidence of CNTs that appear to have protruded from the fracture surface. It follows that those CNTs were embedded within the matrix and were then pulled out as the crack propagated. The process of CNTs separating from the matrix would have dissipated crack propagation energy. Accordingly, we favour the “pull out” mechanism as being the dominant cause of the ductility increases observed. However, the data available cannot rule out a contribution to the ductility enhancement from brittle fracture of a CNT phase and the SEM images of the heat-treated sample (Fig. 4e) showed that an entangled CNT network was present throughout the gel composite.
Conclusions
We have investigated hydrogel composites constructed from colloidal building blocks of MGs and CNTs in this study for the first time. Two MG systems were investigated (MG1 and MG2) and the properties of MG2 have not been previously reported. A new finding is that the use of MA strongly decreased the pKa of the MGs. For constructing the gel composites the MG particles had the dual roles of dispersant and macro-crosslinker; whereas, the CNTs had the dual roles of providing electrical conductivity and modulus enhancement. The ability of the MGs to disperse the CNTs was assisted by attractive hydrophobic MG–CNT interactions and the results shown here extend the range of materials able to be dispersed by MGs. The precursor SX MG/CNT physical gels were injectable and moldable. The DX MG1/CNTs were electrically conductive and possessed a low percolation threshold (0.15%). The DX gel composites were also pH-responsive and had modulus values that increased linearly with MG–CNT interfacial contact area. Remarkably, the ductilities of both the SX MG/CNT and DX MG/CNT gels increased with increasing gel modulus. The ductility increase was ascribed to dissipation associated with the pull out of the CNTs from the matrix. Our new colloidal approach for preparing pH-responsive injectable CNT hydrogel composites was shown to apply to more than one type of MG and should be suitable for use with other MG systems. Our injectable DX MG/CNT systems may have potential for future applications for soft tissue repair and electronic skin.Acknowledgements
BRS and IAK would like to thank the EPSRC for funding this work (EP/K030949/1 and EP/I023879/1). BRS gratefully acknowledges a 5 Year EPSRC Established Career Fellowship (M002020/1). We are grateful for the helpful comments and suggestions provided by one of the referees of this manuscript.References
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- N. Sahiner, Prog. Polym. Sci., 2013, 38, 1329–1356 CrossRef CAS.
- A. Ghoorchian, J. R. Simon, B. Bharti, W. Han, X. Zhao, A. Chilkoti and G. P. López, Adv. Funct. Mater., 2015, 25, 3122–3130 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- H. Therien-Aubin, Z. L. Wu, Z. Nie and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 4834–4839 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- S.-H. Lee and H. Shin, Adv. Drug Delivery Rev., 2007, 59, 339–359 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- J. Li, W. R. K. Illeperuma, Z. Suo and J. J. Vlassak, ACS Macro Lett., 2014, 3, 520–523 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- W. R. K. Illeperuma, J.-Y. Sun, Z. Suo and J. J. Vlassak, Extreme Mech. Lett., 2014, 1, 90–96 CrossRef.
- D. R. King, T. L. Sun, Y. Huang, T. Kurokawa, T. Nonoyama, A. J. Crosby and J. P. Gong, Mater. Horiz., 2015, 2, 584–591 RSC.
- W. Richtering and B. R. Saunders, Soft Matter, 2014, 10, 3695–3702 RSC.
- R. Liu, A. H. Milani, T. J. Freemont and B. R. Saunders, Soft Matter, 2011, 7, 4696–4704 RSC.
- A. H. Milani, A. J. Freemont, J. A. Hoyland, D. J. Adlam and B. R. Saunders, Biomacromolecules, 2012, 13, 2793–2801 CrossRef CAS PubMed.
- T. Yamada, Y. Hayamizu, Y. Yamamoto, Y. Yomogida, A. Izadi-Najafabadi, D. N. Futaba and K. Hata, Nat. Nanotechnol., 2011, 6, 296–301 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun'ko, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- S. K. Peddini, C. P. Bosnyak, N. M. Henderson, C. J. Ellison and D. R. Paul, Polymer, 2014, 55, 258–270 CrossRef CAS.
- J. C. Claussen, A. D. Franklin, A. ul Haque, D. M. Porterfield and T. S. Fisher, ACS Nano, 2009, 3, 37–44 CrossRef CAS PubMed.
- A. A. Bhirde, V. Patel, J. Gavard, G. Zhang, A. A. Sousa, A. Masedunskas, R. D. Leapman, R. Weigert, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 307–316 CrossRef CAS PubMed.
- C. Dong, A. S. Campell, R. Eldawud, G. Perhinschi, Y. Rojanasakul and C. Z. Dinu, Appl. Surf. Sci., 2013, 264, 261–268 CrossRef CAS.
- P. E. Mikael, A. R. Amini, J. Basu, M. Josefina Arellano-Jimenez, C. T. Laurencin, M. M. Sanders, C. Barry Carter and S. P. Nukavarapu, Biomed. Mater., 2014, 9, 035001 CrossRef PubMed.
- S. Zhang, T. Shao, S. S. K. Bekaroglu and T. Karanfil, Environ. Sci. Technol., 2009, 43, 5719–5725 CrossRef CAS PubMed.
- E. Bertels, K. Bruyninckx, M. Kurttepeli, M. Smet, S. Bals and B. Goderis, Langmuir, 2014, 30, 12200–12209 CrossRef CAS PubMed.
- T. Gegenhuber, A. H. Gröschel, T. I. Löbling, M. Drechsler, S. Ehlert, S. Förster and H. Schmalz, Macromolecules, 2015, 48, 1767–1776 CrossRef CAS.
- B. Koh and W. Cheng, Langmuir, 2014, 30, 10899–10909 CrossRef CAS PubMed.
- J. Shi, Z.-X. Guo, B. Zhan, H. Luo, Y. Li and D. Zhu, J. Phys. Chem. B, 2005, 109, 14789–14791 CrossRef CAS PubMed.
- S. Vural, K. B. Dikovics and D. M. Kalyon, Soft Matter, 2010, 6, 3870–3875 RSC.
- S. R. Shin, B. Aghaei-Ghareh-Bolagh, T. T. Dang, S. N. Topkaya, X. Gao, S. Y. Yang, S. M. Jung, J. H. Oh, M. R. Dokmeci, X. S. Tang and A. Khademhosseini, Adv. Mater., 2013, 25, 6385–6391 CrossRef CAS PubMed.
- L. Y. Yan, H. Chen, P. Li, D.-H. Kim and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2012, 4, 4610–4615 CAS.
- S. Hashmi, A. GhavamiNejad, F. O. Obiweluozor, M. Vatankhah-Varnoosfaderani and F. J. Stadler, Macromolecules, 2012, 45, 9804–9815 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- W.-C. Lin, W. Fan, A. Marcellan, D. Hourdet and C. Creton, Macromolecules, 2010, 43, 2554–2563 CrossRef CAS.
- H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- X. Zhang, C. L. Pint, M. H. Lee, B. E. Schubert, A. Jamshidi, K. Takei, H. Ko, A. Gillies, R. Bardhan, J. J. Urban, M. Wu, R. Fearing and A. Javey, Nano Lett., 2011, 11, 3239–3244 CrossRef CAS PubMed.
- P. W. Barone, H. Yoon, R. Ortiz-García, J. Zhang, J.-H. Ahn, J.-H. Kim and M. S. Strano, ACS Nano, 2009, 3, 3869–3877 CrossRef CAS PubMed.
- R. A. MacDonald, C. M. Voge, M. Kariolis and J. P. Stegemann, Acta Biomater., 2008, 4, 1583–1592 CrossRef CAS PubMed.
- T. Lane, J. L. Holloway, A. H. Milani, J. M. Saunders, A. J. Freemont and B. R. Saunders, Soft Matter, 2013, 9, 7934–7941 RSC.
- Z. Cui, A. H. Milani, P. J. Greensmith, J. Yan, D. J. Adlam, J. A. Hoyland, I. A. Kinloch, A. J. Freemont and B. R. Saunders, Langmuir, 2014, 30, 13384–13393 CrossRef CAS PubMed.
- W. Wang, A. H. Milani, L. Carney, J. Yan, Z. Cui, S. Thaiboonrod and B. R. Saunders, Chem. Commun., 2015, 51, 3854–3857 RSC.
- J. Hur, K. Im, S. W. Kim, J. Kim, D.-Y. Chung, T.-H. Kim, K. H. Jo, J. H. Hahn, Z. Bao, S. Hwang and N. Park, ACS Nano, 2014, 8, 10066–10076 CrossRef CAS PubMed.
- L. Bokobza, Polym. Adv. Technol., 2012, 23, 1543–1549 CrossRef CAS.
- A. Jorio, M. A. Pimenta, A. G. Souza-Filho, R. Salto, G. Dresselhaus and M. S. Dresselhaus, New J. Phys., 2003, 5, 139.1–139.17 CrossRef.
- S. Osswald, M. Havel and Y. Gogotsi, J. Raman Spectrosc., 2007, 38, 728–736 CrossRef CAS.
- S. K. Samanta, M. Fritsch, U. Scherf, W. Gomulya, S. Z. Bisri and M. A. Loi, Acc. Chem. Res., 2014, 47, 2446–2456 CrossRef CAS PubMed.
- J. C. Grunlan, A. R. Mehrabi, M. V. Bannon and J. L. Bahr, Adv. Mater., 2004, 16, 150–153 CrossRef CAS.
- D. H. Marsh, G. A. Rance, M. H. Zaka, R. J. Whitby and A. N. Khlobystov, Phys. Chem. Chem. Phys., 2007, 9, 5490–5496 RSC.
- V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge, R. E. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379–1382 CrossRef CAS.
- X. Yao, H. Yao and Y. Li, J. Mater. Chem., 2009, 19, 6516–6520 RSC.
- M. K. Bayazit, L. S. Clarke, K. S. Coleman and N. Clarke, J. Am. Chem. Soc., 2010, 132, 15814–15819 CrossRef CAS PubMed.
- Sudha, B. M. Mishra and D. Kumar, Part. Sci. Technol., 2014, 32, 624–631 CrossRef CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- T. Thanpitcha, A. Sirivat, A. M. Jamieson and R. Rujiravanit, Carbohydr. Polym., 2006, 64, 560–568 CrossRef CAS.
- W. Bauhofer and J. Z. Kovacs, Compos. Sci. Technol., 2009, 69, 1486–1498 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- J. K. W. Sandler, J. E. Kirk, I. A. Kinloch, M. S. P. Shaffer and A. H. Windle, Polymer, 2003, 44, 5893–5899 CrossRef CAS.
- M. Kharaziha, S. R. Shin, M. Nikkhah, S. N. Topkaya, N. Masoumi, N. Annabi, M. R. Dokmeci and A. Khademhosseini, Biomaterials, 2014, 35, 7346–7354 CrossRef CAS PubMed.
- A. Jumahat, C. Soutis, F. R. Jones and A. Hodzic, Plast., Rubber Compos., 2012, 41, 225–232 CrossRef CAS.
- F. L. Acosta, Jr., L. Metz, H. D. Adkisson, J. Liu, E. Carruthers-Liebenberg, C. Milliman, M. Maloney and J. C. Lotz, Tissue Eng., Part A, 2011, 17, 3045–3055 CrossRef PubMed.
- E. Steck, H. Bertram, R. Abel, B. Chen, A. Winter and W. Richter, Stem Cells, 2005, 23, 403–411 CrossRef CAS PubMed.
- N. Lachman, Y. Harel, A. Green, N. Iuster, J.-P. Lellouche and H. D. Wagner, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 957–962 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm00223d |
| This journal is © The Royal Society of Chemistry 2016 |