DOI:
10.1039/C6SC03482A
(Edge Article)
Chem. Sci., 2017,
8, 1337-1343
A metal-mediated base pair that discriminates between the canonical pyrimidine nucleobases†
Received
4th August 2016
, Accepted 12th October 2016
First published on 12th October 2016
Abstract
A nucleoside analogue comprising the ligand 1H-imidazo[4,5-f][1,10]phenanthroline (P) was applied to develop a molecular beacon capable of discriminating the canonical nucleobases cytosine and thymine. The beacon is based on the formation of a stable Ag+-mediated base pair between P and cytosine, whereas the presence of Ag+ strongly destabilizes nucleic acids comprising an artificial base pair between P and thymine. Metal-mediated base pair formation was investigated by temperature-dependent UV spectroscopy and CD spectroscopy and complemented by extensive DFT calculations. The molecular beacon significantly extends the application spectrum of nucleic acids with metal-mediated base pairs. It is of potential use in the detection of single-nucleotide polymorphisms.
Introduction
Nucleic acids represent an optimal scaffold for the introduction of functional molecules.1 The combination of their self-assembling capability with functional moieties renders them ideal building blocks in supramolecular chemistry and nanotechnology.2 Metal-mediated base pairs represent a convenient means for the introduction of metal-based functionality into nucleic acids. In these artificial base pairs, metal ions are located in-between complementary nucleobases, formally replacing the hydrogen bonds found in canonical base pairs.3 Various experimental structures have confirmed that nucleic acid duplexes with metal-mediated base pairs still retain their natural duplex conformation,4 even though a transition to another conformation has been established, too.5 Nucleosides used in metal-mediated base pairs can be either entirely natural,6 entirely artificial,7 or be formally derived from a natural nucleoside.8 Several applications have been proposed and developed for nucleic acids with metal-mediated base pairs.9 While most of these applications are related to the sensing of metal ions (particularly Hg2+ and Ag+),10 investigations toward increasing the charge transfer properties of DNA11 or generating DNA-templated silver nanoparticles12 have also been pursued.
Moreover, the fact that metal-mediated base pairs can be processed by polymerases13 makes them interesting candidates for the use in molecular computing devices.14 Recently, nucleic acids with metal-mediated base pairs have been proposed to be useful in the recognition of nucleic acid sequences.15 We report here the first implementation of this application. Toward this end, the ligand 1H-imidazo[4,5-f][1,10]phenanthroline was used as artificial nucleobase. We could previously show that a nucleoside analogue P (Chart 1) bearing this nucleobase is capable of forming Ag+-mediated homo base pairs16 as well as hetero base pairs17 with an artificial imidazole nucleoside. In this study, we establish its use in the discrimination of the canonical nucleobases cytosine and thymine by developing a turn-on fluorescence sensor.
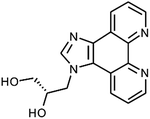 |
| | Chart 1 Artificial nucleoside analogue P based on the ligand 1H-imidazo[4,5-f][1,10]phenanthroline. | |
Results and discussion
Acidity constants of imidazophenanthroline
To evaluate a possible competition between protonation and metalation of the nucleoside analogue P, its acidity constants were determined by pD-dependent 1H NMR spectroscopy. For comparison, the pKa values of the closely related 1-methyl-1H-imidazo[4,5-f][1,10]phenanthroline 1 were determined, too. As can be seen from Fig. 1, S1 and S2,† the experimental data are fitted best under the assumption of three pKa values. Table 1 lists the acidity constants derived from these data.
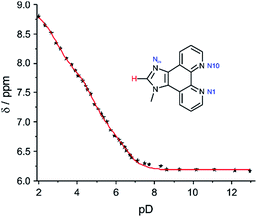 |
| | Fig. 1 Chemical shift of the imidazole hydrogen atom of 1 depending on the pD value, including least-squares fit assuming the presence of three distinct pKa values. | |
Table 1 Acidity constants of the nucleoside analogue P and its methyl derivative 1
| Compound |
pKa1 |
pKa2 |
pKa3 |
|
P
|
5.4 ± 0.2 |
3.6 ± 0.2 |
1.3 ± 0.2 |
|
1
|
6.0 ± 0.1 |
4.3 ± 0.1 |
2.6 ± 0.1 |
The GNA nucleoside P is slightly more acidic than its methyl analogue 1. However, it can be anticipated that the latter data better describe the situation inside a DNA duplex, because the negative charge introduced by the phosphate group typically leads to a decrease in acidity compared to the nucleoside data.18 The NMR data do not allow an assignment of which site is protonated at which pKa value. We therefore calculated proton-binding energies of the three nitrogen atoms in 1H-imidazo[4,5-f][1,10]phenanthroline. These data (Table S1†) indicate that one of the endocyclic nitrogen atoms of the phenanthroline ring is protonated first, as both of them have essentially identical proton-binding energies, exceeding that of the imidazole nitrogen atom by more than 5 kcal mol−1 under aqueous conditions. Hence, the following order of protonation can be assumed: N1 or N10, Nim, N10 or N1. A comparison of the pKa value of 6.0 ± 0.1 with the respective value in unsubstituted 1,10-phenanthroline, which amounts to 4.98,19 shows that the appended imidazole moiety leads to an increase in basicity. Concomitantly, its own basicity decreases from 7.05 for unsubstituted imidazole20 to 4.3 ± 0.1. The pKa values indicate that protonation of the ligand cannot be neglected when working under acidic conditions. The acidity constant pKa2 is similar to that of cytosine (1-methylcytosine, pKa = 4.6; deoxycytidine, pKa = 4.17; 5′-dCMP, pKa = 4.54),18 indicating that when present in the same oligonucleotide, cytosine and the imidazole moiety of P may become protonated concurrently.
Base pairing of P with cytosine and thymine
To investigate the applicability of the nucleoside analogue P in a metal-mediated base pair with a canonical pyrimidine nucleoside, oligonucleotide duplexes of the type depicted in Chart 2 were synthesized. The sequence was chosen because it had previously been used for the characterization of metal-mediated base pairs formed from P:imidazole hetero mispairs, allowing an optimal comparison.17 In the present study, cytosine (C) or thymine (T) were used as the complementary nucleoside denoted X in Chart 2. Information on the stability of the unnatural base pairs was derived from the melting temperatures Tm of the corresponding duplexes, determined by using temperature-dependent UV spectroscopy. All experiments were performed under slightly acidic (pH 5.5), near-neutral (pH 6.8) and slightly alkaline (pH 9.0) conditions, thereby probing also the influence of nucleobase protonation and deprotonation. Silver(I) was chosen as metal ion for metal-mediated base pairs, because previous studies involving P had shown that Ag+ leads to the most significant thermal stabilization. Fig. 2 shows the melting curves of the DNA duplexes in the presence of increasing amounts of AgNO3. Table 2 lists the melting temperatures derived from these curves. They will be discussed in the following two paragraphs.
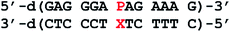 |
| | Chart 2 Oligonucleotide sequence used to determine the base pairing properties of the artificial nucleoside analogue P opposite to a canonical pyrimidine nucleoside X. | |
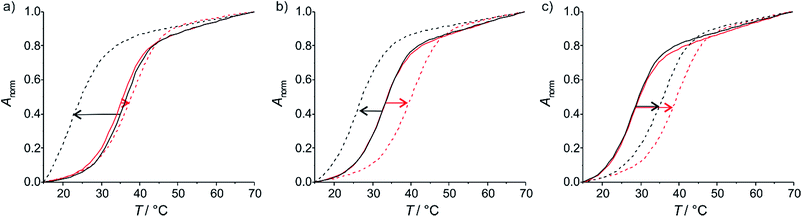 |
| | Fig. 2 Melting curves of the duplexes in the absence (solid lines) and presence (dotted lines) of one equivalent of AgNO3. The red melting curves belong to the duplex comprising a cytosine residue opposite to P, the black ones to that with a thymine residue. (a) pH 5.5, (b) pH 6.8, (c) pH 9.0. | |
Table 2 Melting temperatures (Tm/°C) of the duplexes bearing one P:C or P:T base pair in the absence (Tm,0) and presence (Tm,1) of one equiv. of AgNO3 and change in Tm (ΔTm/°C) upon the addition of one equiv. of AgNO3. References are made to Chart 3 and Fig. 3, which display the proposed base pairing patterns
Hydrogen-bond-mediated base pairs
In the absence of Ag+, the melting temperatures are (within error limits) identical for the duplexes comprising a P:C or a P:T base pair at each of the pH values under investigation. The general trend is that the duplexes are more stable at lower pH. This trend differs from that of the corresponding duplexes with a central A:T or G:C base pair (A, adenine; G, guanine), which are most stable at near-neutral pH (Fig. S4 and Table S2†). Hence, protonation/deprotonation seems to have an important effect on the stability of the metal-free P:C and P:T base pairs. Possible hydrogen-bonded P:C and P:T base pairs are shown in Chart 3. All base pairs contain either two hydrogen bonds (Tm = 32–35 °C, Chart 3a–d) or one bifurcated hydrogen bond (Tm = 28 °C, Chart 3e and f). The structures of the base pairs were confirmed by DFT calculations to represent energy minima (Fig. S5†). The calculations show a good correlation between computed base pairing energies and experimental melting temperature (Table S3†). In an aqueous environment, the base pairing energies of the pairs comprising two hydrogen bonds are around −10 kcal mol−1, coinciding with a Tm in the order of 32–35 °C. The lower melting temperature of 28 °C at pH 9 agrees well with reduced base pairing energies of around −6 kcal mol−1, which can again be explained by the presence of one (bifurcated) hydrogen bond only.
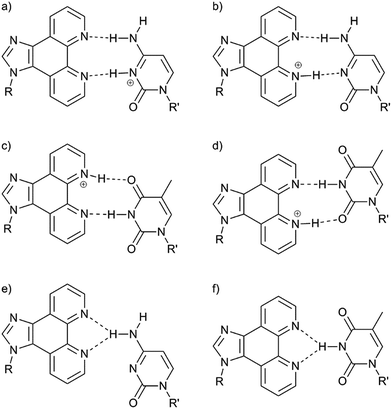 |
| | Chart 3 Possible hydrogen-bonded base pairs. (a) P:C(H)+; (b) P(H(N1))+:C; (c) P(H(N10))+:T; (d) P(H(N1))+:T; (e) P:C; (f) P:T. | |
Metal-mediated base pairs
Upon the addition of AgNO3 to the oligonucleotide duplexes, changes in the melting temperatures can be observed (Fig. 2). In all cases, the addition of the first equivalent of Ag+ influences Tm the most. Excess Ag+ has only minor additional effects on Tm. This behaviour is typical for the formation of a mononuclear metal-mediated base pair.3a Interestingly, the addition of AgNO3 has dramatically different effects depending on whether cytosine or thymine is located opposite to the artificial nucleobase P. In the case of cytosine, the final melting temperature amounts to 40 °C and is (within standard deviation) independent of the pH. This suggests that the same P–Ag+–C base pair (Fig. 3a) is formed under all conditions. In contrast, with thymine a stabilizing base pair is formed under alkaline conditions only, with a Tm of 37 °C. The similar melting temperatures of the duplex comprising this base pair and the duplexes with a P–Ag+–C base pair suggest that these silver(I)-mediated base pairs are of similar composition. Most likely, the elevated pH facilitates the deprotonation of the thymine residue (1-methylthymine, pKa = 10.4; thymidine, pKa = 9.93; 5′-dTMP, pKa = 10.47),18 enabling the formation of a P–Ag+–T–H base pair (Fig. 3b).
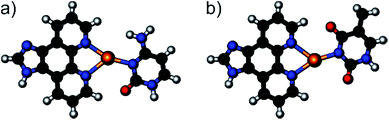 |
| | Fig. 3 Geometry-optimized structures of the silver(I)-mediated base pairs (a) P–Ag+–C and (b) P–Ag+–T–H. | |
At pH 6.8 and 5.5, the presence of AgNO3 effects a significant destabilization of the duplex. The observation that one Ag+ is bound per duplex under all experimental conditions strongly suggests that the bidentate nucleoside P is metalated irrespective of pH. The significant destabilization at pH 6.8 and 5.5 results from the fact that the previously existing P(H(N1))+:T and P(H(N10))+:T base pairs can no longer be formed as a result of the metalation of P. A reasonable assumption is that one of the bases extrudes from the duplex, therefore leading to a decrease in thermal stability.
DFT calculations support the structures of the proposed metal-mediated base pairs. Table 3 lists base pairing energies computed under the assumption that the bidentate P binds Ag+ first, leading to [P–Ag]+ (or [P(H(Im))–Ag]2+ if protonated). The subsequent formation of the Ag+–C or Ag+–T–H bond is presumed to represent the stability-determining step. As can be seen from Table 3, the silver-mediated base pairs P–Ag+–C and P–Ag+–T–H are of very similar stability under aqueous conditions, corroborating the very similar melting temperatures of 37–40 °C. Even a potential protonation of P in P–Ag+–C under acidic conditions would not influence the base pairing energies. No chemically reasonable structures could be computed for a silver(I)-mediated base pair involving P and non-deprotonated T. This finding is in agreement with the proposal that one of the bases extrudes from the duplex when adding AgNO3 to the duplex comprising a central P:T base pair at pH 6.8 or 5.5.
Table 3 Base pairing energies ΔEBP of hydrogen-bond mediated base pairs (kcal mol−1)a
| Entry |
Base pair |
Gas phase |
In water |
|
The following reactions have been used to calculate the base pairing energies. (1) [P(H(Im))–Ag]2+ + C → P(H(Im))+–Ag+–C; (2) [P–Ag]+ + C → P–Ag+–C; (3) [P–Ag]+ + T–H → P–Ag+–T–H.
|
| 1 |
P(H(Im))+–Ag+–C |
−60.6 |
−23.4 |
| 2 |
P–Ag+–C |
−47.0 |
−23.1 |
| 3 |
P–Ag+–T–H |
−125.8 |
−25.8 |
Circular dichroism (CD) spectroscopy was used to investigate the structural similarity of the various duplexes. As can be seen in Fig. 4, the spectra of the duplex comprising a P–Ag+–C base pair are almost identical at all three pH values. This agrees well with identical melting temperatures and base pair formation energies. In contrast, the spectra of the duplex with a central P:T pair differ significantly from one another, which is in agreement with their differing melting temperatures. Interestingly, the CD spectrum of the duplex with a P–Ag+–T–H base pair (black dotted line) is different from the corresponding spectra of the duplex with a P–Ag+–C base pair (red spectra). Hence, despite similar melting temperatures and base pair formation energies, a distinct structural difference must exist. A closer look at the computed structures (Fig. 3) indicates that the thymine residue experiences a certain displacement within the base pair plane, which probably leads to a distortion of the overall duplex structure. In fact, the distance between the two nitrogen atoms involved in the glycosidic bonds amounts to 10.7 Å (P–Ag+–C) and 11.3 Å (P–Ag+–T–H), respectively. All six CD spectra show a small negative Cotton effect at around 300 nm. Cotton effects in this region are normally not due to DNA. As the artificial nucleobase P slightly absorbs light in the visible region (up to ca. 400 nm),16 the Cotton effect must stem from the presence of P within the base pair stack. This in turn means that the nucleobase suggested to extrude from the P:T-containing duplex in the presence of AgNO3 at pH 6.8 and 5.5 is likely to be the thymine residue.
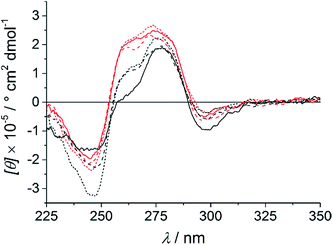 |
| | Fig. 4 CD spectra of the oligonucleotide duplexes with a central cytosine (red) or thymine (black) residue opposite to P in the presence of one equivalent of AgNO3 per duplex. pH 5.5: solid line; pH 6.8: dashed line; pH 9.0: dotted line. | |
Development of a molecular beacon
The fact that the metal-free P:C and P:T base pairs display clearly different affinities towards Ag+ renders them potentially useful for the detection of single-nucleotide polymorphisms (SNPs). SNPs are common variations among the DNA of individuals of the same species. They are expected to be a genetic reason for a varying drug response of different individuals.21 Hence, methods for the detection of SNPs are highly interesting against the background of personalized medicine.22 A molecular beacon is an oligonucleotide probe that fluoresces in the presence of a perfectly matching complementary sequence.23 By using the nucleoside analogue P, a molecular beacon was developed that hybridizes with a target sequence only if the complementary nucleobase is a cytosine (Scheme 1). The sequence used in this study has previously been shown to be ideally suited as a molecular beacon.24
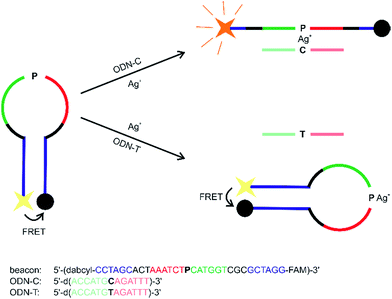 |
| | Scheme 1 Design of a molecular beacon to detect cytosine/thymine SNPs based on the formation of a metal-mediated base pair. | |
As can be seen from the fluorescence spectra shown in Fig. 5, the substantial stability differences of a P:C and P:T base pair in the presence of Ag+ can be successfully exploited to generate a molecular beacon capable of discriminating cytosine and thymine. The fluorescence at 525 nm upon excitation at 495 nm remains essentially unchanged in the presence of Ag+ and the oligonucleotide probe ODN–T with a central thymine residue, whereas it increases by 100% when Ag+ and probe ODN–C with a central cytosine moiety are present. In the absence of Ag+, the beacon is not capable of discriminating ODN–C from ODN–T (Fig. 5), indicating the significance of metal-mediated base pair formation in the sensing process.
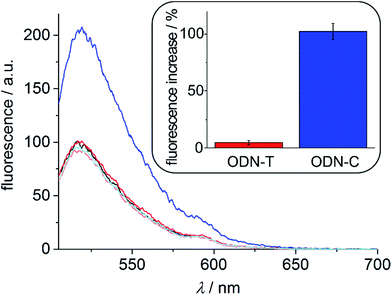 |
| | Fig. 5 Fluorescence spectra of the molecular beacon upon excitation at 495 nm. Molecular beacon ( ); molecular beacon + ODN–T + Ag+ ( ); molecular beacon + ODN–T + Ag+ ( ); molecular beacon + ODN–C + Ag+ ( ); molecular beacon + ODN–C + Ag+ ( ); molecular beacon + ODN–T without Ag+ ( ); molecular beacon + ODN–T without Ag+ ( ); molecular beacon + ODN–C without Ag+ ( ); molecular beacon + ODN–C without Ag+ ( ). The inset shows the average percent increase of the fluorescence at 525 nm in the presence of Ag+ and the respective oligonucleotide probe (including standard deviation of the mean of three independent measurements). All spectra have been normalized with respect to the spectrum of the molecular beacon. ). The inset shows the average percent increase of the fluorescence at 525 nm in the presence of Ag+ and the respective oligonucleotide probe (including standard deviation of the mean of three independent measurements). All spectra have been normalized with respect to the spectrum of the molecular beacon. | |
Conclusions
A molecular beacon that discriminates cytosine from thymine via the formation of a Ag+-mediated base pair has been developed. Towards this end, a nucleoside analogue containing the ligand 1H-imidazo[4,5-f][1,10]phenanthroline P was incorporated into a DNA oligonucleotide. With cytosine in the complementary position, a strongly stabilizing Ag+-mediated base pair is formed. In contrast, the corresponding oligonucleotide sequence containing a thymine instead of the cytosine residue forms a more stable duplex in the absence of Ag+. The molecular beacon makes use of the significantly different acidity constants of cytosine and thymine and the fact that Ag+, which is site-specifically bound by the bidentate ligand P, is not able to replace the thymine N–H proton under neutral or acidic conditions. It can be envisaged that such a molecular beacon may be suitable for the detection of single-nucleotide polymorphisms. Clearly, metal-mediated base pairs capable of discriminating the canonical purine nucleobases need to be developed next.
Experimental section
Determination of the pKa values
The pKa values of 1 and P were determined by recording pD-dependent NMR chemical shifts. pD values of D2O solutions were determined by adding 0.4 to the pH meter reading.25 pKa values in H2O were calculated from the 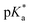 values in D2O according to eqn (1).26
values in D2O according to eqn (1).26| | 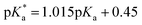 | (1) |
The experimental pD-dependent chemical shift data were fitted with a non-linear least-squares curve fit. The equations used in these fits were taken from the literature or are given in the ESI.†27 The pKa values reported in Table 1 represent weighted means of the individual values determined from the changes in the NMR chemical shifts of the individual hydrogen atom.
Computational details
All calculations have been performed using the Amsterdam Density Functional (ADF) program28 using relativistic density functional theory at the ZORA-BLYP-D/TZ2P for geometry optimization and energies.29 Solvation in water is simulated using the conductor-like screening model (COSMO).30 The stationary points of the monomer and the base pairs were verified to be minima on the potential energy surface (PES) through vibrational analysis. The complexes have also been computed planar to simulate the situation in the DNA helix. See ESI† for more information.
Synthetic procedures
(S)-3-(1H-Imidazo[4,5-f][1,10]phenanthrolin-1-yl)propane-1,2-diol (P) and the artificial phosphoramidite required for the DNA synthesis were prepared as reported previously.16,17 The synthesis of 1 is reported in the ESI.† 6-Carboxyfluorescein (6-FAM CPG), 5′-dabcyl phosphoramidite and the phosphoramidites of the natural nucleosides were purchased from Glen Research. Oligonucleotides were prepared in the DMT-off mode on a K&A Laborgeräte H8 DNA/RNA synthesizer following standard protocols (except for a threefold coupling time for the artificial phosphoramidite) and purified as reported previously.17 Cleavage of the molecular beacon from solid support and deprotection were achieved by treatment with 500 μL of aqueous ammonia for 30 min at room temperature and subsequent treatment with 500 μL of methyl amine. The desalted oligonucleotides were characterized by MALDI-ToF mass spectrometry: 5′-d(GAGGGAPAGAAAG): calcd for [M + H]+: 4148 Da, found: 4148 Da; 5′-d(CTTTCTTTCCCTC): calcd for [M + H]+: 3803 Da, found: 3804 Da; 5′-d(CTTTCTCTCCCTC): calcd for [M + H]+: 3788 Da, found: 3788 Da; beacon: calcd for [M + Na]+: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 546 Da, found: 10548 Da; ODN–T: calcd for [M + H]+: 3947 Da, found: 3947 Da; ODN–C: calcd for [M + H]+: 3933 Da, found: 3933 Da. MALDI-ToF mass spectra were recorded on a Bruker Reflex IV instrument using a 3-hydroxypicolinic acid/ammonium citrate matrix. For quantification of the oligonucleotides, a molar extinction coefficient of ε260 = 10.0 cm2 mmol−1 was used for P.
546 Da, found: 10548 Da; ODN–T: calcd for [M + H]+: 3947 Da, found: 3947 Da; ODN–C: calcd for [M + H]+: 3933 Da, found: 3933 Da. MALDI-ToF mass spectra were recorded on a Bruker Reflex IV instrument using a 3-hydroxypicolinic acid/ammonium citrate matrix. For quantification of the oligonucleotides, a molar extinction coefficient of ε260 = 10.0 cm2 mmol−1 was used for P.
Spectroscopy
UV and CD spectra were recorded of solution containing 1 μM oligonucleotide duplex, 150 mM NaClO4, and 5 mM buffer of MOPS (pH 6.8), MES (pH 5.5), or borate (pH 9.0). UV spectra were recorded on a CARY BIO 100 spectrophotometer. Temperature-dependent denaturation curves were recorded between 10 °C and 70 °C with a heating/cooling rate of 1 °C min−1 and a data interval of 1 °C. UV absorbance was normalized according to Anorm = (A − Amin)/(Amax − Amin) at 260 nm. Melting temperatures were derived from the maxima of the derivatives of the annealing curves. CD spectra were recorded at 10 °C on a Jasco J-815 spectrometer. The spectra were smoothed, and a manual baseline correction was applied. Fluorescence spectra were recorded on a Jasco FP-6500 instrument with excitation at 495 nm and an emission range of 505–700 nm at a PMT voltage of 520 V. The fluorescence intensity of the molecular beacon was measured in solutions containing 0.05 μM beacon, 50 mM NaClO4, 2.5 mM Mg(ClO4)2, 5 mM MES buffer (pH 5.5) at room temperature before and after right after adding ODN–C or ODN–T along with 10 equiv. of AgNO3.
Acknowledgements
Support by COST Action CM1105, the DFG (SFB 858), and the NWO is gratefully acknowledged. Biswarup Jash is a recipient of an NRW Graduate School of Chemistry fellowship. We thank Prof. Bart Jan Ravoo for access to the fluorescence spectrometer and Dipl.-Ing. Jutta Kösters for solving the crystal structure.
References
- T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138 RSC
 .
.
-
E. Stulz and G. H. Clever, DNA in Supramolecular Chemistry and Nanotechnology, John Wiley & Sons, Chichester, 2015 Search PubMed .
-
(a) J. Müller, Eur. J. Inorg. Chem., 2008, 3749 CrossRef ;
(b) Y. Takezawa and M. Shionoya, Acc. Chem. Res., 2012, 45, 2066 CrossRef CAS PubMed ;
(c) G. H Clever, C. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226 CrossRef PubMed ;
(d) Y. Tanaka, J. Kondo, V. Sychrovský, J. Šebera, T. Dairaku, H. Saneyoshi, H. Urata, H. Torigoe and A. Ono, Chem. Commun., 2015, 51, 17343 RSC .
-
(a) S. Kumbhar, S. Johannsen, R. K. O. Sigel, M. P. Waller and J. Müller, J. Inorg. Biochem., 2013, 127, 203 CrossRef CAS PubMed ;
(b) S. Johannsen, N. Megger, D. Böhme, R. K. O. Sigel and J. Müller, Nat. Chem., 2010, 2, 229 CrossRef CAS PubMed ;
(c) J. Kondo, Y. Tada, T. Dairaku, H. Saneyoshi, I. Okamoto, Y. Tanaka and A. Ono, Angew. Chem., Int. Ed., 2015, 54, 13323 CrossRef CAS PubMed ;
(d) H. Yamaguchi, J. Šebera, J. Kondo, S. Oda, T. Komuro, T. Kawamura, T. Dairaku, Y. Kondo, I. Okamoto, A. Ono, J. V. Burda, C. Kojima, V. Sychrovský and Y. Tanaka, Nucleic Acids Res., 2014, 42, 4094 CrossRef CAS PubMed ;
(e) J. Kondo, T. Yamada, C. Hirose, I. Okamoto, Y. Tanaka and A. Ono, Angew. Chem., Int. Ed., 2014, 53, 2385 CrossRef CAS PubMed ;
(f) C. Kaul, M. Müller, M. Wagner, S. Schneider and T. Carell, Nat. Chem., 2011, 3, 794 CrossRef CAS PubMed .
- S. Atwell, E. Meggers, G. Spraggon and P. G. Schultz, J. Am. Chem. Soc., 2001, 123, 12364 CrossRef CAS PubMed .
- A. Ono, H. Torigoe, Y. Tanaka and I. Okamoto, Chem. Soc. Rev., 2011, 40, 5855 RSC .
-
(a) X. Tan, S. Litau, X. Zhang and J. Müller, Langmuir, 2015, 31, 11305 CrossRef CAS PubMed ;
(b) K. Tanaka, A. Tengeiji, T. Kato, N. Toyama and M. Shionoya, Science, 2003, 299, 1212 CrossRef CAS PubMed ;
(c) G. H. Clever, K. Polborn and T. Carell, Angew. Chem., Int. Ed., 2005, 44, 7204 CrossRef CAS PubMed ;
(d) M. Su, M. Tomás-Gamasa and T. Carell, Chem. Sci., 2015, 6, 632 RSC .
-
(a) E.-K. Kim and C. Switzer, Org. Lett., 2014, 16, 4059 CrossRef CAS PubMed ;
(b) B. D. Heuberger, D. Shin and C. Switzer, Org. Lett., 2008, 10, 1091 CrossRef CAS PubMed ;
(c) C. Switzer, S. Sinha, P. H. Kim and B. D. Heuberger, Angew. Chem., Int. Ed., 2005, 44, 1529 CrossRef CAS PubMed ;
(d) I. Sinha, C. Fonseca Guerra and J. Müller, Angew. Chem., Int. Ed., 2015, 54, 3603 CrossRef CAS PubMed ;
(e) D. A. Megger, C. Fonseca Guerra, J. Hoffmann, B. Brutschy, F. M. Bickelhaupt and J. Müller, Chem.–Eur. J., 2011, 17, 6533 CrossRef CAS PubMed ;
(f) H. Mei, I. Röhl and F. Seela, J. Org. Chem., 2013, 78, 9457 CrossRef CAS PubMed ;
(g) S. K. Jana, X. Guo, H. Mei and F. Seela, Chem. Commun., 2015, 51, 17301 RSC ;
(h) H. Yang, H. Mei and F. Seela, Chem.–Eur. J., 2015, 21, 10207 CrossRef CAS PubMed ;
(i) D. Ukale, V. S. Shinde and T. Lönnberg, Chem.–Eur. J., 2016, 22, 7917 CrossRef CAS PubMed ;
(j) Y. Takezawa, S. Yoneda, J.-L. H. A. Duprey, T. Nakama and M. Shionoya, Chem. Sci., 2016, 7, 3006 RSC ;
(k) N. Santamaría-Díaz, J. M. Méndez-Arriaga, J. M. Salas and M. A Galindo, Angew. Chem., Int. Ed., 2016, 55, 6170 CrossRef PubMed .
- P. Scharf and J. Müller, ChemPlusChem, 2013, 78, 20 CrossRef CAS .
- A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300 CrossRef CAS PubMed .
-
(a) S. Liu, G. H. Clever, Y. Takezawa, M. Kaneko, K. Tanaka, X. Guo and M. Shionoya, Angew. Chem., Int. Ed., 2011, 50, 8886 CrossRef CAS PubMed ;
(b) T. Ehrenschwender, W. Schmucker, C. Wellner, T. Augenstein, P. Carl, J. Harmer, F. Breher and H.-A. Wagenknecht, Chem.–Eur. J., 2013, 19, 12547 CrossRef CAS PubMed .
- J. C. Léon, L. Stegemann, M. Peterlechner, S. Litau, G. Wilde, C. A. Strassert and J. Müller, Bioinorg. Chem. Appl., 2016, 7485125 Search PubMed .
-
(a) H. Urata, E. Yamaguchi, T. Funai, Y. Matsumura and S.-i. Wada, Angew. Chem., Int. Ed., 2010, 49, 6516 CrossRef CAS PubMed ;
(b) T. Kobayashi, Y. Takezawa, A. Sakamoto and M. Shionoya, Chem. Commun., 2016, 52, 3762 RSC .
- K. S. Park, C. Jung and H. G. Park, Angew. Chem., Int. Ed., 2010, 49, 9757 CrossRef CAS PubMed .
-
(a) S. Taherpour, O. Golubev and T. Lönnberg, Inorg. Chim. Acta, 2016, 452, 43 CrossRef CAS ;
(b) S. Taherpour, O. Golubev and T. Lönnberg, J. Org. Chem., 2014, 79, 8990 CrossRef CAS PubMed ;
(c) S. Taherpour, H. Lönnberg and T. Lönnberg, Org. Biomol. Chem., 2013, 11, 991 RSC .
- B. Jash, J. Neugebauer and J. Müller, Inorg. Chim. Acta, 2016, 452, 181 CrossRef CAS .
- P. Scharf, B. Jash, J. A. Kuriappan, M. P. Waller and J. Müller, Chem.–Eur. J., 2016, 22, 295 CrossRef CAS PubMed .
- B. Lippert, Prog. Inorg. Chem., 2005, 54, 385 CrossRef CAS .
- W. A. E. McBryde, Can. J. Chem., 1965, 43, 3472 CrossRef CAS .
- H. Walba and R. W. Isensee, J. Org. Chem., 1961, 26, 2789 CrossRef CAS .
- J. J. McCarthy and R. Hilfiker, Nat. Biotechnol., 2000, 18, 505 CrossRef CAS PubMed .
- J. Wang, G. S. Y. Pang, S. S. Chong and C. G. L. Lee, Curr. Drug Metab., 2012, 13, 978 CrossRef CAS PubMed .
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303 CrossRef CAS PubMed .
- R. Yang, J. Jin, L. Long, Y. Wang, H. Wang and W. Tan, Chem. Commun., 2009, 322 RSC .
- R. Lumry, E. L. Smith and R. R. Glantz, J. Am. Chem. Soc., 1951, 73, 4330 CrossRef CAS .
- R. B. Martin, Science, 1963, 139, 1198 CAS .
- R. Tribolet and H. Sigel, Eur. J. Biochem., 1987, 163, 353 CrossRef CAS PubMed .
-
(a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS ;
(b)
ADF2013, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed .
-
(a) S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed ;
(b) E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783 CrossRef CAS .
-
(a) A. Klamt, J. Phys. Chem., 1995, 99, 2224 CrossRef CAS ;
(b) C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Determination of the pKa values of 1; determination of the pKa values of P; melting curves of duplexes with a central P:X base pair; melting curves of duplexes with a central A:T, G:C or C:T base pair; geometry-optimized structures of possible hydrogen-bonded base pairs; normalized proton-binding energies of P and C; melting temperatures of the duplexes comprising a central A:T, G:C or C:T base pair; base pairing energies of hydrogen-bond mediated base pairs; equations used to fit the pD-dependent chemical shift data; full computational details; Cartesian coordinates and energies of geometry-optimized structures; MALDI spectra of oligonucleotides, synthesis and characterization of 1; characterization of P. CCDC 1489603. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03482a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Philipp
Scharf
a,
Nikolas
Sandmann
a,
Célia
Fonseca Guerra
ab,
Philipp
Scharf
a,
Nikolas
Sandmann
a,
Célia
Fonseca Guerra


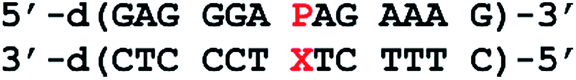






 ); molecular beacon + ODN–T + Ag+ (
); molecular beacon + ODN–T + Ag+ ( ); molecular beacon + ODN–C + Ag+ (
); molecular beacon + ODN–C + Ag+ ( ); molecular beacon + ODN–T without Ag+ (
); molecular beacon + ODN–T without Ag+ ( ); molecular beacon + ODN–C without Ag+ (
); molecular beacon + ODN–C without Ag+ ( ). The inset shows the average percent increase of the fluorescence at 525 nm in the presence of Ag+ and the respective oligonucleotide probe (including standard deviation of the mean of three independent measurements). All spectra have been normalized with respect to the spectrum of the molecular beacon.
). The inset shows the average percent increase of the fluorescence at 525 nm in the presence of Ag+ and the respective oligonucleotide probe (including standard deviation of the mean of three independent measurements). All spectra have been normalized with respect to the spectrum of the molecular beacon. values in D2O according to eqn (1).26
values in D2O according to eqn (1).26