
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new approach to the asymmetric Mannich reaction catalyzed by chiral N,N′-dioxide–metal complexes†
Xiangjin
Lian
,
Lili
Lin
,
Kai
Fu
,
Baiwei
Ma
,
Xiaohua
Liu
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: xmfeng@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
First published on 3rd October 2016
Abstract
A highly efficient asymmetric Mannich-type reaction between α-tetralone-derived β-keto esters/amides and 1,3,5-triaryl-1,3,5-triazinanes was realized in the presence of chiral N,N′-dioxide–Ni(II) or Mg(II) complex. A variety of optically active β-amino compounds with all-carbon quaternary stereocenters were obtained in good yields with excellent enantioselectivities. A possible transition state was proposed based on these experiments and previous reports.
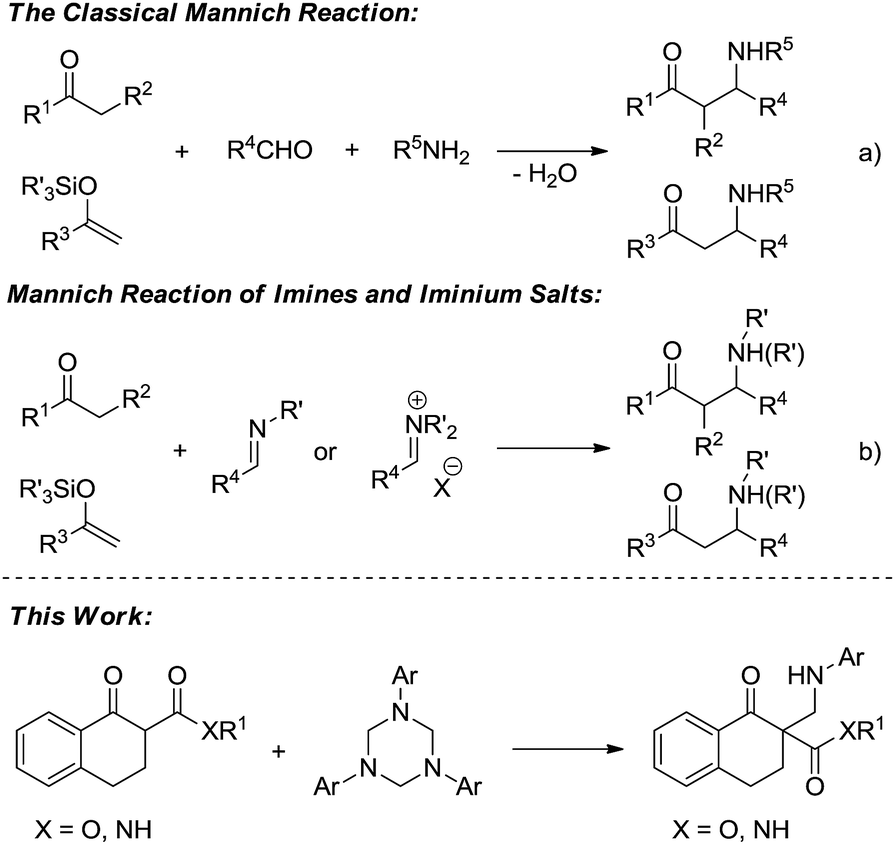
Because the resulting nitrogen-containing compounds are widely distributed in nature and include many biologically important molecules,1 the Mannich reaction has received a lot of attention since its discovery in the early 20th century (Scheme 1a).2 It has become one of the most efficient methods to construct C–C bonds.3 Despite its important synthetic value, the development of the classical intermolecular Mannich reaction has been plagued by a number of serious disadvantages such as the undesired side products formed in many cases, and the ability to control the regio- and stereoselectivity is generally unsatisfactory.4 The first catalytic enantioselective approach was reported by Kobayashi using a novel chiral zirconium catalyst in 1997.5 To overcome the drawbacks of the classical Mannich reaction, preformed Mannich reagents such as imines and iminium salts have been developed (Scheme 1b).6 Subsequently, the catalytic asymmetric Mannich reaction has received a certain amount of development.7 However, such preformed Mannich reagents also have some defects such as low activity, sensitivity to moisture and instability, and therefore the development of new Mannich reagents is desirable.
 | ||
| Scheme 1 Classical Mannich-type reaction and the new approach. | ||
1,3,5-Triaryl-1,3,5-triazinanes, which are conveniently prepared through the condensation of paraformaldehyde and aromatic amines,8 can generate the corresponding imines in solvent, which can be used as Mannich reagents. Very recently, Krische reported investigations on the hydroaminomethylation of allenes and 1,3-dienes with 1,3,5-triaryl-1,3,5-triazinanes catalyzed by ruthenium.9 Inspired by Krische's work, we think that the in situ generated imines from 1,3,5-triaryl-1,3,5-triazinanes might be used as Mannich reagents. On the other hand, all-carbon quaternary stereocenters are widely present in natural products and to build such structures is still a challenge, especially in a catalytic enantioselective manner.10 In recent years, our group has been committed to utilizing N,N′-dioxide–metal complexes as catalysts and has achieved a series of catalytic asymmetric reactions, including the construction of compounds with chiral all-carbon quaternary stereocenters.11 Herein, we report the first asymmetric Mannich reaction employing 1,3,5-triaryl-1,3,5-triazinanes as new Mannich reagents catalyzed by N,N′-dioxide–metal complexes, and a variety of optically active β-amino compounds, each with an all-carbon quaternary stereocenter, were obtained.
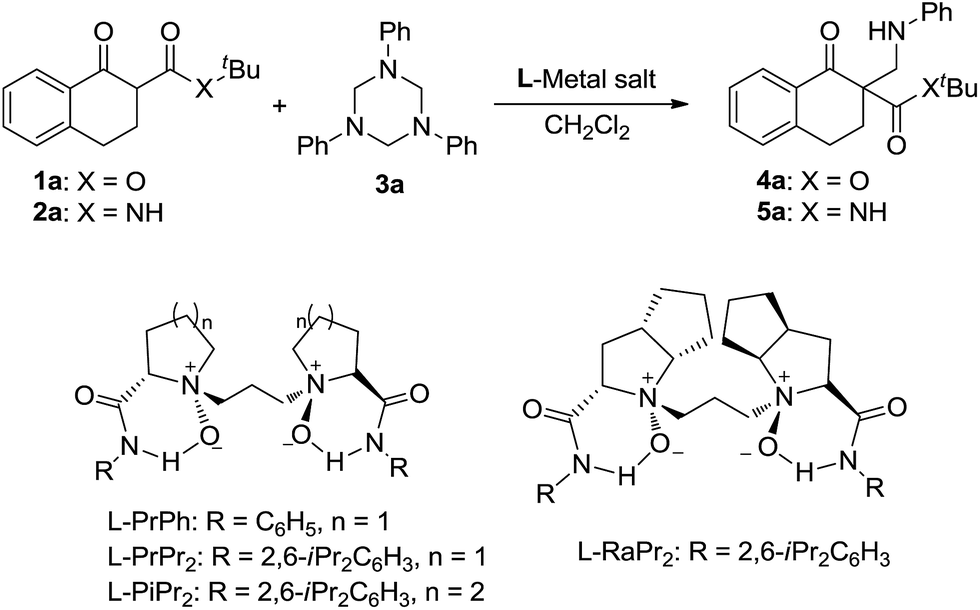
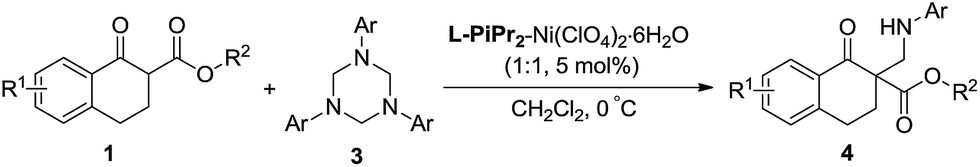
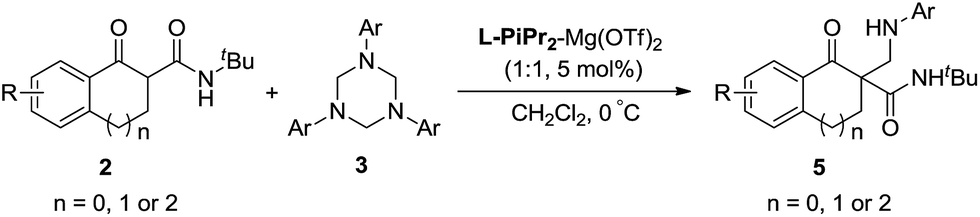
In our preliminary screening, the α-tetralone-derived β-keto ester 1a and 1,3,5-triphenyl-1,3,5-triazinane 3a were chosen as the model substrates to optimize the reaction conditions (Table 1). Initially, the performance of various metal salts was evaluated when combined with the chiral N,N′-dioxide ligand L-PrPh, which is derived from L-proline, and the reactions were performed in CH2Cl2 at 30 °C (Table 1, entries 1–5). Lanthanides, the N,N′-dioxide complexes of which have proved to be efficient catalysts for many reactions,11 can only provide the desired product 4a with low ee values or as a racemate, although the yields were good (Table 1, entries 1–3). The complex of Mg(OTf)2 could give the desired product in 85% yield but with only 18% ee (Table 1, entry 4). To our delight, the complex of Ni(ClO4)2·6H2O provided 4a with a better ee value (44% ee, Table 1, entry 5 versus entries 1–4). Increasing the steric hindrance of the amide substituents on the chiral N,N′-dioxide ligand further improved the enantioselectivity. Chiral N,N′-dioxide L-PrPr2 with a more sterically hindered i-Pr at the ortho-positions of aniline improved the enantioselectivity to 53% ee (Table 1, entry 6 versus entry 5). Then we investigated the effect of the chiral backbone moiety, the (S)-pipecolic acid derived N,N′-dioxide L-PiPr2 (Table 1, entry 8) was superior to L-proline derived L-PrPr2 and L-ramipril-derived L-RaPr2 (Table 1, entries 6 and 7), giving the product in 94% yield with 96% ee. In addition, lowering the temperature to 0 °C improved the enantioselectivity to 99% ee albeit with a lower yield (Table 1, entry 9). Remarkably, upon reducing the catalyst loading to 5 mol% the yield improved to 97% with the enantioselectivity maintained (Table 1, entry 10). When the α-tetralone-derived β-keto amide 2a was employed in this reaction instead of 1a, the desired product 5a was obtained in good yield but with unsatisfactory enantioselectivity (Table 1, entry 11). Then we replaced the metal salt with Mg(OTf)2 and got comparable results (Table 1, entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Substrate | Metal salt | Ligand | Yieldb (%) | eec (%) |
| a Unless otherwise noted, the reactions were performed with 1a or 2a (0.10 mmol), 3a (0.034 mmol), ligand (0.01 mmol), and metal salt (0.01 mmol) in 1.0 mL CH2Cl2 at 30 °C for 8 h. b Isolated yield of the product. c Determined by HPLC analysis on a chiral stationary phase. d The reaction was performed at 0 °C for 12 h. e 5 mol% L-PiPr2 (0.005 mmol) and 5 mol% Ni(ClO4)2·6H2O (0.005 mmol) were used. f The reaction was performed with L-PiPr2 (0.005 mmol) and Mg(OTf)2 (0.005 mmol). | |||||
| 1 | 1a | Sc(OTf)3 | L-PrPh | 83 | 0 |
| 2 | 1a | Yb(OTf)3 | L-PrPh | 84 | 0 |
| 3 | 1a | La(OTf)3 | L-PrPh | 90 | 13 |
| 4 | 1a | Mg(OTf)2 | L-PrPh | 85 | 18 |
| 5 | 1a | Ni(ClO4)2·6H2O | L-PrPh | 97 | 44 |
| 6 | 1a | Ni(ClO4)2·6H2O | L-PrPr2 | 67 | 53 |
| 7 | 1a | Ni(ClO4)2·6H2O | L-RaPr2 | 87 | 87 |
| 8 | 1a | Ni(ClO4)2·6H2O | L-PiPr2 | 94 | 96 |
| 9d | 1a | Ni(ClO4)2·6H2O | L-PiPr2 | 87 | 99 |
| 10d,e | 1a | Ni(ClO4)2·6H2O | L-PiPr2 | 97 | 99 |
| 11d,e | 2a | Ni(ClO4)2·6H2O | L-PiPr2 | 95 | 61 |
| 12d,f | 2a | Mg(OTf)2 | L-PiPr2 | 98 | 97 |
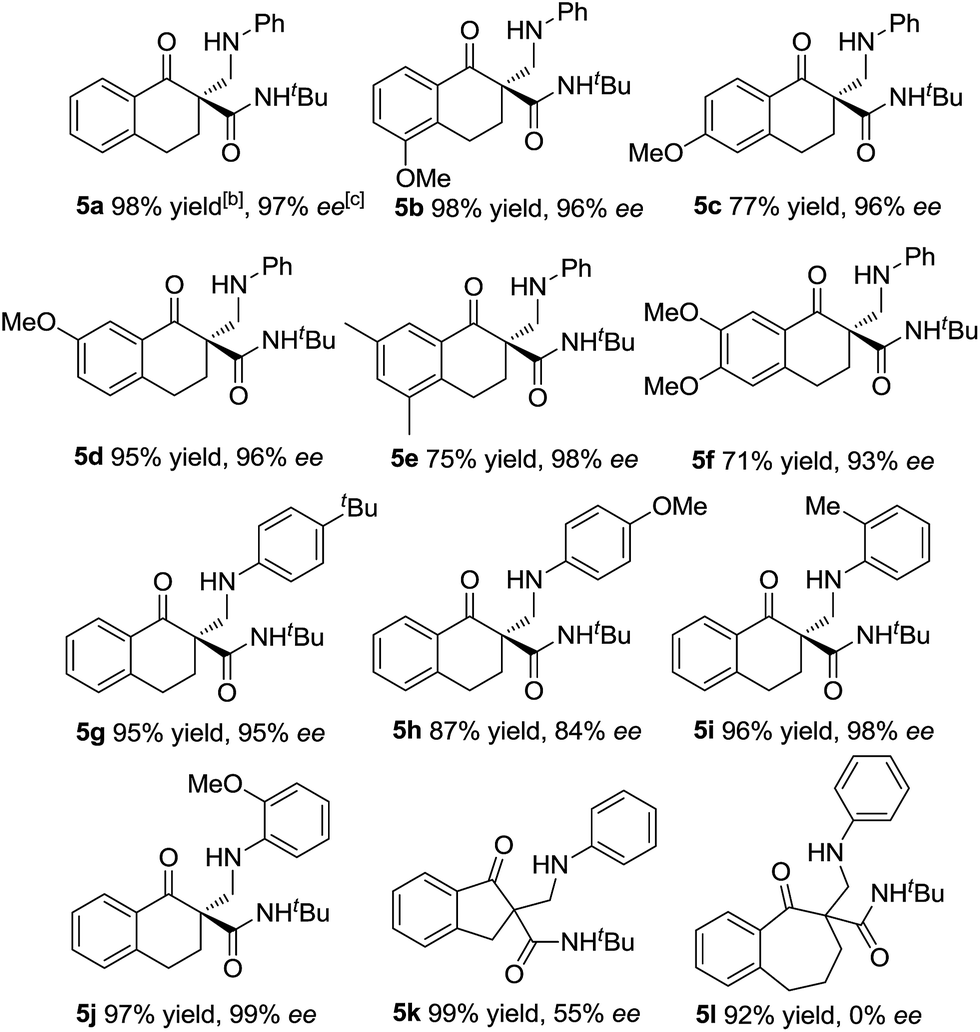
With the optimized reaction conditions in hand, we firstly investigated the scope of the reactions between α-tetralone-derived β-keto esters and 1,3,5-triaryl-1,3,5-triazinanes (Table 2). Delightfully, the electronic nature and the positions of the substituents on the β-keto esters had little influence on both the yields and enantioselectivities (83–98% yield, 81–99% ee; 4a–4f). Next, the 1,3,5-triaryl-1,3,5-triazinanes were varied. As it shown in Table 2 (4g–4k), the positions of the substituents have a certain influence on the yields, but the enantioselectivities were good in all cases. Generally, the 2-substituted 1,3,5-triaryl-1,3,5-triazinanes showed a slight decrease in yield compared with the 4-substituted ones. What's more, 1-adamantanol substituted β-keto ester 1l was also a suitable substrate for this reaction and the corresponding product 4l was obtained in 99% yield with 93% ee (Table 2, 4l). Additionally, the absolute configuration of 4a was determined to be R by X-ray crystallography12 and the configurations of the others were determined to be R by circular dichroism (for details see the ESI†).

Subsequently, we turned our attention to investigate the substrate scope of the reactions between α-tetralone-derived β-keto amides and 1,3,5-triaryl-1,3,5-triazinanes (Table 3). To our delight, a variety of β-keto amides with different substituents were tolerated and gave the corresponding products with excellent enantioselectivities (Table 3, 93–98% ee; 5a–5f). Then the scope of 1,3,5-triaryl-1,3,5-triazinanes was examined. The results are different from the results for the reactions of the β-keto esters, and both 2- and 4-substituted 1,3,5-triaryl-1,3,5-triazinanes afforded the corresponding products in excellent yields and enantioselectivities (95–99% yields, 95–99% ee, 5g, 5i and 5j) except the 4-MeO substituted 1,3,5-tris(4-methoxyphenyl)-1,3,5-triazinane, which gave the corresponding product in 84% ee. Besides this, five- and seven-membered β-keto amide substrates were also examined. Unfortunately, the five-membered β-keto amide gave the corresponding product 5k with only 55% ee, while the seven-membered β-keto amide gave a racemic product 5l though the yields were excellent under the standard conditions. A cyclohexanone-derived β-keto amide was also tested under the standard reaction conditions, but the reaction didn't occur. Meanwhile, the absolute configuration of 5a was determined to be R by X-ray crystallography analysis12 and configurations of the others were also determined to be R by circular dichroism (for details see the ESI†).
To evaluate the synthetic value of this catalytic system, gram-scale reactions were performed (Scheme 2). In the presence of the L-PiPr2–Ni(ClO4)2·6H2O complex (5 mol%), the starting material 1a (4.0 mmol) reacted with 3a (1.3 mmol, 1.0 equivalent) smoothly, and the corresponding product 4a was obtained in 92% yield with 99% ee (Scheme 2a). In the system of α-tetralone-derived β-keto amides and 1,3,5-triaryl-1,3,5-triazinanes, the reaction between 0.98 g 2a and 0.42 g 3a was performed under the optimized reaction conditions, affording 1.34 g (95% yield) of the corresponding product 5a with 97% ee (Scheme 2b).
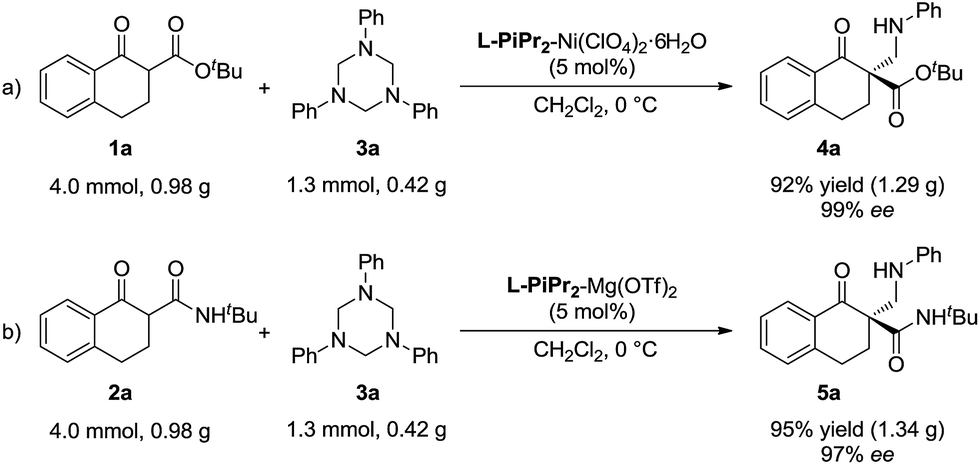 | ||
| Scheme 2 Gram-scale version of the reaction. | ||
On the other hand, the product 4a could be efficiently converted into useful β-hydroxyl ester 6 through reduction using NaBH4 as a reducing agent (Scheme 3). The diastereomer of the product 6 was determined to be trans- using NOESY spectra (see the ESI† for details). The product 4h could be converted into N-Boc-β-amino ester 7 by deprotection with cerium ammonium nitrate (CAN) followed by Boc protection of the amino group with Boc2O (see the ESI† for details).
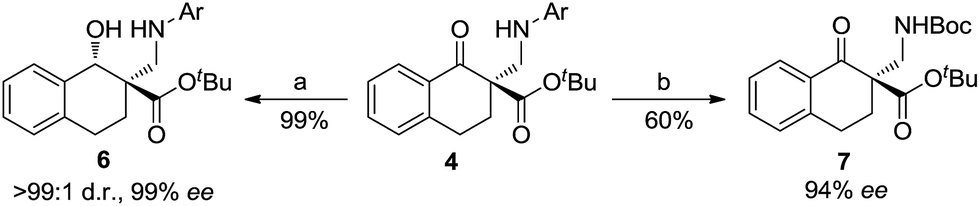 | ||
Scheme 3 Transformations of the product 4 into other derivatives; reaction conditions: (a) NaBH4 and MeOH/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0 °C (4a: Ar = Ph, 99% ee); (b) CAN, CH3CN/H2O; then Et3N and Boc2O (4h: Ar = 4-MeOC6H4, 94% ee). Boc = tert-butyloxycarbonyl. :1), 0 °C (4a: Ar = Ph, 99% ee); (b) CAN, CH3CN/H2O; then Et3N and Boc2O (4h: Ar = 4-MeOC6H4, 94% ee). Boc = tert-butyloxycarbonyl. | ||
To gain insight into the mechanism, the relationship between the ee value of the ligand L-PiPr2 and that of 4a was investigated under the optimal reaction conditions.13 A linear effect was observed (see the ESI† for details), which suggested that a monomeric catalyst may be the main catalytically active species in the reaction system. Based on the experiments and our previous work11 as well as the absolute configuration of the products, a possible transition state model is proposed in Fig. 1 to elucidate the origin of the asymmetric induction. In the transition state, the oxygens of the N,N′-dioxides and the amide oxygens coordinate to Ni(II) in a tetradentate manner. The β-keto ester 1a could be activated after coordinating to the nickel atom in a bidentate fashion. The Si-face of β-keto ester 1a is effectively shielded by the amide moiety and the piperidine ring on the underside of the ligand L-PiPr2. In contrast, the Re-face is located in a relatively open space. The highly selective approach of the in situ generated N-methyleneaniline toward the Re-face of the bidentate-coordinated β-keto ester leads to the desired product with an R configuration, which is consistent with the observed absolute configuration of the product.
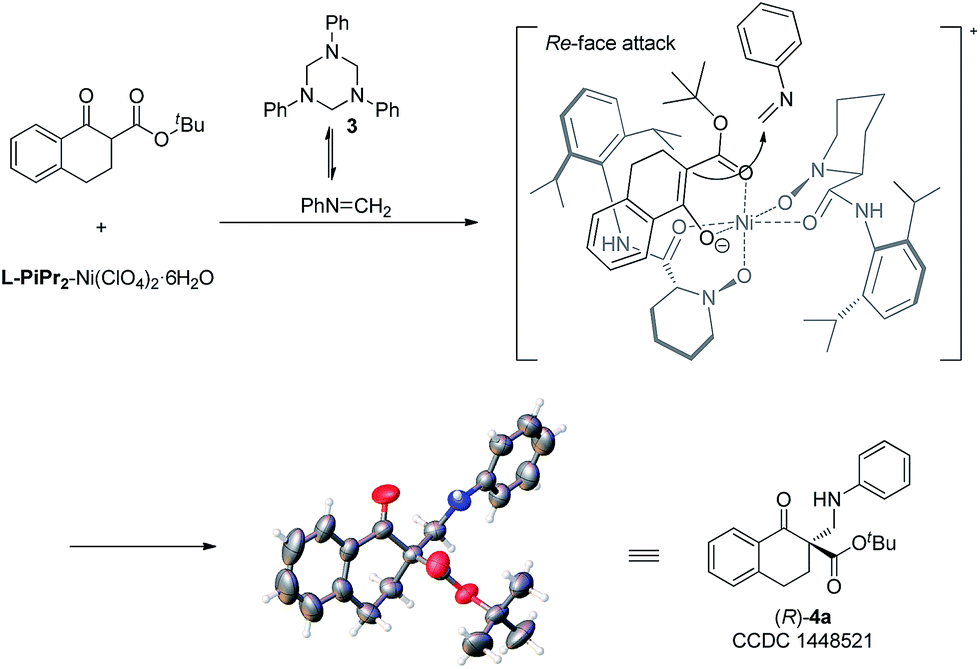 | ||
| Fig. 1 Proposed transition state and the absolute configuration of 4a. | ||
Conclusions
In summary, a highly enantioselective Mannich-type reaction between α-tetralone-derived β-keto esters/amides and 1,3,5-triaryl-1,3,5-triazinanes was realized. In the presence of chiral N,N′-dioxide–Ni(II) or N,N′-dioxide–Mg(II) complex, a variety of corresponding β-amino compounds each with an all-carbon quaternary stereocenter were obtained in good to excellent enantioselectivities (up to 99% ee) and good to excellent yields (up to 99%). In particular, this is the first time that 1,3,5-triaryl-1,3,5-triazinanes were used as electrophilic reagents in the catalytic asymmetric Mannich reaction. Further studies focused on the reactions of 1,3,5-triaryl-1,3,5-triazinanes are under way.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21372162, 21432006 and 21321061) for financial support.Notes and references
- M. Arend, B. Westermann and N. Rish, Angew. Chem., Int. Ed., 1998, 37, 1044 CrossRef.
- C. Mannich and W. Krosche, Arch. Pharm., 1912, 250, 647 CrossRef CAS.
- M. Tramontini and L. Angiolini, Tetrahedron, 1990, 46, 1791 CrossRef CAS.
- For reviews, see: (a) M. Tramontini and L. Angiolini, Mannich-Bases, Chemistry and Uses, CRC, F. L. Boca Raton, 1994 Search PubMed; (b) M. Tramontini, L. Angiolini and N. Ghedeni, Polymer, 1988, 29, 771 CrossRef CAS; (c) M. Tramontini, Synthesis, 1973, 703 CrossRef CAS; (d) H. Hellmann and G. Opitz, α-Aminoalkylierung, Verlag Chemie, Weinheim, 1960 Search PubMed; (e) B. Reichert, Die Mannich reaktion, Springer, Berlin, 1959 Search PubMed; (f) F. F. Blicke, Org. React., 1942, 1, 303 Search PubMed.
- (a) H. Ishitani, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 1997, 119, 7153 CrossRef; (b) For an early review, see: S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS PubMed.
- For examples, see: (a) H. J. Ha and Y. G. Ahn, Synth. Commun., 1995, 25, 969 CrossRef CAS; (b) A. R. Katritzky, S. Rachwal and G. J. Hitchings, Tetrahedron, 1991, 47, 2683 CrossRef CAS; (c) S. Kobayashi, H. Ishitani, S. Komiyama, D. C. Oniciu and A. R. Katritzky, Tetrahedron Lett., 1996, 37, 3731 CrossRef CAS; (d) P. C. B. Page, S. M. Allin, E. W. Collington and R. A. E. Carr, J. Org. Chem., 1993, 58, 6902 CrossRef CAS; (e) A. R. Katritzky, N. Shobana and P. A. Harris, Tetrahedron Lett., 1990, 31, 3999 CrossRef CAS; (f) A. R. Katritzky and P. A. Harris, Tetrahedron, 1990, 46, 987 CrossRef; (g) D. Enders, D. Ward, J. Adam and G. Raabe, Angew. Chem., Int. Ed., 1996, 35, 981 CrossRef CAS; (h) U. Jahn and W. Schroth, Tetrahedron Lett., 1993, 34, 5863 CrossRef CAS.
- For reviews of asymmetric Mannich reactions, see: (a) A. Cordova, Acc. Chem. Res., 2004, 37, 102 CrossRef CAS PubMed; (b) A. G. Wenzel and E. N. Jacobsen, Enantioselective Synthesis of β-Amino Acids, ed. E. Juaristi and V. Soloshonok, Wiley-VCH, New York, 2005, ch. 4 Search PubMed; (c) A. Ting and S. E. Schaus, Eur. J. Org. Chem., 2007, 2007, 5797 CrossRef; (d) J. M. M. Verkade, L. J. C. von Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC; (e) B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656 RSC; (f) B. Karimi, D. Enders and E. Jafari, Synthesis, 2013, 45, 2769 CrossRef CAS.
- (a) C. A. Bischoff and F. Reinfeld, Chem. Ber., 1903, 36, 41 CrossRef CAS; (b) A. G. Giumanini, G. Verardo, E. Zangrando and L. Lassiani, J. Prakt. Chem., 1987, 329, 1087 CrossRef CAS; (c) A. G. Giumanini, N. Toniutti, G. Verardo and M. Merli, Eur. J. Org. Chem., 1999, 141 CrossRef CAS; (d) H. J. Ha, C. J. Choi and W. K. Lee, Synth. Commun., 2002, 32, 1495 CrossRef CAS; (e) M. I. P. Reis, G. A. Romeiro, R. Damasceno, F. de C. da Silva and V. F. Ferreira, Rev. Virtual Quim., 2013, 5, 283 CAS; (f) G. O. Jones, J. M. García, H. W. Horn and J. L. Hedrick, Org. Lett., 2014, 16, 5502 CrossRef CAS PubMed.
- (a) S. Oda, B. Sam and M. J. Krische, Angew. Chem., Int. Ed., 2015, 54, 8525 CrossRef CAS PubMed; (b) S. Oda, J. Franke and M. J. Krische, Chem. Sci., 2016, 7, 136 RSC.
- (a) K. Fuji, Chem. Rev., 1993, 93, 2037 CrossRef CAS; (b) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef; (c) J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS; (d) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS PubMed; (e) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473 CrossRef CAS; (f) B. M. Trost and C. Jiang, Synthesis, 2006, 369 CrossRef CAS; (g) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS; (h) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (i) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC.
- For reviews of N,N′-dioxide–metal complexes in asymmetric reactions, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) K. Zheng, L. L. Lin and X. M. Feng, Acta Chim. Sin., 2012, 70, 1785 CrossRef CAS; (c) “Chiral Scandium Complexes in Catalytic Asymmetric Reactions”: X. M. Feng and X. H. Liu, Scandium: Compounds, Productions and Applications, ed. V. A. Greene, Nova Science, New York, 2011, p. 1 Search PubMed; (d) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC . For recent examples, see: ; (e) W. Li, F. Tan, X. Y. Hao, G. Wang, Y. Tang, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 1608 CrossRef CAS PubMed; (f) J. L. Zhang, Y. L. Zhang, L. L. Lin, Q. Yao, X. H. Liu and X. M. Feng, Chem. Commun., 2015, 51, 10554 RSC.
- CCDC 1448521 (4a) and CCDC 1480808 (5a) contain the supplementary crystallographic data for this paper.
- (a) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456 CrossRef CAS PubMed; (b) D. Guillaneux, S. H. Zhao, O. Samuel, D. Rainford and H. B. Kagan, J. Am. Chem. Soc., 1994, 116, 9430 CrossRef CAS; (c) C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1448521 and 1480808.For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03902b |
| This journal is © The Royal Society of Chemistry 2017 |