
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA resorcinarene for inhibition of Aβ fibrillation†
Xu
Han
a,
Jiyong
Park
 b,
Wei
Wu
c,
Andres
Malagon
d,
Lingyu
Wang
c,
Edgar
Vargas
d,
Athula
Wikramanayake
c,
K. N.
Houk
*b and
Roger M.
Leblanc
*a
b,
Wei
Wu
c,
Andres
Malagon
d,
Lingyu
Wang
c,
Edgar
Vargas
d,
Athula
Wikramanayake
c,
K. N.
Houk
*b and
Roger M.
Leblanc
*a
aDepartment of Chemistry, Cox Science Center, University of Miami, Coral Gables, Florida 33146, USA. E-mail: rml@miami.edu
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: houk@chem.ucla.edu
cDepartment of Biology, Cox Science Center, University of Miami, Coral Gables, Florida 33146, USA
dDepartamento de Quimica, Universidad de los Andes, Cr. 1 No. 18A 10, Bogota 111711, Colombia
First published on 17th November 2016
Abstract
Amyloid-β peptides (Aβ) fibrillation is the hallmark of Alzheimer's disease (AD). However, it has been challenging to discover potent agents in order to inhibit Aβ fibrillation. Herein, we demonstrated the effect of resorcinarene on inhibiting Aβ fibrillation in vitro via experimental and computational methods. Aβ were incubated with different concentrations of resorcinarene so as to monitor the kinetics by using thioflavin T binding assay. The results, which were further confirmed by far-UV CD spectroscopy and atomic force microscopy, strongly indicated that the higher concentration of resorcinarene, the more effective the inhibition of Aβ fibrillation. A cytotoxicity study showed that when sea urchin embryos were exposed to the resorcinarene, the majority survived due to the resorcinarene low toxicity. In addition, when the resorcinarene was added, the formation of toxic Aβ 42 species was delayed. Computational studies of Aβ fibrillation, including docking simulations and MD simulations, illustrated that the interaction between inhibitor resorcinarene and Aβ is driven by the non-polar interactions. These studies display a novel strategy for the exploration of promising antiamyloiddogenic agents for AD treatments.
Introduction
Alzheimer's disease (AD), the most common form of dementia, causes progressive memory loss, behavior, and thinking problems.1,2 It has become a major threat for human beings around the globe. Approximately 24.3 million people suffer from AD in the world nowadays.2,3 In accordance with a recent report,4 the number of the AD patients is expected to increase up to 81.1 million by 2040. Over the past 30 years, the etiology of the disease has been attributed to the accumulation of the extracellular plaques, which are mainly composed of amyloid-β peptides (Aβ), and intracellular tangles formed by tau proteins.5,6 Much scientific research7–12 has proven that sequentially cleavage of amyloid precursor protein (APP) by BACE1 and β- and γ-secretases can produce Aβ monomer, and the aggregation of Aβ monomer into amyloid fibrils is strongly associated with AD.13 In other words, Aβ plays a central role in the pathogenesis of AD. Both Aβ 42 and Aβ 40 are primary isoforms of Aβ, where Aβ 42 is considered as the most toxic form,14 and Aβ 40 is the most abundant form.15 The development of therapeutic agents targeting upstream secretases had been unsuccessful.16 Thus the development of an inhibitor for early stage Aβ fibrillation could be a promising treatment of AD.17The major Aβ contains 40 or 42 amino acids with residues from 18 to 42 forming two parallel β sheets.18 Significant efforts and progress have gone into developing new amyloid fibrillation inhibitors. Aβ fibrillation inhibitors are mainly designed in two different ways: peptides or peptide mimetics19–23 and organic compounds.10,24–27 However, the challenge to design small molecules for Aβ fibrillation is because protein–protein interaction regions, approximately 20 nm2, are larger than the protein-small molecule interaction regions which are around 3–10 nm2.28 Most small molecules, therefore, cannot afford adequate steric hindrance to prevent the aggregation. Supramolecular strategy to block the aggregation process exhibits another option for potent AD therapies. Lee et al. have demonstrated such a strategy by using cucurbit[7]uril (CB[7]).29 However, the efficiency of the designed inhibitor is low. More inhibitors – the reported lowest effective concentration ratio of the inhibitor to Aβ 42 is approximately 100 – to prevent amyloid fibrillation are needed (Table S1†). In addition to less powerful performance on blocking Aβ aggregation, the cytotoxicity of Aβ 42 fibrils cannot be delayed or inhibited by most other designed inhibitors (Table S1†). It is, thus, in dire need to develop inhibitors with sufficient potency to delay the onset of Aβ fibrillation and cytotoxicity of Aβ 42.
The utility of resorcinarene has expanded greatly in the field of supramolecular chemistry and biochemistry.30–32 Not only the semi-rigid structure and the π-electron rich cavity, but also the hydrogen bonding sites of the molecule, provide an excellent platform for the host–guest complexation.33,34 These compounds can be functionalized in diverse manners for different applications, such as recognition of amino acids as well as delivery tool in pharmacology.35–37 Inspired by such vital applications in host–guest chemistry, we turned to the functionalized resorcinarene as a potential strategy towards the inhibition of Aβ amyloid fibrillation for the first time. Fig. 1 shows the Aβ fibrillation pathway, where the partial denaturation of the peptide monomer initiates the fibrillation route. The denatured monomers self-assemble into oligomers and in turn form fibrils via protofibril formation. In a set of sequential events, resorcinarene is expected to interact with Aβ in a way that increases the steric hindrance for fibril aggregation and abolishes the Aβ fibrillation process which may delay the toxic Aβ 42 as well. Herein, we studied such a hypothesis by experimental method, including thioflavin T (ThT) binding assay, circular dichroism spectroscopy (CD), atomic force microscopy (AFM), and cytotoxicity assay, as well as theoretical approach using docking calculations and MD simulations. These results demonstrate a great potential application of resorcinarene in retarding Aβ fibrillation, and propose a new possibility for further optimization of the inhibitor design. In particular, resorcinarene as a delivery system with recognition and encapsulation properties will emerge as a promising agent for therapies of Alzheimer's disease.
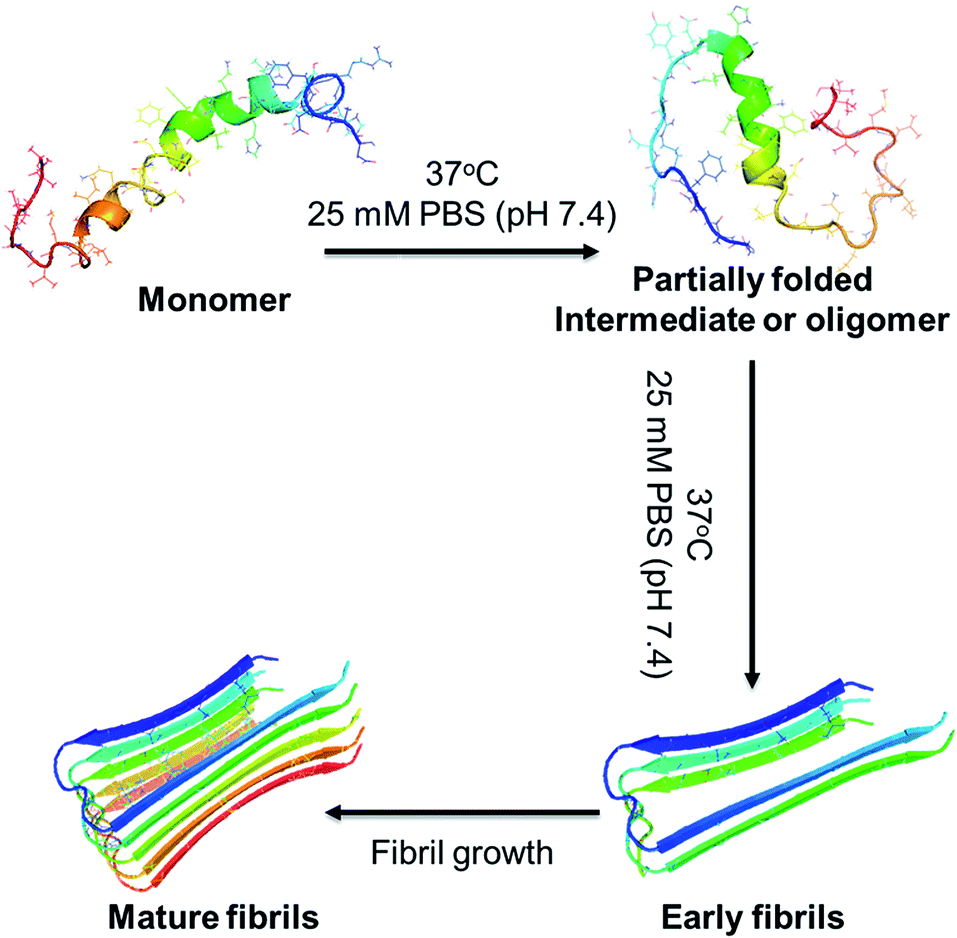 | ||
| Fig. 1 A schematic overview in vitro study of Aβ fibrillation, which includes the conformational transition from monomer to partially folded intermediate or oligomer to mature fibrils. | ||
Experimental section
Synthesis of resorcinarene
The resorcinarene was prepared according to our previous published literature procedures.38 All characterizations, including NMR, mass-spec, and X-ray crystallography, are consistent with previous reported results. Briefly, the resorcinarene precursor was added to a solution which contained formaldehyde, sodium sulfide, and water, and stirred for 4 h at 90–95 °C. The mixture was cooled down to room temperature, and neutralized with hydrochloric acid. Acetonitrile was added to precipitate sulfonated resorcinarene. Finally, the resulting product 1 (Scheme 1) was recrystallized and washed in the same solvent acetonitrile.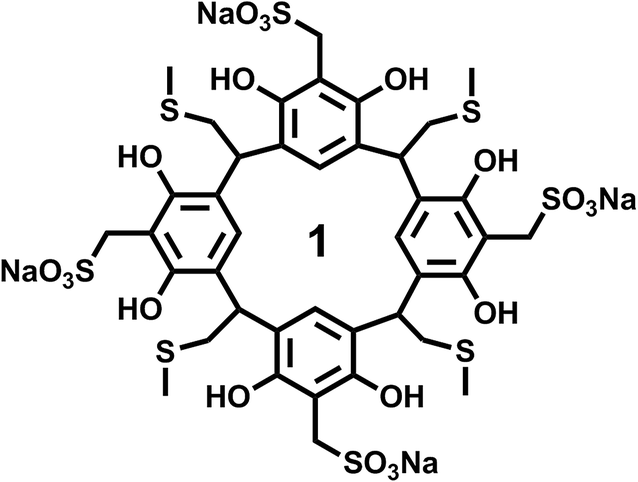 | ||
| Scheme 1 The structure of resorcinarene. | ||
Thioflavin T (ThT) fluorescence assay
The kinetics of Aβ 42 and Aβ 40 fibrillation can be characterized by the fluorescence increase of fibril-specific dye of thioflavin T (ThT). To accomplish this, different concentrations of resorcinarene, including the ratio of peptides to the resorcinarene at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 1:0.1, 1:1, and 1:5, were prepared for examination. At first, Aβ 42 and Aβ 40 were dissolved in 1,1,1,3,3,3-hexafluoroisopropanol to stabilize the α-helix structure of the peptides. After evacuating the solvent, the solution was filtered through a 0.2 μm membrane to remove the pre-fibrils. Different concentrations of resorcinarene were added to 10 μM Aβ in 25 mM PBS buffer solution (pH 7.4), respectively. Thereafter, the mixture were incubated at 37 °C, and aliquots of incubation solutions at different time points were collected and then diluted two times into ThT solution (25 μM ThT in 25 mM PBS buffer, pH 7.6). ThT fluorescence was measured by a fluorescence spectrophotometer (LS-55, Perkin Elmer) at 25 °C with a slit width of 5 nm for both excitation and emission. ThT emission was monitored at 480 nm with the excitation at 440 nm.
:0, 1:0.1, 1:1, and 1:5, were prepared for examination. At first, Aβ 42 and Aβ 40 were dissolved in 1,1,1,3,3,3-hexafluoroisopropanol to stabilize the α-helix structure of the peptides. After evacuating the solvent, the solution was filtered through a 0.2 μm membrane to remove the pre-fibrils. Different concentrations of resorcinarene were added to 10 μM Aβ in 25 mM PBS buffer solution (pH 7.4), respectively. Thereafter, the mixture were incubated at 37 °C, and aliquots of incubation solutions at different time points were collected and then diluted two times into ThT solution (25 μM ThT in 25 mM PBS buffer, pH 7.6). ThT fluorescence was measured by a fluorescence spectrophotometer (LS-55, Perkin Elmer) at 25 °C with a slit width of 5 nm for both excitation and emission. ThT emission was monitored at 480 nm with the excitation at 440 nm.
Circular dichroism (CD) spectroscopy
To gain insights into the effects of resorcinarene on the conformational transition of Aβ, far-UV CD spectroscopy (JASCO J-810) were utilized to monitor the secondary structure of Aβ over time.39 Similar to the fluorescence characterization, aliquots of incubation solutions described above were collected for CD spectra as well. The spectra was recorded between 190 and 260 nm at room temperature by a 2 mm optical path length quartz cell.Atomic force microscopy (AFM)
AFM images of samples withdrawn at different incubation times can be studied to directly observe and monitor the formation of fibrils. Tapping mode was used to observe morphologies during the fibrillation process. The cantilever has a resonance frequency of approximately 170 kHz with typical force constant of 7.5 N m−1. To scan the AFM images, aliquots of samples withdrawn at different incubation times were diluted with pure water, drop-coated on a freshly cleaved mica surface, and allowed to dry for at least 1 h.Cytotoxicity assay
Toxicity tests were performed in a new 24-well cell culture plate. In each well, health fertilized eggs were incubated in 2 ml control sea water or 2 ml dilution of 1 in sea water. For the 1 cytotoxicity test, the plate with fertilized eggs (Strongylocentrotus purpuratus sea urchins) was incubated at 15 °C for 48 hours until they reach prism stage embryos. Aβ 42 fibrils and different Aβ 42–1 complex were collected from incubated solutions. The plate with fertilized egg (Lytechinus variegatus sea urchins) to test the inhibiting effect of 1 on Aβ 42 toxic species were performed at room temperature for 24 hours until they reach prism stage embryos. 100 of embryos were examined and rate of embryo with normal development was calculated. Each experiment was repeated with similar results. The error bars indicate the standard error of the mean.Ligand docking and molecular dynamics simulations
Possible binding modes of the resorcinarene to Aβ 42 amyloid fibrils were simulated using Potential Energy Landscape Explorer web-server (PELE: http://pele.bsc.es/pele.wt).40 We hypothesized that the resorcinarene can bind to the ends of the filament and delay the amyloid fiber growth by interfering the monomer addition. Accordingly, two most populated conformations from the PELE simulations with the ligand bound on the top or the bottom of the filament were selected for the initial configurations of the subsequent molecular dynamics simulations. Molecular dynamics (MD) simulations of the ligand bound amyloid fibrils were carried out using Amber14 software.41 The molecular mechanics parameters of the resorcinarene was prepared using the Generalized Amber Force Fields (GAFF).42 Atomic partial charges of the ligand were computed using the AM1-BCC charge model embedded in the antechamber program (Table S2†). The amyloid filament was modelled using the AmberFF14SB force fields.43 The MD simulations were initiated from the two most populated structures found from the PELE simulations. Each initial structure was solvated in an octahedral box filled with TIP3P water molecules, where the solute molecule has 11 Å margin from the solvation boundary. Counter ions were added to guarantee the charge neutrality. Each solvated system was equilibrated at 300 K for 6 ns, while keeping positions of all carbon atoms at initial values. 100 ns production runs were followed, while restraining alpha carbons of the filament at initial positions using weak harmonic potentials (spring constant was 0.1 kcal mol−1 Å−2).Results and discussion
Inhibitory effect of resorcinarene on Aβ fibrillation
The cationic benzothiazole dye, thioflavin T (ThT), is well known for its specific staining of amyloid fibrils, which enhances emission fluorescence. Fig. 2a shows the aggregation kinetics of Aβ fibril formation at different concentrations of 1 by the time dependent ThT fluorescence assay. When incubating 10 μM Aβ 42 alone, the ThT fluorescent profile presented a lag phase within 3 h, a slow elongation, and a saturated phase after 3 days, while Aβ 40 displayed a similar but faster fibrillation process with an almost negligible lag phase which maintained less than 1 h, an elongation phase within 3 h, and a saturated phase after 4 h. To evaluate the inhibition activity of 1 against Aβ fibrillation, Aβ were present with different concentrations of 1 at the ratio of the resorcinarene to peptides of 0.1, 1, and 5, respectively. At a Aβ 42/1 ratio of 0.1, suppression of Aβ 42 aggregation was observed. The elongation phase did not start until incubating for 12 h. In contrast to this, it took Aβ 42 3 h to enter the same phase in the absence of 1. Adding more 1, the fluorescent intensity decreased by over 50% at the plateau. At Aβ 42/1 ratio of 5, almost no fibrils were observed as evidenced by the nearly flat curve comparing to others. Overall, ThT profiles demonstrated that 1 inhibited Aβ 42 fibrillation in a dose dependent manner. 1 displayed a similar but more effectively inhibition on Aβ 40. 1 began to exhibit inhibition effect at Aβ 40/1 ratio of 0.1. When the Aβ 40/1 ratio increased up to 1, the lag phase period extended to approximately 4 h. If the concentration of 1 was further increased, 1 showed a stronger effect of fibrillation inhibition, which retained active for 5 days. As control experiments, resorcinarene was incubated alone at 37 °C for 24 h, and the fluorescence spectra of not only resorcinarene (Fig. S1†), also resorcinarene with 20 μM ThT (Fig. S2†) showed no disturb towards ThT assay tests above. To note, it is surprising to observe that Aβ 40 aggregated faster than Aβ 42. It may attribute to the protein purity since impure protein tends to aggregate more slowly. However, in accordance with Table S1† and results above, 1 exhibited excellent potential for effectively inhibiting both Aβ 42 and Aβ 40 fibrillation. The other reason leading to the faster Aβ 40 aggregation might be that the prepared amyloid-β peptides may contain seeds which can accelerate the fibrillation process.44–46 To further confirm our hypothesis, we carried out additional series of measurements with regard to the aggregation of kinetics of Aβ 42 and 40. Preformed fibrils (seeds)47 were added to Aβ 42 and Aβ 40 at the beginning of incubation, respectively. By recording ThT assay of aggregation kinetics, the lag phase time of both amyloid-β peptides with additional seeds was found to decrease comparing to that without added seeds. In other words, both Aβ 42 and Aβ 40 with additional seeds aggregate faster than those without seeds.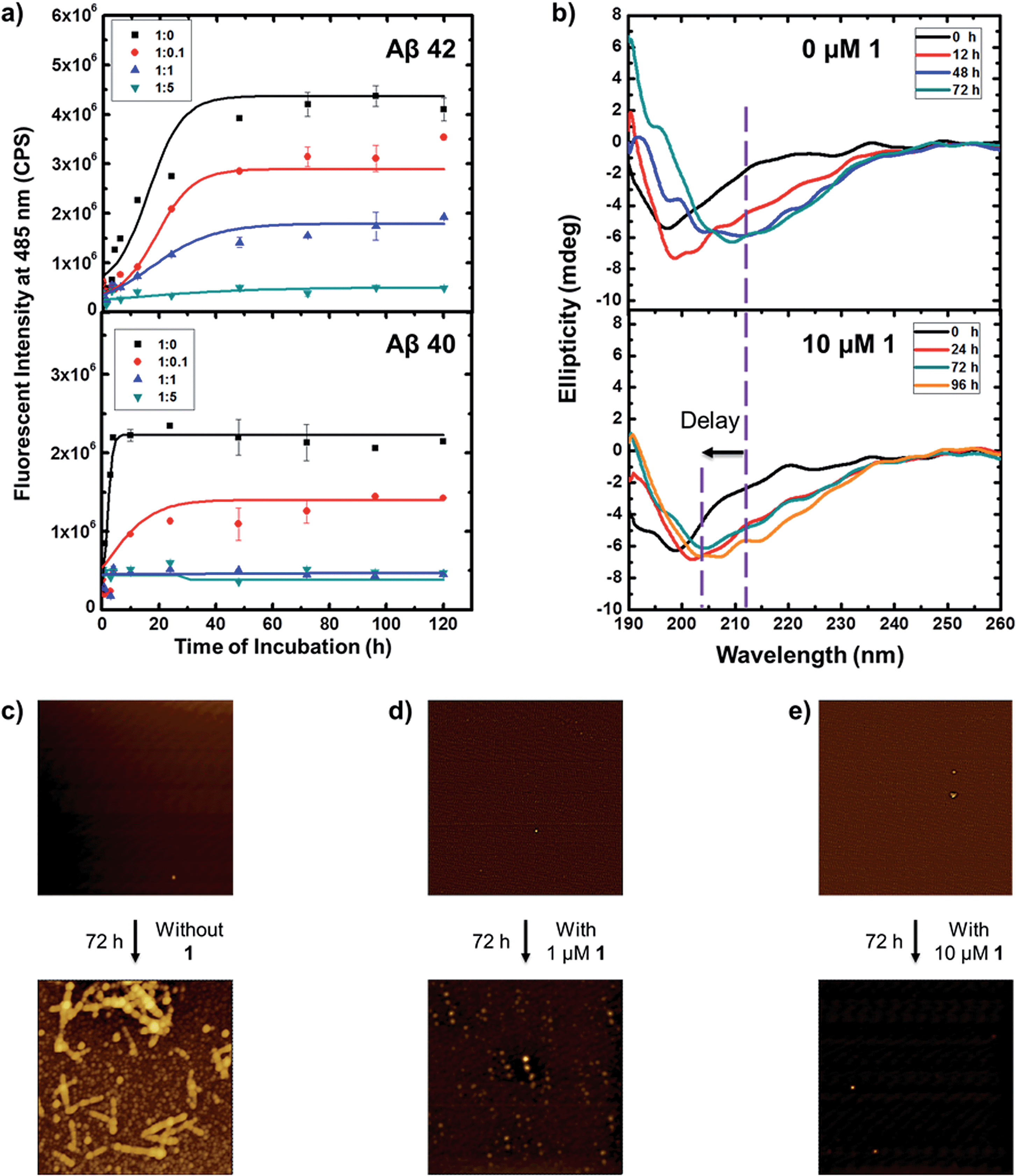 | ||
| Fig. 2 (a) Kinetics of 10 μM of Aβ 42 and Aβ 40 fibrillation: fluorescence intensity of thioflavin T (ThT) at 485 nm as a function of incubation time at 37 °C in 25 mM PBS, pH 7.4 with the ratio of Aβ to 1 at 1:0, 1:0.1, 1:1, and 1:5, respectively. The final concentration was a 2-fold dilution with 20 μM ThT. Baseline was corrected against the spectra of 1 (Fig. S2†). The ThT fluorescence was obtained for three repeats of each sample. The error bars indicate the standard error of the mean. (b) Far-UV circular dichroism spectra of 10 μM Aβ 42 alone (top panel) in 25 mM pH = 7.4 PBS at 0, 12, 48, 72 h, and 10 μM Aβ 42 incubated with 10 μM 1 (bottom panel), in 25 mM pH = 7.4 PBS at 0, 24, 48, 96 h. AFM images (size: 2.5 × 2.5 μm) of 10 μM Aβ 42 incubated at 37 °C in 25 mM PBS, pH 7.4 with (c) 0 μM, (d) 1 μM, and (e) 10 μM 1, respectively. | ||
The Aβ peptides are mainly random coil or α-helical in the native conformation. They undergo a conversion to β-strand during the fibril formation, which is consistent with ThT assay examination. To attain a clear perception of the effect of 1 on the conformational conversion of Aβ 42 and 40 upon aggregation, far-UV CD spectroscopy was employed to monitor the secondary structure of the Aβ peptides over time (Fig. 2b). Aliquots of Aβ 42 and 40 in the absence and presence of inhibitor were collected for CD spectra at different time, respectively. Even though peptides were incubated in PBS buffer which has an influence on the CD spectra, the spectra obtained is still distinguishable. As shown in Fig. 2b, Aβ 42 alone at incubation time 0 had a negative band at around 197 nm, indicating that the initial secondary structure of Aβ 42 adopted a random coil form. After incubating 12 hours, this peak diminished and shifted slightly to approximately 200 nm, suggesting the conformational change of Aβ 42. Upon peptides aggregation for 48 hours, a negative valley appeared at 213 nm, which is assigned to β sheet of mature peptide fibril. When present with 10 μM 1, Aβ 42 showed a similar structure transition with adopting mainly random coil conformations as evidenced from Fig. 2b. However, Aβ 42 did not completely convert to β sheet conformation even after incubating 96 hours comparing to the study of Aβ 42 in the absence of 1. That is, 1 can potentially delay the conformational transition of Aβ 42. In the case of Aβ 40, Fig. S5† showed a mixture of random coil, β sheet, and β turn forms. Upon peptide aggregation, bands shrink demonstrating conformational change severely where a significant decrease in the negative absorption around 200 nm. The final content of β strand conformers at 4 h was much more than that of initial Aβ 40. In the presence of 1, it displayed a similar random coil dominated conformations, but undergoing no transition with 24 h incubation, suggesting a delay of fibrillation process (Fig. S6†). It can be concluded that the inhibitory effect of 1 on conformational transition of Aβ aggregation from random coils to β strand.
To directly observe and monitor the formation of these Aβ 40 and 42 fibrils, AFM images were obtained in the absence and presence of 1. The inhibition effect of 1 on Aβ peptides fibrillation is consistent with the previous observations. As illustrated in Fig. 2c, Aβ 42 fibrils were apparent after 72 h incubation in the absence of 1, whereas when present with 1, especially with higher concentration, no long fibrils showed up during the incubation. Aβ 40 went through a similar fibrillation process with a higher rate. Fibrils can be observed clearly only after 24 h incubation (Fig. S7†), in contrast to the fact that no long fibrils but a few protofibrils were recognized when incubating with the inhibitor. To further confirm the contents of the ThT endpoint samples, a pellet assay with an SDS-PAGE gel was prepared.47 Fig. S4† showed that fibrils from the solution without 1 are much larger than that with 1, which again reveals that 1 can retard the aggregation of both amyloid-β peptides.
To note, all the tested Aβ and resorcinarene mixtures contained the same amount of Aβ, and any increase in the fluorescent intensity from ThT assay profile may indicate more fibrils were formed. It is interesting to observe that the final ThT intensity of resorcinarene mixed Aβ solution decreased and stabilized much lower than that of Aβ alone, meaning less fibrils were produced. The amyloid fibril formation is a multi-stage process that depends on the formation of small oligomeric species.9,48,49 With regard to the inhibition effect, we speculate that resorcinarene interact with Aβ filament at early stage. The bound resorcinarene impede inter oligomers association by steric hindrance and delay the onset of fibril growth. The hypothesis is in accordance with CD and AFM study where Aβ fibrils were apparent in the absence of resorcinarene while a few oligomers were formed in the presence of the inhibitor.
Cytotoxicity test of resorcinarene to sea urchin embryos
The effective inhibition of Aβ fibril formation by 1 suggests the potential capability of 1 for therapy of AD application. A series of cell viability tests were conducted to evaluate the cytotoxicity of 1 in the cellulous environment in which sea urchin embryos were selected as a model system due to its extremely sensitive to hazardous materials. Also sea urchin embryos have been widely used for static acute toxicity of many chemicals test. Strongylocentrotus purpuratus sea urchins (Marinus Scientific, Long Beach, CA) were utilized in this study to test the toxicity of 1, and adult animals with ripe gonads were used to collect gametes. Fresh eggs were washed for 3 times with cold filtered artificial sea water and mixed with sperm to examine fertilization rate. Only eggs with a fertilization rate greater than 95% were used for the toxicity tests. Three biological replicates using three individual male–female pairings were performed. Fig. 3a showed 90% of embryos retain a normal morphology after 20 h of incubation with the presence of 20 μg ml−11, in which concentration already exhibited significant effect on inhibition of Aβ fibrillation, indicating low cytotoxicity of 1 to cells. Unfortunately, since sea urchins are extremely sensitive to invading materials, when the concentration of 1 increasing up to 1000 μM, most of the cells failed to survive under the incubation environment. The effective inhibition of Aβ 42 cytotoxicity by 1 can suggest potential utility for therapeutic applications. A series of cell viability tests were performed to measure such effects induced by 1. Pure Aβ 42 fibril reduced the viability of sea urchins cells (Lytechinus variegatus) in normal forms to 0%, whereas the Aβ 42–1 complex delayed the formation of Aβ 42 toxic species, where the rate of normal embryos increased to 81% with 50 μM 1 (Fig. 3b). In addition to the normal form of sea urchin cells, there exist some cells alive but unable to swim. In Fig. S8,† 92% of embryos are alive but in abnormal forms when present with 10 μM pure Aβ 42 fibrils. Applying more 1, the rate decreased, which indicated the reducing toxicity of Aβ 42 fibrils induced by 1.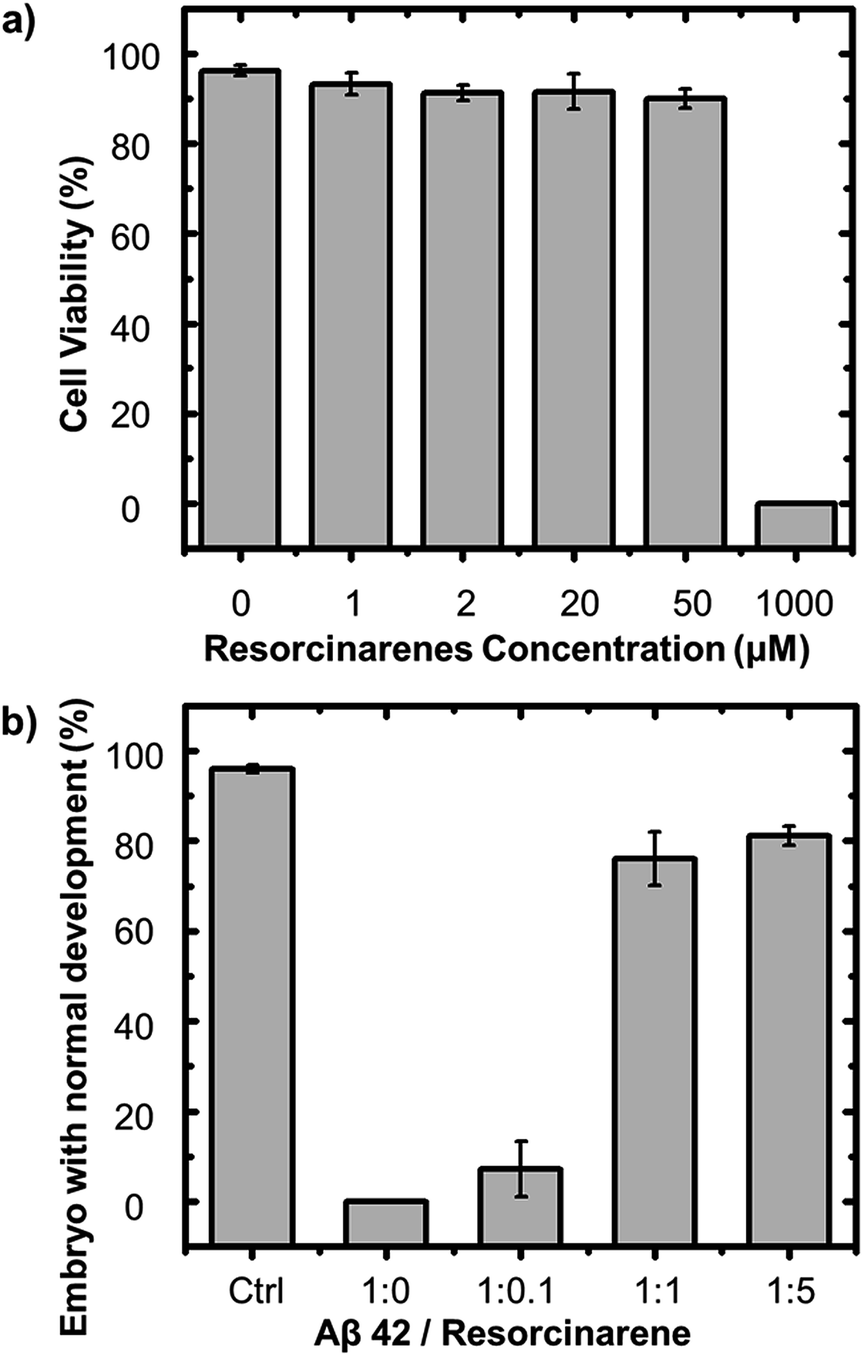 | ||
| Fig. 3 (a) Cytotoxicity test of resorcinarene to seaurchin embryos. (b) The inhibitory effect of resorcinarene on the cytotoxicity of Aβ 42 fibrils at different molar ratios of Aβ 42 to resorcinarene. | ||
Resorcinarene binds to the top and the bottom of the Aβ 42 filament
The detailed interactions between the resorcinarene and the amyloid filament were studied using molecular docking and molecular dynamics (MD) simulations. The MD simulations were initiated with bound resorcinarene on the top or the bottom of the amyloid fibril (Fig. S6†). The bound conformations were identified from the results of the docking simulations using the PELE software. Fig. 4 shows two of the binding modes of the resorcinarene resulting from the 100 ns MD simulations. After 80 ns of MD simulations, the root-mean-squared deviation (RMSD) of all heavy atoms of the bound resorcinarene converged to <3.0 Å from the last configuration at 100 ns. Of note, the RMSD to the initial docked conformation increased to >4.0 Å as MD simulation progressed, that suggests a conformational relaxation took place during the course of MD simulations. The solvent accessible surface area of the ligand-amyloid fiber complex was always smaller than that of the isolated ligand and amyloid fiber. The finding indicates the ligand is always in contact with the amyloid fibril. The averaged buried solvent accessible surface area (ΔSASA) over the last 20 ns was −767 Å2 for the mode 1 and −845 Å2 for the mode 2. We also computed surface area dependent non-polar hydrophobic energies using the formula proposed by Honig's group: Ehphobic = γ × ΔSASA, where γ is 0.05 kcal mol−1 Å−2.50 The computed non-polar hydrophobic energies averaged over the last 30 ns MD simulations are −38 kcal mol−1 and −42 kcal mol−1 for the mode 1 and the mode 2, respectively. The amount of the non-polar interaction energies are sufficient to overcome translational–rotational entropic penalty that is +26 kcal mol−1 less favorable for binding. We analyzed favorable interactions across the interface of the bound resorcinarene and the amyloid fibril. In the first binding mode (Fig. 4a), thiomethyl groups of the resorcinarene were in contact with methyl group of Ala21 in the amyloid bilayer interface. Also a sp2 carbon of resorcinol is in contact with the sidechain of Leu34 and Phe19. Likewise, in the second binding mode (Fig. 4b), thiomethyl groups make non-polar contacts with Val36 and Leu34. A sp2 carbon of resorcinol is in contact with the sidechain of Phe19.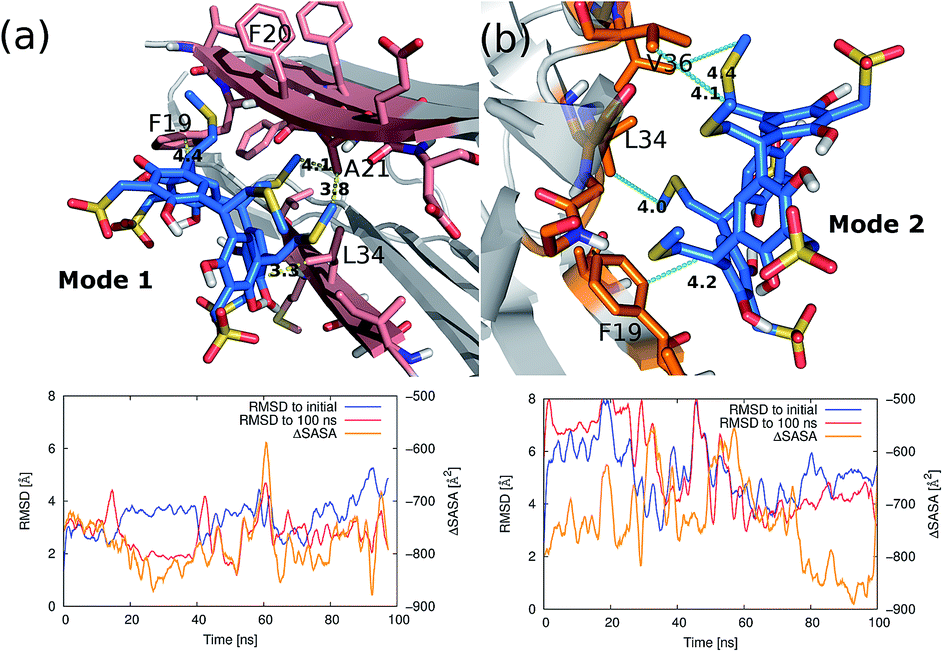 | ||
| Fig. 4 Predicted interactions of the resorcinearene and the Aβ 42 amyloid filament. Two of the relatively stable interactions patterns are predicted using the PELE and the MD simulations: (a) and (b). The last snapshots from the MD simulations are illustrated. Root-mean-squared deviations from the initial docked structure (blue), the last configuration of the MD simulation (red), and the buried surface area of the ligand–protein interface (orange) of each are plotted on bottom. | ||
Conclusions
In summary, the effective inhibition of Aβ fibrillation by resorcinarene was demonstrated in vitro by the ThT assay, the circular dichroism spectroscopy, and the atomic force microscopy. Using the computational methods, including docking simulations and MD simulations, the binding of resorcinarene was shown to be mediated by non-polar interactions. The advantages that could arise with resorcinarene that is significantly better than others at Aβ fibrillation inhibition include: (1) lower concentration required to block Aβ aggregation, (2) low toxicity to seaurchin embryo, which is extremely sensitive to hazardous materials, and (3) the formation of Aβ toxic species can be delayed, which all make our designed inhibitor a promising candidate for the treatment of Alzheimer's disease. To our knowledge, it is the first time for the application of resorcinarene on the Aβ fibrillation. Thus, with the remarkable inhibitory function towards Aβ fibrillation and its capability in encapsulation, resorcinarene presented in this work shows great potential to be applied in pharmaceutical industry or other areas for the treatment of Alzheimer's disease.Acknowledgements
R. M. L. gratefully acknowledges the support of the National Science Foundation under Grant 1355317. J. P. and K. N. H. are grateful for utilizing the computational resources of Hoffman2 cluster from UCLA Institute for Digital Research and Education and XSEDE program (TGCHE040013N). A. H. W. is funded by a grant from NSF (IOS1257967).Notes and references
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777 CrossRef CAS PubMed.
- B. Duthey, A Public Health Approach to Innovation, 2013, p. 1 Search PubMed.
- M. P. Mattson, Nature, 2004, 430, 631 CrossRef CAS PubMed.
- C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Q. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer, M. Scazufca and A. D. Intl, Lancet, 2005, 366, 2112 CrossRef.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353 CrossRef CAS PubMed.
- B. A. Yankner, L. K. Duffy and D. A. Kirschner, Science, 1990, 250, 279 CAS.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384 CrossRef CAS PubMed.
- M. Zhang, X. B. Mao, Y. Yu, C. X. Wang, Y. L. Yang and C. Wang, Adv. Mater., 2013, 25, 3780 CrossRef CAS PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188 CrossRef CAS PubMed.
- J. Habchi, P. Arosio, M. Perni, A. R. Costa, M. Yagi-Utsumi, P. Joshi, S. Chia, S. I. A. Cohen, M. B. D. Müller, S. Linse, E. A. A. Nollen, C. M. Dobson, T. P. J. Knowles and M. Vendruscolo, Sci. Adv., 2016, 2, e1501244 Search PubMed.
- C. Geula, C. K. Wu, D. Saroff, A. Lorenzo, M. L. Yuan and B. A. Yankner, Nat. Med., 1998, 4, 827 CrossRef CAS PubMed.
- P. Arosio, M. Vendruscolo, C. M. Dobson and T. P. Knowles, Trends Pharmacol. Sci., 2014, 35, 127 CrossRef CAS PubMed.
- N. Suzuki, T. T. Cheung, X. D. Cai, A. Odaka, L. Otvos, C. Eckman, T. E. Golde and S. G. Younkin, Science, 1994, 264, 1336 CAS.
- S. I. A. Cohen, P. Arosio, J. Presto, F. R. Kurudenkandy, H. Biverstal, L. Dolfe, C. Dunning, X. T. Yang, B. Frohm, M. Vendruscolo, J. Johansson, C. M. Dobson, A. Fisahn, T. P. J. Knowles and S. Linse, Nat. Struct. Mol. Biol., 2015, 22, 207 CAS.
- D. J. Selkoe, Nature, 1999, 399, A23 CrossRef CAS PubMed.
- P. T. Lansbury, Curr. Opin. Chem. Biol., 1997, 1, 260 CrossRef CAS PubMed.
- R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030 CrossRef CAS PubMed.
- D. B. Teplow, N. D. Lazo, G. Bitan, S. Bernstein, T. Wyttenbach, M. T. Bowers, A. Baumketner, J. E. Shea, B. Urbanc, L. Cruz, J. Borreguero and H. E. Stanley, Acc. Chem. Res., 2006, 39, 635 CrossRef CAS PubMed.
- S. A. Funke and D. Willbold, Curr. Pharm. Des., 2012, 18, 755 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Naslund, F. Lindqvist, J. Johansson, A. R. Karlstrom, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545 CrossRef CAS PubMed.
- M. M. Gessel, C. Wu, H. Y. Li, G. Bitan, J. E. Shea and M. T. Bowers, Biochemistry, 2012, 51, 108 CrossRef CAS PubMed.
- N. Xiong, X. Y. Dong, J. Zheng, F. F. Liu and Y. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 5650 CAS.
- S. A. Sievers, J. Karanicolas, H. W. Chang, A. Zhao, L. Jiang, O. Zirafi, J. T. Stevens, J. Munch, D. Baker and D. Eisenberg, Nature, 2011, 475, 96 CrossRef CAS PubMed.
- X. Han, S. H. Li, Z. L. Peng, A. R. O. Al-Yuobi, A. S. O. Bashammakh, M. S. EI-Shahawi and R. M. Leblanc, J. Oleo Sci., 2016, 65, 1 CrossRef CAS PubMed.
- L. E. Scott, M. Telpoukhovskaia, C. Rodriguez-Rodriguez, M. Merkel, M. L. Bowen, B. D. G. Page, D. E. Green, T. Storr, F. Thomas, D. D. Allen, P. R. Lockman, B. O. Patrick, M. J. Adam and C. Orvig, Chem. Sci., 2011, 2, 642 RSC.
- Q. M. Wang, X. Yu, K. Patal, R. D. Hu, S. Chuang, G. Zhang and J. Zheng, ACS Chem. Neurosci., 2013, 4, 1004 CrossRef CAS PubMed.
- X. Y. Zheng, D. Liu, F. G. Klarner, T. Schrader, G. Bitan and M. T. Bowers, J. Phys. Chem. B, 2015, 119, 4831 CrossRef CAS PubMed.
- Q. Nie, X. G. Du and M. Y. Geng, Acta Pharmacol. Sin., 2011, 32, 545 CrossRef CAS PubMed.
- H. H. Lee, T. S. Choi, S. J. C. Lee, J. W. Lee, J. Park, Y. H. Ko, W. J. Kim, K. Kim and H. I. Kim, Angew. Chem., Int. Ed., 2014, 53, 7461 CrossRef CAS PubMed.
- S. H. Ma, D. M. Rudkevich and J. Rebek, J. Am. Chem. Soc., 1998, 120, 4977 CrossRef CAS.
- M. H. K. Ebbing, M. J. Villa, J. M. Valpuesta, P. Prados and J. de Mendoza, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4962 CrossRef CAS PubMed.
- H. Mansikkamaki, M. Nissinen and K. Rissanen, Angew. Chem., Int. Ed., 2004, 43, 1243 CrossRef PubMed.
- S. D. Bian, S. B. Zieba, W. Morris, X. Han, D. C. Richter, K. A. Brown, C. A. Mirkin and A. B. Braunschweig, Chem. Sci., 2014, 5, 2023 RSC.
- X. Han, Y. T. Zheng, C. J. Munro, Y. W. Ji and A. B. Braunschweig, Curr. Opin. Biotechnol., 2015, 34, 41 CrossRef CAS PubMed.
- R. V. Rodik, V. I. Boyko and V. I. Kalchenko, Curr. Med. Chem., 2009, 16, 1630 CrossRef CAS PubMed.
- B. Mokhtari and K. Pourabdollah, J. Inclusion Phenom. Macrocyclic Chem., 2012, 73, 1 CrossRef CAS.
- L. Mutihac, J. H. Lee, J. S. Kim and J. Vicens, Chem. Soc. Rev., 2011, 40, 2777 RSC.
- E. Sanabria, M. A. Esteso, A. Perez-Redondo, E. Vargas and M. Maldonado, Molecules, 2015, 20, 9915 CrossRef CAS PubMed.
- Z. Peng, S. Li, X. Han, A. Al-Youbi, A. Bashammakh, M. El-Shahawi and R. M. Leblanc, Anal. Chim. Acta, 2016, 937, 113 CrossRef CAS PubMed.
- A. Madadkar-Sobhani and V. Guallar, Nucleic Acids Res., 2013, 41, W322 CrossRef PubMed.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham, I. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696 CrossRef CAS PubMed.
- P. Arosio, R. Cukalevski, B. Frohm, T. P. J. Knowles and S. Linse, J. Am. Chem. Soc., 2014, 136, 219 CrossRef CAS PubMed.
- E. Hellstrand, B. Boland, D. M. Walsh and S. Linse, ACS Chem. Neurosci., 2010, 1, 13 CrossRef CAS PubMed.
- D. M. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, FEBS J., 2009, 276, 1266 CrossRef CAS PubMed.
- See ESI† for details.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758 CrossRef CAS PubMed.
- S. I. A. Cohen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, J. Mol. Biol., 2012, 421, 160 CrossRef CAS PubMed.
- A. Nicholls, K. A. Sharp and B. Honig, Proteins, 1991, 11, 281 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04854d |
| This journal is © The Royal Society of Chemistry 2017 |