
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous interfacial gels assembled from small molecule supramolecular polymers†
Alexander S.
Groombridge‡
,
Aniello
Palma‡
,
Richard M.
Parker
 ,
Chris
Abell
and
Oren A.
Scherman
*
,
Chris
Abell
and
Oren A.
Scherman
*
Department of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: oas23@cam.ac.uk; Fax: +44 (0) 1223 334866; Tel: +44 (0) 1223 331508
First published on 11th October 2016
Abstract
The self-assembly of a stimuli-responsive aqueous supramolecular hyperbranched polymer from small molecules is reported. This system is composed of ditopic and tritopic guest-functionalised molecules that are able to form heteroternary supramolecular complexes with the macrocyclic host cucurbit[8]uril (CB[8]). We demonstrate that the supramolecular hyperbranched polymer formed is responsive to both photo- and chemical stimuli, exhibiting reversibility. Furthermore, this system is shown to assemble at liquid–liquid interfaces, which upon gelation, is observable on the micrometre scale. This self-healing supramolecular network can act as a soft matter barrier for aqueous microdroplets, inhibiting their coalescence.
1 Introduction
Hyperbranched polymers are a class of highly-branched polymers, similar in their properties to dendrimers but with a more facile synthetic pathway to high molecular weight species.1,2 In contrast to dendrimers that are composed of only branched and terminal units, hyperbranched polymers include some unconsumed linear units. The higher the resultant degree of branching, the more compact the polymer structure will be, which results in unique properties such as decreased viscosity of solutions and small molecule encapsulation.1Supramolecular polymers are of great interest due to their inherent adaptability and dynamic nature. Such polymers are assembled by non-covalent interactions, often exploiting the versatility of host–guest complexation.2,3 More recently, there has been a growing interest towards the preparation of supramolecular analogues of hyperbranched polymers to form stimuli-responsive and self-healing ‘smart’ hyperbranched architectures.
Within the literature there exist a few examples of supramolecular hyperbranched-like polymers (SHPs) that have been prepared via host–guest complexation, primarily with low binding constants (Ka < 103 M−1) and in organic or mixed solvents.4–14 Efforts towards assembling SHPs in water have thus far focussed upon the cucurbit[8]uril (CB[8]) macrocyclic host exploiting ternary host–guest complexation, on account of high binding constants in aqueous media (typically >109 M−2).13,15–17 As supramolecular polymerisation is dependent on concentration and binding constants, the higher the Ka between subunits, the larger the potential supramolecular polymer.2,3 CB[8]-based SHPs have been utilised in the self-assembly of complex advanced materials from aqueous media including 2D supramolecular organic frameworks16 and branched materials with advanced tuneable optical properties.13
The specific supramolecular architecture will greatly affect the macroscopic properties of the polymer, determining the degree of branching and the amount of cross-linking available.1 Ideally, a single branching monomer can be employed, resulting in a truly hyperbranched architecture where intra- and inter-polymeric bonds are severely inhibited by steric hindrance. For example, the AB2 monomer demonstrated by Huang and Gibson where host (crown ether) and guests (viologen derivative) are present on the same molecule.4 However, it is synthetically easier to prepare one or more monomers that can be coupled in a separate host macrocycle. One such architecture is composed of A2 and B3 molecules where B3 acts as a branching point, and the A functionality can only bind to the B functionality.5,10,12,13 In this case, tendency towards a hyperbranched architecture will be observed at low conversions (ρ). As the polymer grows, however, both intra- and inter-molecular bonds will form, in the former case resulting in particles and in the latter, an extended cross-linked network (i.e. a gel). This resembles classical A2–B3 condensation chemistry.1,18
Herein we report the novel fabrication of a multi-stimuli responsive SHP in water that can be self-assembled into a macroscopic gel. There are limited examples of such a gelation occurring by host–guest chemistry.19 We have focused our efforts on a ternary complex inspired by the well-described20,21 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 heteroternary complex between methyl viologen (MV), azobenzene, and CB[8] in order to successfully generate a light and chemical responsive SHP. Aqueous droplets were used as templates for self-assembly of the SHP into a gel, which was shown to be elastic and self-healing. This interfacial gel was found to stabilise a microemulsion.
:1:1 heteroternary complex between methyl viologen (MV), azobenzene, and CB[8] in order to successfully generate a light and chemical responsive SHP. Aqueous droplets were used as templates for self-assembly of the SHP into a gel, which was shown to be elastic and self-healing. This interfacial gel was found to stabilise a microemulsion.
2 Results and discussion
2.1 Host–guest interactions
In order to obtain an extended SHP, the ditopic monomer (A2) and tritopic monomer (B3) were synthesised (Fig. 1a, see ESI† for synthetic methods). In the presence of CB[8], each MV moiety present on A2 binds exclusively to the azobenzene moiety on B3, yielding a heteroternary complex.20,21 The binding constant of the initial MV–CB[8] complex has been reported to be Ka = 5.0 × 105 M−1,20,21 with sequential binding to the azobenzene imidazolium salt derivative reported to be Ka = 3.5 × 104 M−1, yielding an overall Ka of 1.8 × 1010 M−2.20,21 This ternary complex is only formed when the azobenzene is present as the E isomer.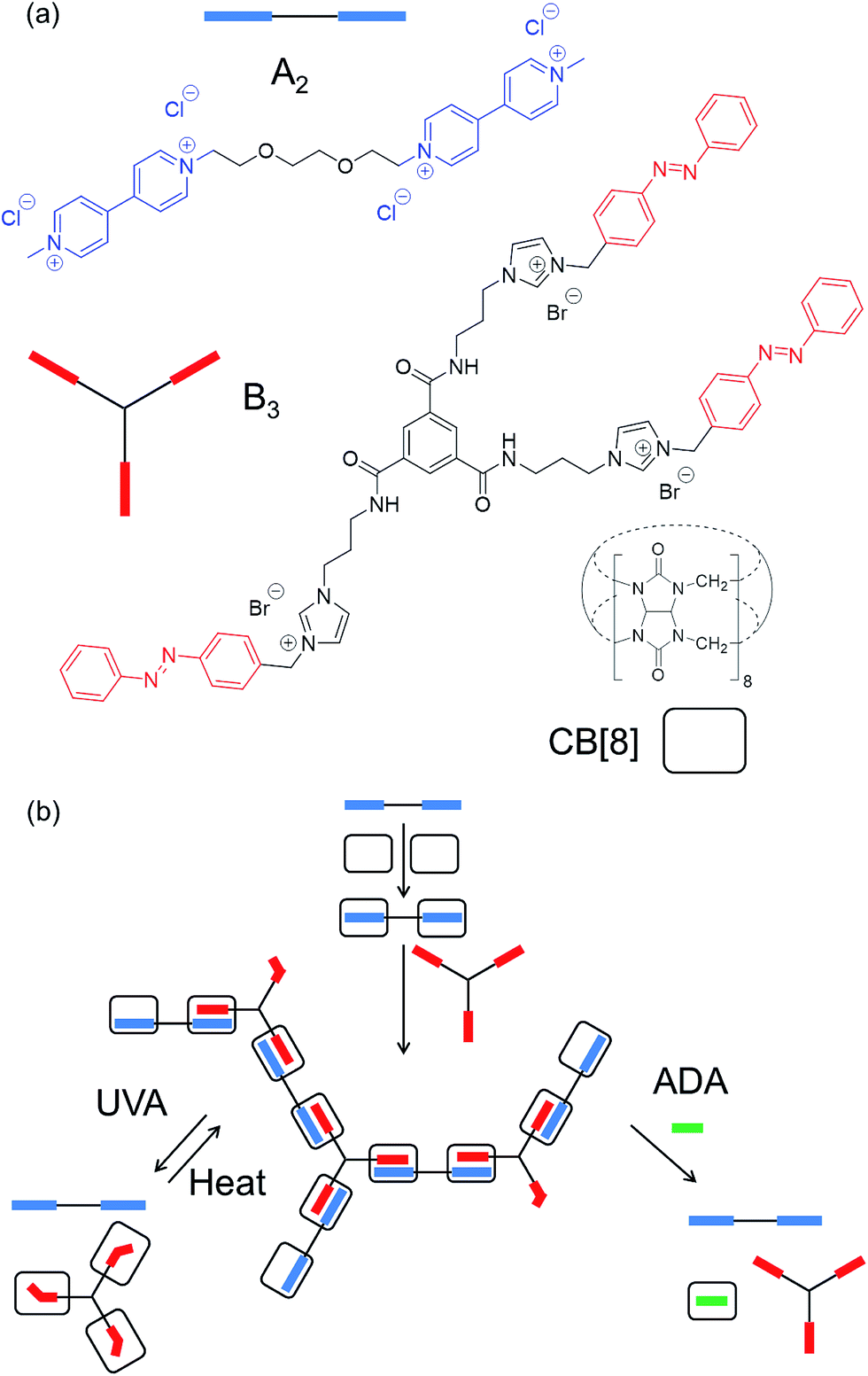 | ||
| Fig. 1 (a) Chemical structures and schematic representations of CB[8], and the ditopic (A2) and tritopic (B3) monomers used in this work. (b) Overview of the proposed assembly process; A2 is first complexed with CB[8] (2 eq.), and then mixed with B3 (0.67 eq.) to form the SHP. This SHP can be disassembled in response to either (i) reversible photoisomerisation of the azobenzene (left), or (ii) introduction of a competitive guest (right). | ||
The properties of the spacers between the guest moieties on monomers A2 and B3 were carefully selected for optimal supramolecular polymerisation. In particular, the spacers chosen for the two monomers possess minimal length and thus limited flexibility, as these properties will suppress the formation of intra-chain cross-links, and the effect of long chain entanglement (inducing the formation of a premature 3D network).1,5,8,9,13
2.2 Supramolecular polymerisation
The A2 and B3 monomers were first studied by 1H NMR spectroscopy, before combining with CB[8] to form a SHP. The binding of A2 to 1 and 2 equivalents of CB[8] in D2O was shown to be consistent with literature, with 2 CB[8] molecules bound to the terminal viologen derivatives (Fig. S1†).22 The 1H NMR spectrum of B3 in D2O evidenced the formation of self-associating aggregates; these aggregates were not detected when subsequent spectra were recorded in deaggregating solvents, such as d6-DMSO (Fig. S2†). Such higher order structures are known to be associated with the benzene 1,3,5-tricarboxamide (BTA) moiety in cases where long hydrophobic alkyl chains are introduced, and may be further induced here by the presence of the azobenzene imidazolium salt moiety.3,23,24 A similar structure without the imidazolium salt present has been reported for the assembly of dendritic rotaxanes in D2O with CB[8] exhibiting no self-association in the NMR spectra.25 In order to overcome aggregation, B3 was dissolved in a minimal amount of d6-DMSO and thereafter diluted with D2O (resultant solutions 0.7 v/v% d6-DMSO in D2O; Fig. S3†). As a result, the A2–CB[8] (2:1) complex was pre-assembled in water, before introduction of B3.
Upon combining a solution of A2–CB[8] (2:1) with a solution of B3 to give a final composition of A2:B3:CB[8] = 1.5:1:3 ratio ([A2] = 0.55 mM), the formation of an extended polymer was observed in the 1H NMR spectrum, as shown in Fig. 1b. In all cases the peaks in the 1H NMR spectrum have shifted significantly, which is indicative of host–guest interactions between CB[8] and the guest moieties on A2 and B3 (Fig. 2c).21 In addition, the broadening of all peaks is attributed to the formation of an extended supramolecular polymer. Of particular interest is the significant broadening observed for the peaks corresponding to CB[8], evidencing the formation of a supramolecular polymer. The A2–B3–CB[8] solution obtained was slightly turbid with precipitates observed at high concentration ([A2] = 1.10 mM; Fig. S3†), this is most likely attributed to the decrease in solubility of the growing supramolecular polymer.
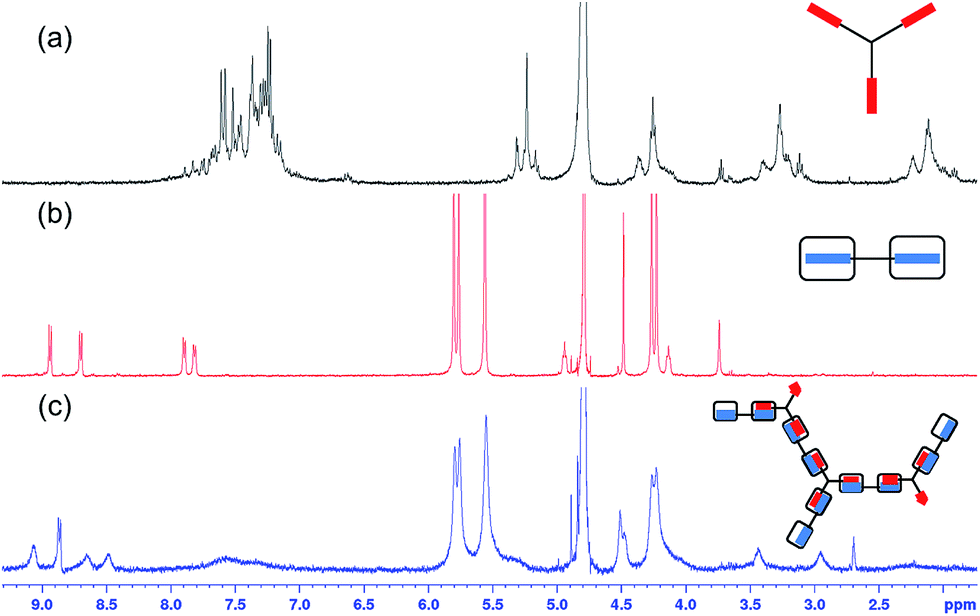 | ||
| Fig. 2
1H NMR spectra in D2O of (a) B3, (b) A2–CB[8] (1:2), (c) A2–B3–CB[8] (1.5:1:3) SHP. Concentrations in all cases were A2 = 0.55 mM, B3 = 0.37 mM, CB[8] = 1.11 mM. | ||
2.3 Stimuli-responsiveness
In order to fully understand the potential stimuli-responsiveness of the SHP, experiments were first carried out to characterise the photo-physical properties of B3. From the 1H NMR spectrum in d6-DMSO, it was calculated that the isomeric E–Z ratio of B3 under equilibrium at room temperature is 80% (E)-B3. Upon exposure to UVA light (λmax = 360 nm, 1 h) a photostationary state of 17% (E)-B3 could be reached (Fig. S4†). Upon heating this solution at 80 °C, >99% conversion to the (E)-B3 isomer was achieved, which then relaxed back to the original equilibrium state over 12 h at room temperature. Photoisomerisation in water was confirmed by UV-vis spectroscopy (Fig. S5†). With this information in hand, the SHP was illuminated with UVA in order to photoisomerise the (E)-B3 to (Z)-B3 and consequently disrupt the heteroternary complex, as tracked by 1H NMR spectroscopy (Fig. 3). | ||
| Fig. 3 1H NMR spectra in D2O of (a) A2–B3–CB[8] hyperbranched supramolecular polymer in its equilibrium state; (b) after 1 h of UVA exposure; (c) after subsequent heating at 80 °C for 6 h. Note, upon heating the azobenzene is >99% E isomer, higher than that originally present in (a). | ||
Upon exposure to UVA, the solution was observed to lose its characteristic orange colour, attributed to precipitation and sedimentation of the resultant (Z)-B3–CB[8] binary complex. This complex is formed due to the proximity of the cationic imidazolium salts present on the B3 monomer favouring binding to the carbonyl groups of the CB[8].21 Notably, precipitation does not occur in the absence of CB[8]. Disassembly of the SHP is fully reversible, with isomerisation back to (E)-B3 achievable upon exposure to blue light (λmax ≈ 450 nm), or heat. Heating the (Z)-B3–CB[8] dispersion to 80 °C resulted in the dissolution of the precipitate and reformation of the SHP (Fig. 3c). UV-vis spectroscopy was also performed to follow this isomerisation (Fig. S6†).
The ability of the SHP to respond to a chemical stimulus is demonstrated, as shown schematically in Fig. 1b. Titrating an increasing amount of the competitive guest adamantylamine HCl salt (ADA·HCl) resulted in the chemical shifts of the peaks associated with the A2 and B3 monomers returning to their complex-free value as shown in Fig. 4. In concert, the CB[8] and ADA·HCl peaks shift and sharpen (Fig. S7†). Moreover, the solution is visibly free of precipitates. These results confirm the complete disassembly of the SHP upon addition of ADA·HCl to the system by displacement of the guests within CB[8].
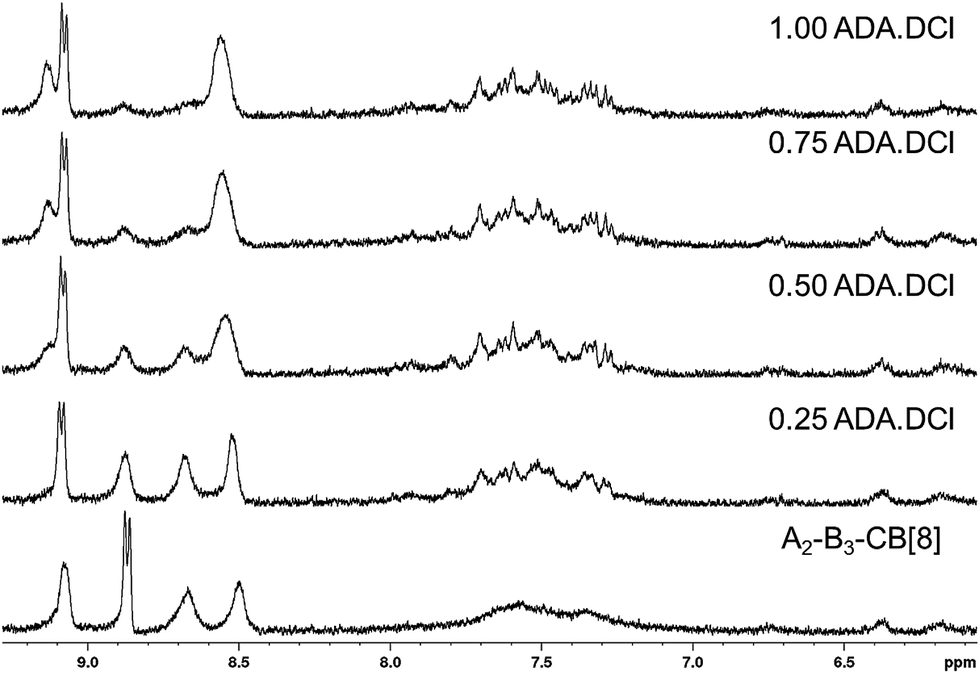 | ||
| Fig. 4 1H NMR spectra in D2O of A2–B3–CB[8] hyperbranched supramolecular polymer upon titration with up to 1 molar equivalent of ADA·DCl relative to CB[8]. | ||
2.4 Supramolecular polymers at liquid–liquid interfaces
Whilst the formation of a SHP has been shown in dilute solutions, an increase in concentration results in precipitation. This is attributed to a decrease in solubility of growing SHPs, which prevents formation of longer architectures. In order to circumvent precipitation and drive formation of an extended 3D cross-linked network, a liquid–liquid interface was used as a template for assembly from dilute aqueous solution.Microfluidic microemulsions make ideal candidates for studying the self-assembly processes in a controlled environment. There have been many advances in recent years in their application across the sciences, from studying single cells, to controlling chemical reactions in liquid microreactors.26,27
In this experiment, the concentration dependence of supramolecular polymerisation (i.e. the adaptability of supramolecular polymers) was exploited, with a schematic overview of the assembly process shown in Fig. 5. By loading aqueous microfluidic droplets with a dilute solution of the SHP, we can then control self-assembly at the microdroplet interface by electrostatic assembly,28 as the component monomers bear multiple conjugated cationic charges. This results in a rapid increase in concentration and density directly from solution. The surface charge of the droplet can be controlled by addition of perfluoropolyether dopants bearing either a pendant carboxylic acid group (Krytox 157-FSL, K−), or a synthesised amine-derivative (K+, see ESI for synthetic details†), structures of which are shown in the ESI.† This methodology has been previously employed in the assembly of supramolecular microcapsules from CB[8] crosslinked linear polymer networks (<5 mol% guest present on the polymer backbone).29 It has recently been shown that after evaporative concentration of the droplet, the interfacial polymer–CB[8] network undergoes a phase transition to a physically crosslinked gel.30
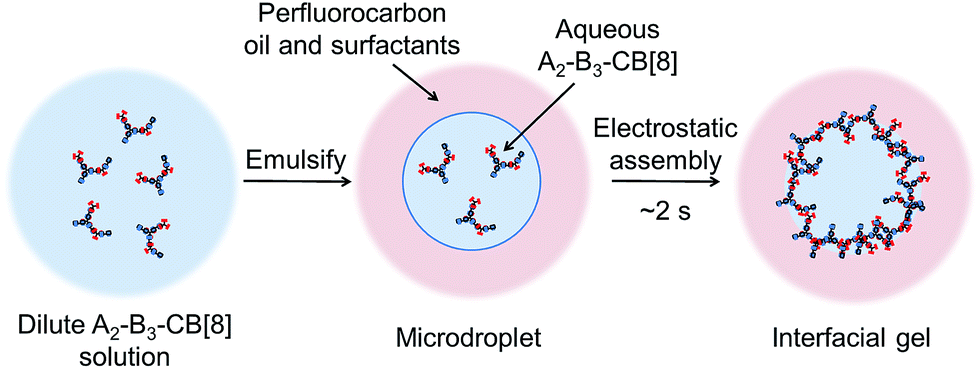 | ||
| Fig. 5 A schematic outline of the assembly process from dilute solution to an interfacial gel. At dilute solution there are no precipitates as only small polymeric species are present. After emulsification and subsequent electrostatic attraction to the liquid–liquid interface, the concentration and density of the polymer rapidly increases over ca. 2 s, leading to inter-polymeric crosslinking and gelation to occur (Fig. 6b). | ||
Here, we have achieved a similar result from branched small molecules. As a result of the rapid (ca. 2 s) assembly of the SHP at the interface from electrostatic accumulation, the SHP quickly grows to much larger sizes than in bulk preparation via simple dissolution. This results in inter-chain crosslinks readily forming, resulting in an elastic and self-healing physically crosslinked gel at the interface composed of entirely small molecules.
This is illustrated in Fig. 6b, where microdroplets have undergone a phase transition to form ‘buckled’ droplets in the presence of K−, indicating the formation of a viscoelastic gel.30 If CB[8] is absent from the droplet, this buckling transition does not occur and instead the droplets simply evaporate until the high charge density destabilises the system, and the droplets burst (Fig. S8†). Similarly, if CB[7] is used, which will only accommodate one MV guest, then the droplets again destabilise upon evaporation (Fig. S8†). If a positively-charged perfluorocarbon dopant is employed, assembly at the interface is disfavoured and the droplets shrink to a smaller size resulting in a microparticle structure upon complete loss of water (Fig. 6c). During this process, precipitation occurs as observed on the bulk scale. Initially these precipitates are free-flowing inside the droplet, and then halt motion due to a phase transition to a mixed particle-gelled state at high concentration. Fig. 6b highlights how directed assembly to the interface prevents precipitation from occurring.
 | ||
| Fig. 6 Transmission optical micrographs. (a) An example flow-focussing junction resulting in monodisperse microdroplets, typically with flow rates of 150 and 100 μL h−1 for the oil and water flows respectively; (b) top panel shows aqueous microdroplets containing A2–B3–CB[8] and continuous FC-40 oil containing 2 wt% XL171 and 1 wt% K−, and bottom panel is at higher magnification; (c) top panel shows aqueous microdroplets containing A2–B3–CB[8] and continuous FC-40 oil containing 2 wt% XL171 and 1 wt% K+, and bottom panel is at higher magnification. | ||
In order to demonstrate how the SHP network can stabilise the high surface energy of the microdroplet interface, perfluorooctanol (PFOH) was used as a de-emulsifier. PFOH is a very poor fluorosurfactant in comparison to XL171, and upon addition of excess PFOH microdroplets typically will coalesce (Video S1, ESI†). However, when PFOH is added to microdroplets containing the SHP, this interfacial gel is sufficient to inhibit coalescence, stabilising the microdroplet in the absence of effective fluorosurfactants (Video S2, ESI†). The network is not acting as a surfactant as there is no distinct amphiphilic structure present, but as a soft matter barrier once it has passed through its phase transition, allowing the droplet to become non-uniform in shape (i.e. non-spherical). This is analogous to recent works investigating ‘armoured bubbles’,31 and is illustrated in Fig. 7 whereupon aqueous droplets are stable to coalescence after the addition of PFOH. Note that in order to prevent rapid evaporation occurring after removal of surfactants, 500 kDa dextran was used to increase the viscosity.
 | ||
| Fig. 7 Transmission optical micrographs of microfluidic microemulsions deposited onto a glass slide. (a) Aqueous microdroplets containing 500 kDa dextran and (b) aqueous microdroplets containing A2–B3–CB[8] and 500 kDa dextran. Both continuous phases comprised FC-40 containing 2 wt% XL171 and 1 wt% K−. Upon addition of PFOH complete coalescence is observed over a few seconds in case (a), and in case (b) the network stabilises the microdroplet interface to coalescence. | ||
2.5 Pendant droplet interfacial tension
In order to study the interfacial supramolecular polymer network in an environment where evaporation is not a disruptive factor, measurements on stimuli-responsiveness were carried out in a pendant droplet. Interfacial tension measurements have been shown to be a good approximation of a microdroplet for observing phenomena and calculating material properties.30As the perfluorocarbon oil is denser than water, the liquid–liquid interface was inverted relative to the microfluidic experiments, with a hanging perfluorocarbon droplet in a continuous aqueous phase. The concentration of the SHP was lower than that used in the microfluidic environment (20×). This configuration also required a lower concentration of K− to drive interfacial assembly (0.001 wt% versus 1 wt%) and was carried out in the absence of XL171 surfactant, as the ability to shear flows forming microdroplets was no longer required. The interfacial tension (IFT) measured for a droplet of K− in FC-40, suspended in water alone was 45.9 ± 2.1 mN m−1.
When the aqueous phase contains the SHP, the IFT decreases over 45 min reflecting electrostatic accumulation of the dilute SHP to the interface, reaching a value of 34.4 ± 1.3 mN m−1 (see Fig. S9†). Upon reduction of the droplet volume, buckling can be clearly observed in Fig. 8a. This indicates that the system has undergone a phase change to a gelled state – as in the microdroplet environment. Once gelled, the droplet can be expanded beyond its initial volume and then contracted to reach the same buckled state with no equilibration time (Fig. 8b). This remarkable result highlights the elasticity and self-healing nature of the membrane as is characteristic of physically crosslinked polymer networks. If the buckled droplet is observed for a minute, the membrane will also flatten and smooth out (i.e. self-heal) to recover a spheroidal shape.
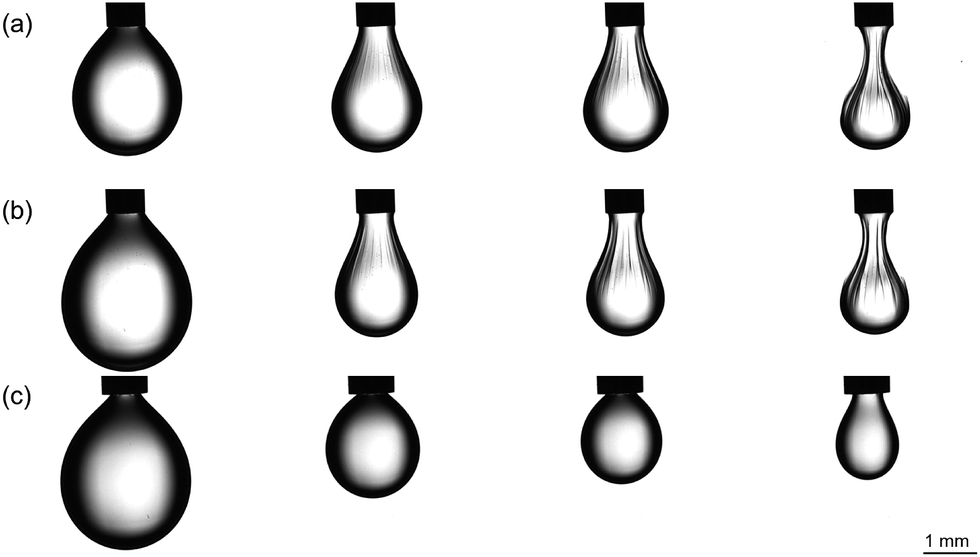 | ||
| Fig. 8 Transmission optical micrographs of the pendant droplet during IFT measurements of continuous aqueous A2–B3–CB[8], and droplet of 0.001 wt% K− in FC-40. From left to right, (a) equilibrated 5 μL droplet pumped in and exhibiting a buckled phase change; (b) droplet after subsequent pumping out to 8.5 μL followed by immediate pumping in; (c) droplet containing 0.01 wt% K+ in FC-40 and subsequent pumping in. | ||
Control measurements containing only A2 and B3 exhibited a lower IFT than that of pure water (25.0 ± 0.5 mN m−1, Fig. S10†), confirming that the electrostatic-based accumulation of monomers at the interface occurs to reduce the IFT. However, in the absence of CB[8], upon reduction of the volume of the droplet no phase change was observed (Fig. S10†). Similarly, a control with the opposing charge of fluorosurfactant (K+) did not show evidence of buckling upon reduction of the droplet volume (Fig. 8c), mimicking the microfluidic experiments. Furthermore, as the cationic charge inhibits initial assembly of A2 and B3 at the interface, the equilibrium IFT was higher at 42.0 ± 0.5 mN m−1, similar to the ‘water-only’ value vide supra.
Upon addition of ADA·HCl as a chemical stimulus, which was previously shown to break apart the network in NMR experiments, buckling is not observed after a reduction in volume (Fig. S10†), illustrating how the SHP network retains its chemical responsiveness. This occurs as the ADA outcompetes the MV and azobenzene guest moieties, breaking apart the network into its constituent small molecules. In contrast, photo-triggered disassembly did not prevent interfacial gelation, and is discussed in the ESI (Fig. S10 and S11†). In addition, diluting the SHP solution by orders of magnitude still displayed buckling, highlighting the large electrostatic accumulation effect occuring from dilute solution to form an interfacial network (Fig. S12†).
3 Conclusions
A multi-stimuli responsive hyperbranched supramolecular polymer has been assembled from small molecules in an aqueous environment. Light and chemical responsiveness of the assemblies were demonstrated. An extended physically crosslinked supramolecular network can be formed at the interface of a droplet, which can act as a soft matter barrier to inhibit water droplet coalescence upon displacement of surfactants. Future work will be based upon developing architectures of monomers responsive to different stimuli without interfering precipitation.Acknowledgements
A. S. G. acknowledges support from the EPSRC Cambridge NanoDTC, EP/G037221/1, A. P. and O. A. S. acknowledge support from the ERC starting investigator grant (ASPiRe 240629) and EPSRC Programme Grant (NOtCH, EP/L027151/1).References
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- F. Huang and H. W. Gibson, J. Am. Chem. Soc., 2004, 126, 14738–14739 CrossRef CAS PubMed.
- H. Li, X. Fan, W. Tian, H. Zhang, W. Zhang and Z. Yang, Chem. Commun., 2014, 50, 14666–14669 RSC.
- J. Zhang, J. Zhu, C. Lu, Z. Gu, T. He, A. Yang, H. Qiu, M. Zhang and S. Yin, Polym. Chem., 2016, 7, 4317–4321 RSC.
- Y.-K. Tian, Z.-S. Yang, X.-Q. Lv, R.-S. Yao and F. Wang, Chem. Commun., 2014, 50, 9477–9480 RSC.
- H. Li, X. Fan, X. Shang, M. Qi, H. Zhang and W. Tian, Polym. Chem., 2016, 7, 4322–4325 RSC.
- X. Yan, T. R. Cook, J. B. Pollock, P. Wei, Y. Zhang, Y. Yu, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 4460–4463 CrossRef CAS PubMed.
- V. H. Soto Tellini, A. Jover, J. Carrazana García, L. Galantini, F. Meijide and J. Vázquez Tato, J. Am. Chem. Soc., 2006, 128, 5728–5734 CrossRef PubMed.
- Z. Ge, H. Liu, Y. Zhang and S. Liu, Macromol. Rapid Commun., 2011, 32, 68–73 CrossRef CAS PubMed.
- R. Dong, Y. Liu, Y. Zhou, D. Yan and X. Zhu, Polym. Chem., 2011, 2, 2771–2774 RSC.
- H. Yang, Z. Ma, Z. Wang and X. Zhang, Polym. Chem., 2014, 5, 1471–1476 RSC.
- C. Zhou, J. Tian, J.-L. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 341–345 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. Del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- K. D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C. H. Sue, T. Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z. T. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef CAS PubMed.
- R. Fang, Y. Liu, Z. Wang and X. Zhang, Polym. Chem., 2013, 4, 900–903 RSC.
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718–2723 CrossRef CAS.
- X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362–369 CrossRef CAS PubMed.
- J. del Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760–11763 CrossRef CAS PubMed.
- C. S. Y. Tan, J. del Barrio, J. Liu and O. A. Scherman, Polym. Chem., 2015, 6, 7652–7657 RSC.
- S. Deroo, U. Rauwald, C. V. Robinson and O. A. Scherman, Chem. Commun., 2009, 644–646 RSC.
- J. Leenders, M. B. Baker, I. Pijpers, R. Lafleur, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, Soft Matter, 2016, 12, 2887–2893 RSC.
- C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- S. Y. Kim, Y. H. Ko, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Chem.–Asian J., 2007, 2, 747–754 CrossRef CAS PubMed.
- X. C. I. Solvas and A. DeMello, Chem. Commun., 2011, 47, 1936–1942 RSC.
- A. J. DeMello, Nature, 2006, 442, 394–402 CrossRef CAS PubMed.
- R. M. Parker, J. Zhang, Y. Zheng, R. J. Coulston, C. A. Smith, A. R. Salmon, Z. Yu, O. A. Scherman and C. Abell, Adv. Funct. Mater., 2015, 25, 4091–4100 CrossRef CAS PubMed.
- Z. Yu, J. Zhang, R. J. Coulston, R. M. Parker, F. Biedermann, X. Liu, O. A. Scherman and C. Abell, Chem. Sci., 2015, 6, 4929–4933 RSC.
- A. R. Salmon, R. M. Parker, A. S. Groombridge, A. Maestro, R. J. Coulston, J. Hegemann, J. Kierfield, P. Cicuta, O. A. Scherman and C. Abell, Langmuir, 2016 DOI:10.1021/acs.langmuir.6b03011.
- A. Bala Subramaniam, M. Abkarian, L. Mahadevan and H. A. Stone, Nature, 2005, 438, 930 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, videos, and figures referenced throughout the text. See DOI: 10.1039/c6sc04103e. Additional data related to this publication is available at the University of Cambridge data repository (http://dx.doi.org/10.17863/CAM.4875) |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |