
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable α/δ phase junction of formamidinium lead iodide perovskites for enhanced near-infrared emission†
Fusheng
Ma
,
Jiangwei
Li
,
Wenzhe
Li
,
Na
Lin
,
Liduo
Wang
and
Juan
Qiao
*
Key Lab of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, PR. China. E-mail: qjuan@mail.tsinghua.edu.cn
First published on 15th September 2016
Abstract
Although formamidinium lead iodide (FAPbI3) perovskite has shown great promise in the field of perovskite-based optoelectronic devices, it suffers the complications of a structural phase transition from a black perovskite phase (α-FAPbI3) to a yellow non-perovskite phase (δ-FAPbI3). Generally, it is pivotal to avoid δ-FAPbI3 since only α-FAPbI3 is desirable for photoelectric conversion and near-infrared (NIR) emission. However, herein, we firstly exploited the undesirable δ-FAPbI3 to enable structurally stable, pure FAPbI3 films with a controllable α/δ phase junction at low annealing temperature (60 °C) through stoichiometrically modified precursors (FAI/PbI2 = 1.1–1.5). The α/δ phase junction contributes to a striking stabilization of the perovskite phase of FAPbI3 at low temperature and significantly enhanced NIR emission at 780 nm, which is markedly different from pure α-FAPbI3 (815 nm). In particular, the optimal α/δ phase junction with FAI/PbI2 = 1.2 exhibited preferable long-term stability against humidity and high PLQY of 6.9%, nearly 10-fold higher than that of pure α-FAPbI3 (0.7%). The present study opens a new approach to realize highly stable and efficient emitting perovskite materials by utilizing the phase junctions.
Introduction
Organic–inorganic hybrid perovskites, especially CH3NH3PbI3 (MAPbI3) and its mixed halide analogues, have attracted tremendous attention as light absorbers for solar cells. The power conversion efficiencies (PCEs) of perovskite solar cells (PSCs) have been boosted from 3.8% in 2009 to over 20% in 2016.1–6 Recently, these hybrid perovskite materials also have shown great promise as light emitters for lasers and light-emitting diodes because of their very high color purity, low material cost and simply tunable band gaps.7,8 Bright photoluminescence (PL) and electroluminescence (EL) in the near-infrared (NIR) and visible region have been demonstrated based on solution-processed MAPbX3 perovskites by tuning the halide composition at room temperature.9–19 Comparing with the prototype MAPbI3, formamidinium lead iodide (FAPbI3) has recently emerged as a promising candidate for use in PSCs because of its favorable band gap (1.47 eV) and superior photo- and thermal stability.20–22 Moreover, FAPbI3 perovskite demonstrates a true NIR emission around 815 nm,23,24 also making it a preferable candidate for NIR emitters in terms of achieving more robust perovskite light-emitting diodes.However, FAPbI3 was reported to be unstable in phase structure.20–29 It can crystallize into either a black trigonal perovskite phase (α-FAPbI3) or a yellow hexagonal non-perovskite phase (δ-FAPbI3) depending on the heat-treatment temperature. It usually requires a very high phase transition temperature (up to 160 °C) to obtain black α-FAPbI3, which is desirable for photoelectric conversion and NIR emission.26–28 Unfortunately, such high temperature can result in partial decomposition of FAPbI3 into PbI2.21,30 Moreover, the obtained black α-FAPbI3 can turn into the undesirable yellow δ-FAPbI3 under an ambient humid atmosphere.24,26–28 To date, most work on the FA-based perovskites focuses on the stabilization of black α-FAPbI3 at relatively low temperature. Very recently, several groups have demonstrated that mixed cations such as MA/FA or Cs/FA can fully avoid the formation of δ-FAPbI3.25,27–29 But as far as we know, the stabilization of the pure FA phase has not been conclusively demonstrated.
Herein, we for the first time exploited the undesirable δ-FAPbI3 to achieve structurally stable FAPbI3 perovskite films at low annealing temperature (60 °C) through fine and controllable stoichiometry modification (FAI/PbI2 = 1.1–1.5). All the obtained films demonstrated a unique α/δ phase junction together with a significantly enhanced phase stability and NIR emission at 780 nm, which is markedly different from the pure α-FAPbI3 (815 nm). The hypochromic emission was attributed to a slight bandgap widening due to band bending at the α/δ phase junction. Moreover, such a phase junction can substantially decrease the dielectric constant and grain size of the films, thus effectively increasing the exciton binding energy and confining the excitons in the nanograins for high emission. Most notably, the optimal α/δ phase junction with FAI/PbI2 = 1.2 exhibited superior long-term stability even under an ambient humid atmosphere and the highest PL quantum efficiency (PLQY = 6.9%), nearly one order of magnitude higher than that of pure α-FAPbI3 (PLQY = 0.7%).
Results and discussion
Firstly, the phase structure of FAPbI3 was systematically investigated by varying the molar ratio of FAI/PbI2 in the precursor solution from 1 to 3. All the films were readily prepared and characterized by X-ray diffraction. For clarity, we name the as-prepared film x M-FAI in the following discussion, x being the molar ratio of FAI/PbI2 in the precursor solution. In Fig. 1a, we show XRD patterns zoomed in the 2θ peak from 8 to 24° (the complete diffraction patterns are shown in Fig. S1 and S2†). For the 1 M-FAI film annealed at 60 °C, the diffraction peaks at 11.8° (010) and 16.3° (011) show the formation of pure δ-FAPbI3.24 However, when the annealing temperature reached 170 °C, the observed peaks at 13.9° (−111) and 19.7° (012) indicate the formation of pure α-FAPbI3.24,26 Interestingly, as the precursor solution changed from 1.1 M-FAI to 1.5 M-FAI, all the obtained films at a low annealing temperature of 60 °C exhibited two obvious diffraction peaks at 13.9° and 11.8°. The former corresponds to α-FAPbI3, while the latter belongs to δ-FAPbI3. Moreover, along with the increase of excess FAI, the diffraction peak at 13.9° gradually increased while the peak at 11.8° relatively decreased, suggesting a growing volume fraction of α-FAPbI3. When FAI/PbI2 = 2 (2 M-FAI), the (010) diffraction corresponding to δ-FAPbI3 disappeared and a new peak appeared at 10.4°. Although the exact phase formed with 2 M-FAI has not been identified yet, this small angle diffraction peak might be related to a large interplanar spacing of some layered intermediate phase.31 In the case of 3 M-FAI, the diffraction peaks at 10.3° and 9.9° could be assigned to a reported one-dimensional compound FA3PbI5 (Fig. S3†).32 It should be noted that the excess FAI is not detected in all of the XRD patterns (Fig. S4†), which is presumably due to the low crystallinity of FAI in the as-deposited films as previously reported.33 Another possible reason is that the excess FAI can insert into the crystal lattice and form some low dimensional perovskites as in the case of 2 M-FAI and 3 M-FAI. We also fabricated the film from an under-stoichiometric precursor with FAI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PbI2 = 0.9:1, which gave a mixture of PbI2 and δ-FAPbI3 (XRD pattern shown in Fig. S5†). Overall, by carefully tuning the molar ratio of FAI/PbI2 between 1.1 and 1.5 in the precursor solution, we are able to obtain a series of mixed-phase FAPbI3 films with controllable α/δ composition.
:PbI2 = 0.9:1, which gave a mixture of PbI2 and δ-FAPbI3 (XRD pattern shown in Fig. S5†). Overall, by carefully tuning the molar ratio of FAI/PbI2 between 1.1 and 1.5 in the precursor solution, we are able to obtain a series of mixed-phase FAPbI3 films with controllable α/δ composition.
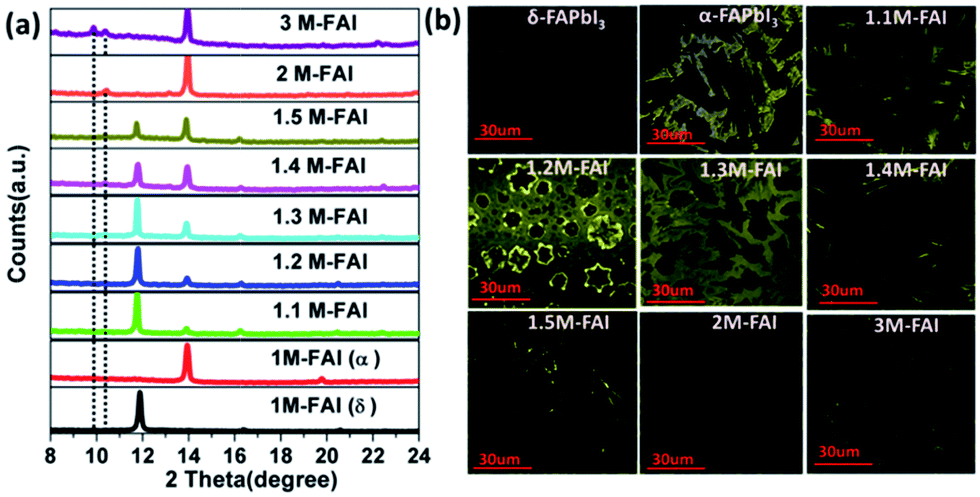 | ||
| Fig. 1 XRD patterns (a) and confocal fluorescence microscopy images (b) of the FAPbI3 films formed with different FAI/PbI2 molar ratios from 1 to 3. All the films were annealed at low temperature (60 °C), except for pure α-FAPbI3 at 170 °C. For confocal fluorescence microscopy, all the films were excited at 485 nm, except for δ-FAPbI3 at 405 nm. | ||
Confocal fluorescence microscopy was then utilized to characterize the crystal morphology and optical properties of the obtained FAPbI3 films.34 As shown in Fig. 1b, the δ-FAPbI3 (P63mc) crystals are in the shape of a hexagon, while the α-FAPbI3 crystals (P3m1) look like a triangle (regular or distorted).24 Generally, the shape of crystals mainly comes from the symmetry of the lattice structure. When excess FAI is present, the shape of the crystals exhibits a gradual transition from triangles for 1.1 M-FAI to regular polygons for 1.2 M-FAI and then to distorted polygons for 1.3 M-FAI to 1.5 M-FAI, all of which were quite different from those of pure α or δ-FAPbI3. In the cases of 2 M-FAI and 3 M-FAI, the crystals become round and hexagon-like, respectively. Concomitantly, the images also qualitatively reflect on the brightness of each film. Notably, the mixed-phase films look brighter in the microscopy images, clearly indicating their higher PL intensity, especially for the film with 1.2 M-FAI. We then conducted steady state PL measurements on these films (Fig. 2a). For the FAPbI3 films of the pure phases, the PL spectra were in accordance with the previous observation, a broad emission from 400 to 650 nm for δ-FAPbI3 and a maximum peak at 815 nm for α-FAPbI3.23,24 In comparison, all the mixed-phase films demonstrated distinct emission profiles with the same maximum peak around 780 nm, which is very different from either of the pure α or δ phases or the simple mixture of the α and δ phase. The films with 2 M-FAI and 3 M-FAI showed much more blue-shifted emissions due to their relatively larger band gaps of low dimensional perovskites.31,32
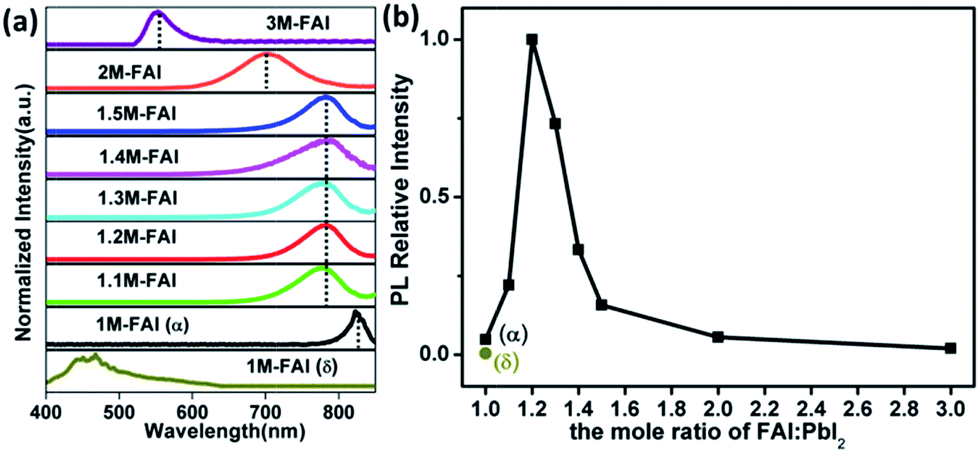 | ||
| Fig. 2 (a) The normalized PL spectra of the FAPbI3 films formed with stoichiometrically modified precursor solutions. All the films were excited at 466 nm, except for δ-FAPbI3 at 300 nm. (b) The relative PL intensity of different FAPbI3 films measured with the integral area of their PL spectra. | ||
Apart from the tunable emission peak wavelength, the relative PL intensity increases and reaches its maximum at 1.2 M-FAI and then decreases with more FAI towards 3 M-FAI (Fig. 2b), which was consistent with the observed brightness change by the confocal fluorescence microscopy analysis. It was very striking that all the as-prepared mixed-phase FAPbI3 showed much higher PL intensities than the pure phases. Especially for the 1.2 M-FAI film, the relative PL intensity was nearly 20-fold and 218-fold higher than that of pure α-FAPbI3 and δ-FAPbI3, respectively. The absolute PL quantum yield (PLQY) of the 1.2 M-FAI film was measured to be 6.9%, which is comparable to that of the MAPbI3 film,34 but nearly one order of magnitude higher than those of pure α-FAPbI3 (0.7%) and δ-FAPbI3 (0.3%). We further performed fluorescence lifetime imaging microscopy (FLIM) on these films (Fig. S6†). The PL decay of the 1.2 M-FAI film exhibited the longest lifetime of 6.67 ns in all cases, which indicated a much lower defect concentration.35,36 The distinct optical properties clearly indicated that the as-prepared mixed-phase FAPbI3 films, in particular for the 1.2 M-FAI film, should have unique microstructures and electronic structures. Here, it is worth noting the residue of excess FAI in the mixed-phase FAPbI3 films. Although it is not detected by XRD, it becomes interesting to wonder what role the FAI residue plays in the PL properties of the mixed-phase film. We further conducted XRD and PL measurements on the film from the same over-stoichiometric precursor at 1.2 M-FAI but annealed at 170 °C. The XRD pattern of the film clearly showed that δ-FAPbI3 vanished because of the high annealing temperature and only α-FAPbI3 remained mixed with the residue of excess FAI (Fig. S7a†). However, this film gave very similar PL spectra and comparable PLQY around 0.7% to that of the pure α-FAPbI3 film from the stoichiometric 1 M-FAI precursor (Fig. S7b†). This result indicated that the residue of excess FAI may not play a role in affecting the PL properties of α-FAPbI3. In comparison, for the 1.2 M-FAI film annealed at a low temperature of 60 °C, δ-FAPbI3 was involved and mixed with α-FAPbI3. Consequently, great changes took place in the PL properties of the film, which showed markedly hypochromic emission centered at 780 nm and 10-fold enhanced PLQY. These results strongly suggested that the involved δ-FAPbI3 and concomitant co-existence of α-FAPbI3 and δ-FAPbI3 would be highly correlated with the distinct PL properties of the mixed-phase films.
To visualize such mixed-phase structures, the 1.2 M-FAI film was investigated by high resolution transmission electron microscopy (HRTEM). As shown in Fig. 3a, well defined lattice fringes with a separation of 3.17 Å and 2.38 Å could be well indexed to the (−222) plane of α-FAPbI3 and the (031) plane of δ-FAPbI3, respectively.24 Such HRTEM results (other images shown in Fig. S8†), clearly showed that a junction structure between the α and δ phases is formed in the as-prepared FAPbI3 films. When excited with different wavelengths of light (Fig. S9a†), the 1.2 M-FAI film always exhibits the same PL emission peak around 780 nm, regardless of whether the excitation energy was strong enough to excite δ-FAPbI3 individually. There is a 35 nm blue shift compared to that of the pure α phase. These results clearly reveal that the existence of the α/δ phase junction does modify the band structure of the as-prepared mixed-phase films, and the amplitude is observable.
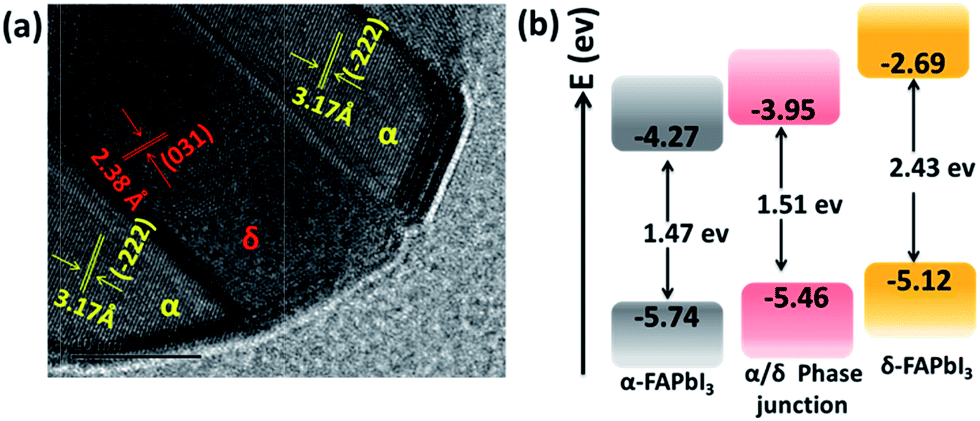 | ||
| Fig. 3 (a) HRTEM of the 1.2 M-FAI film with the α/δ phase junction. (b) Schematic energy level diagrams of the films of pure α-FAPbI3, pure δ-FAPbI3 and the α/δ phase junction for the 1.2 M-FAI film. | ||
To confirm these observations, we performed ultraviolet-visible absorption and ultraviolet photoelectron spectroscopy (UPS) measurements on the as-prepared film with 1.2 M-FAI in comparison with that of the pure α and δ phase. For the films of pure α-FAPbI3 and δ-FAPbI3, the absorption onset is located around 840 nm and 510 nm (Fig. S9b†), corresponding to an optical band gap energy (Eg) of 1.47 and 2.43 eV, respectively, which is in good agreement with the literature reported.24 For the 1.2 M-FAI film, the absorption onset has a blue shift of 20 nm and lies at 820 nm, indicating a slightly larger Eg (1.51 eV) compared to that of pure α-FAPbI3. The valence band maximum (VBM) of the films was then determined by UPS measurements. As shown in Fig. S10,† the VBM is determined to be −5.74, −5.12, and −5.46 eV for α-FAPbI3, δ-FAPbI3 and the 1.2 M-FAI film, respectively. The schematic energy level diagram is shown in Fig. 3b, which clearly demonstrates that both the conduction band minimum (CBM) and VBM of the 1.2 M-FAI film are just between those of α-FAPbI3 and δ-FAPbI3. It was reported that band bending would occur in the phase junction through Fermi level lining up when they are in contact with each other.37 Accordingly, the band edge of the α/δ phase junction could be attributed to the valence band upward bending of α-FAPbI3 and the conduction band downward bending of δ-FAPbI3 in their junction area. Such an α/δ phase junction exists in all of the films from 1.1 M-FAI to 1.5 M-FAI, so these films should have the same bandgap, which is in good agreement with their similar PL spectra.
For the perovskite films, the high PL intensity is strongly related to the high exciton binding energy (EB) because the thermal ionization of excitons with a low EB could induce PL quenching, and also involve the decrease of grain size, hence spatially confining the excitons.11,38,39 The exciton binding energy, EB, is a modified Rydberg energy given by a static solution to the Wannier equation:40
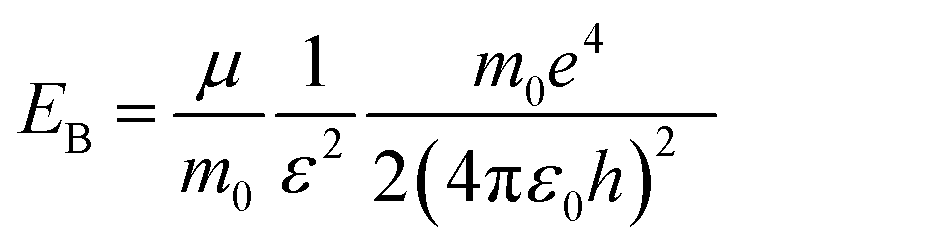 | (1) |
Herein, μ is the exciton effective mass, m0 the electron mass, e the unit charge, ε the dielectric constant and ε0 the vacuum permittivity. To understand the greatly enhanced PL from the 1.2 M-FAI film with the α/δ phase junction, we then measured the dielectric constant of the film in comparison to the films with the pure α and δ phase (Fig. S11†). The dielectric constant of the 1.2 M-FAI film was measured to be 4.8, much lower than those of both the pure α phase (6.6) and pure δ phase (6.9). Such a markedly reduced dielectric constant can be roughly described by Lichtenecker's model for the dielectric function of a two-phase composite according to a serial mixing rule,41 in which two phases alternate or randomly connect in parallel as evidenced by the HRTEM of the 1.2 M-FAI film. Since the exciton binding energy is proportional to 1/ε2 as eqn (1) states, if we roughly estimated the same exciton effective mass, the exciton binding energy of the 1.2 M-FAI film might be nearly two-fold higher than that of the pure α-FAPbI3 and δ-FAPbI3. In accordance with this, the reduced dielectric constant would help increase the exciton binding energy, thus avoiding luminescence quenching.11 In the meantime, scanning electron microscopy (SEM) was used to measure the average grain size of these films. As shown in Fig. 4, the average grain sizes of the pure α-FAPbI3 and δ-FAPbI3 are 139 and 261 nm, respectively, whereas that of the 1.2 M-FAI film is greatly reduced to 64 nm, only one third or one fourth of that of the pure phases (Fig. S12†). This substantial reduction of the average grain size, on the one hand, can lead to a reduction of coherence length of the ferroelectric coupling between local dipoles and a diminished macroscopic polarization, thus causing a reduction in the dielectric constant,42 on the other hand, it can spatially confine the excitons, thus increasing the radiative recombination in the nanograins.11 In addition, the volume fraction of α-FAPbI3 in the 1.2 M-FAI film is considerably small due to low annealing temperature, which can help reduce the re-absorption by α-FAPbI3, and thereby likewise contribute to high PL intensity.
 | ||
| Fig. 4 Top-view scanning electron microscope (SEM) images of the films: δ-FAPbI3, α-FAPbI3 and 1.2 M-FAI with the α/δ phase junction. | ||
To reveal the role of the precursor stoichiometry in the formation of such unique α/δ phase junctions, we then investigated the colloidal properties of these precursor solutions. The absorbance edge of all of these precursor solutions demonstrated a gradual red shift along with the precursor varying from 1 M-FAI to 3 M-FAI (Fig. S13a†). Meanwhile, dynamic light scattering tests were conducted to characterize the colloidal size distribution associated with the formed lead polyiodide compounds in different precursor solutions (Fig. S13b†). Results with a shrinking variation trend came out showing a gradually decreased colloidal size when increasing the molar ratio of FAI/PbI2. The average size of the colloid in the 1 M-FAI precursor solution was estimated to be ∼1262 nm, while the value of the 1.2 M-FAI precursor solution remarkably decreased to ∼354 nm. This trend revealed that excess FAI in the precursor solution can bring about more coordination effects and thus reduce the formation of large colloids,31 which was consistent with the above observed small grains for the 1.2 M-FAI film. Generally, δ-FAPbI3 is found to be more thermodynamically favorable and stable compared to α-FAPbI3 at low temperature due to a lower formation energy.43 However, when the size of the colloid particles decreases to sufficiently low values, the total free energy (including surface and bulk) of δ-FAPbI3 may be equal to or even higher than that of α-FAPbI3 and the relative phase stability may reverse, thus facilitating the formation of α-FAPbI3 even at a low annealing temperature and further allowing the formation of an α/δ phase junction.44,45 On the basis of these results, we proposed a possible mechanism concerning the perovskite crystal growth with excess FAI in the precursor solution (as shown in Fig. 5). Therefore, even when annealed at a temperature as low as 60 °C, the FAPbI3 films with an appropriate excess of FAI could form the α and δ phase synchronously, which results in the formation of an α/δ phase junction.
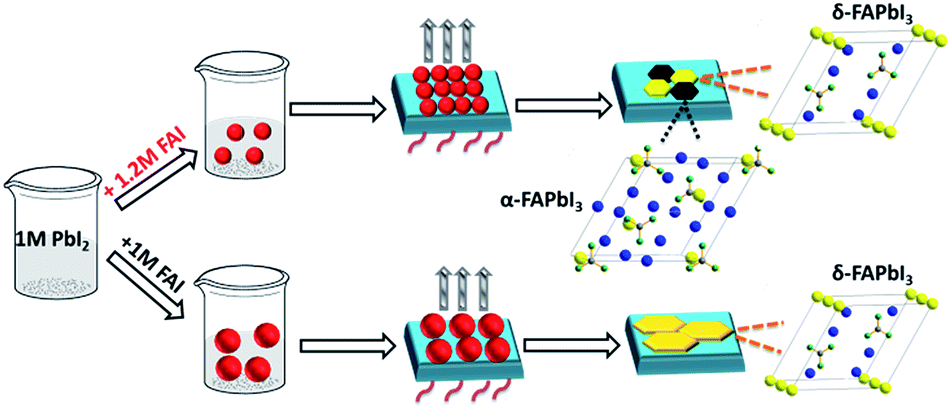 | ||
| Fig. 5 Schematic view of the possible crystal growth process of FAPbI3 films via controllable precursors. The unit cell of FAPbI3: yellow: Pb; gray: C; green: N; blue: I. | ||
Finally, we investigated the preliminary stability of the as-prepared FAPbI3 perovskite films with an α/δ phase junction in comparison with that of the pure α-FAPbI3. All the films were stored in an ambient environment without encapsulation (at 25 °C and relative humidity <50%). Unexpectedly, even after 15 days, the XRD pattern of the 1.2 M-FAI film with the α/δ phase junction remained unchanged without showing any observable decomposition or phase transition (Fig. 6a). In contrast, for the as-prepared film with pure α-FAPbI3, the XRD pattern (Fig. 6b) showed the peak at 12.6° corresponding to PbI2, indicating that FAPbI3 does decompose when annealed at high temperature.21,30 Within only 24 hours, the peak at 11.8°belonging to δ-FAPbI3 appeared, indicating that part of the α phase turned into the δ phase. After 15 days, α-FAPbI3 had fully turned into δ-FAPbI3. Therefore, these results clearly demonstrated that the obtained films with the α/δ phase junction showed long-term stability against humidity. As evidenced by the XRD patterns in Fig. 1, we can roughly estimate that there is only a small volume fraction of α-FAPbI3 in the 1.2 M-FAI film. Considering the thermodynamics, such superior air stability could be mainly attributed to the majority of the film being thermodynamically stable δ-FAPbI3 at low temperature and concomitant stabilization from the mixing entropy of the α/δ phase.28 In addition, such stabilization can be rationalized in terms of miscibility of the α/δ phase due to almost the same volume per stoichiometric unit of the two crystals (∼256 Å3).24,28 To gain more insight into the role of the α/δ phase junction in air stability, further investigation through in situ characterization and theoretical calculation is required.
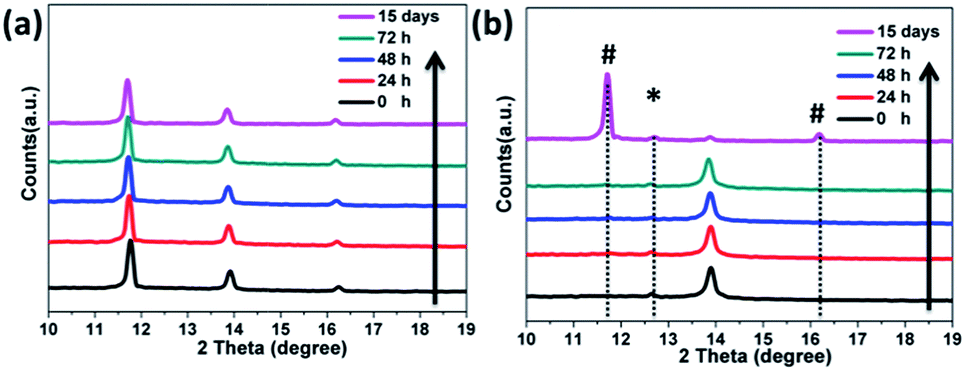 | ||
| Fig. 6 XRD spectra of the films as a function of time under ambient humid conditions (temperature at 25 °C and relative humidity <50%). (a) The 1.2 M-FAI film with the α/δ phase junction, and (b) pure α-FAPbI3 annealed at 170 °C. The reflections indicated by * and # represent the Bragg reflections associated with PbI2 and δ-FAPbI3, respectively. | ||
Conclusions
In summary, we have realized the first pure FAPbI3 perovskite films with controllable α/δ phase junctions by using stoichiometrically modified precursors, which demonstrated desirable humidity stability and significantly enhanced NIR emission. The enhanced NIR emission was found to be related to the unique α/δ phase junction and the reduced grain size. Interestingly, such α/δ phase junctions can be readily obtained at low annealing temperature (60 °C) and demonstrated long-term phase stability against humidity. The stable and controllable phase-junction presented herein would pave a new way for developing highly stable and efficient perovskite light emitting materials and devices. We are currently carrying out studies on the perovskite light-emitting diodes with this aim.Acknowledgements
This work was supported by the National Key Basic Research and Development Program of China (973 program, Grant No. 2015CB655002 and 2016YFB0401003) founded by MOST, National Nature Science Foundation of China (Grants No. 51473086 and 91433205) and Tsinghua University Initiative Scientific Research Program.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591–597 Search PubMed
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D.-D. Jarausch, R. Higler, S. Hüttner, T. Leijtens, S. D. Stranks, H. J. Snaith, M. Atatüre, R. T. Phillips and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS PubMed
- Y.-H. Kim, H. Cho, J. H. Heo, T.-S. Kim, N. Myoung, C.-L. Lee, S. H. Im and T.-W. Lee, Adv. Mater., 2015, 27, 1248–1254 CrossRef CAS PubMed
- J. Byun, H. Cho, C. Wolf, M. Jang, A. Sadhanala, R. H. Friend, H. Yang and T.-W. Lee, Adv. Mater., 2016, 28, 7515–7520 CrossRef CAS PubMed
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T.-W. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed
- J. Wang, N. Wang, Y. Jin, J. Si, Z.-K. Tan, H. Du, L. Cheng, X. Dai, S. Bai, H. He, Z. Ye, M. L. Lai, R. H. Friend and W. Huang, Adv. Mater., 2015, 27, 2311–2316 CrossRef CAS PubMed
- O. A. Jaramillo-Quintero, R. S. Sanchez, M. Rincon and I. Mora-Sero, J. Phys. Chem. Lett., 2015, 6, 1883–1890 CrossRef CAS PubMed
- R. L. Z. Hoye, M. R. Chua, K. P. Musselman, G. Li, M.-L. Lai, Z.-K. Tan, N. C. Greenham, J. L. MacManus-Driscoll, R. H. Friend and D. Credgington, Adv. Mater., 2015, 27, 1414–1419 CrossRef CAS PubMed
- G. Li, Z.-K. Tan, D. Di, M. L. Lai, L. Jiang, J. H.-W. Lim, R. H. Friend and N. C. Greenham, Nano Lett., 2015, 15, 2640–2644 CrossRef CAS PubMed
- J. C. Yu, D. B. Kim, G. Baek, B. R. Lee, E. D. Jung, S. Lee, J. H. Chu, D.-K. Lee, K. J. Choi, S. Cho and M. H. Song, Adv. Mater., 2015, 27, 3465 CrossRef
- J. Li, S. G. R. Bade, X. Shan and Z. Yu, Adv. Mater., 2015, 27, 5196–5202 CrossRef CAS PubMed
- J. C. Yu, D. B. Kim, E. D. Jung, B. R. Lee and M. H. Song, Nanoscale, 2016, 8, 7036–7042 RSC
- S. G. R. Bade, J. Li, X. Shan, Y. Ling, Y. Tian, T. Dilbeck, T. Besara, T. Geske, H. Gao, B. Ma, K. Hanson, T. Siegrist, C. Xu and Z. Yu, ACS Nano, 2016, 10, 1795–1801 CrossRef CAS PubMed
- T. M. Koh, K. Fu, Y. Fang, S. Chen, T. C. Sum, N. Mathews, S. G. Mhaisalkar, P. P. Boix and T. Baikie, J. Phys. Chem. C, 2014, 118, 16458–16462 CAS
- S. Pang, H. Hu, J. Zhang, S. Lv, Y. Yu, F. Wei, T. Qin, H. Xu, Z. Liu and G. Cui, Chem. Mater., 2014, 26, 1485–1491 CrossRef CAS
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 CAS
- D.-J. Seol, J.-W. Lee and N.-G. Park, ChemSusChem, 2015, 8, 2414–2419 CrossRef CAS PubMed
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed
- N. Pellet, P. Gao, G. Gregori, T.-Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Gratzel, S. M. Zakeeruddin, U. Rothlisberger and M. Gratzel, Energy Environ. Sci., 2016, 9, 656–662 CAS
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Hörantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed
- S. Lv, S. Pang, Y. Zhou, N. P. Padture, H. Hu, L. Wang, X. Zhou, H. Zhu, L. Zhang, C. Huang and G. Cui, Phys. Chem. Chem. Phys., 2014, 16, 19206–19211 RSC
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, J. Am. Chem. Soc., 2015, 137, 4460–4468 CrossRef CAS PubMed
- S. Wang, D. B. Mitzi, C. A. Feild and A. Guloy, J. Am. Chem. Soc., 1995, 117, 5297–5302 CrossRef CAS
- M. Yang, Y. Zhou, Y. Zeng, C.-S. Jiang, N. P. Padture and K. Zhu, Adv. Mater., 2015, 27, 6363–6370 CrossRef CAS PubMed
- D. W. de Quilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed
- B. Sun, A. V. Vorontsov and P. G. Smirniotis, Langmuir, 2003, 19, 3151–3156 CrossRef CAS
- I. B. Koutselas, L. Ducasse and G. C. Papavassiliou, J. Phys.: Condens. Matter, 1996, 8, 1217–1227 CrossRef CAS
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS
- Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2014, 9, 106–112 CrossRef
- K. Lichtenecker, Phys. Z., 1926, 27, 58–115 Search PubMed
- V. Brize, G. Gruener, J. Wolfman, K. Fatyeyeva, M. Tabellout, M. Gervais and F. Gervais, Mater. Sci. Eng., B, 2006, 129, 135–138 CrossRef CAS
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2016, 28, 284–292 CrossRef CAS
- H. Zhang and J. F. Banfield, J. Mater. Chem., 1998, 8, 2073–2076 RSC
- A. Orendorz, A. Brodyanski, J. Lösch, L. H. Bai, Z. H. Chen, Y. K. Le, C. Ziegler and H. Gnaser, Surf. Sci., 2007, 601, 4390–4394 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03542f |
| This journal is © The Royal Society of Chemistry 2017 |