
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The stereodivergent formation of 2,6-cis and 2,6-trans-tetrahydropyrans: experimental and computational investigation of the mechanism of a thioester oxy-Michael cyclization†
Kristaps
Ermanis
 ,
Yin-Ting
Hsiao
,
Uğur
Kaya
,
Alan
Jeuken
and
Paul A.
Clarke
*
,
Yin-Ting
Hsiao
,
Uğur
Kaya
,
Alan
Jeuken
and
Paul A.
Clarke
*
Department of Chemistry, University of York, Heslington, York, North Yorkshire YO10 5DD, UK. E-mail: paul.clarke@york.ac.uk
First published on 30th August 2016
Abstract
The origins of the stereodivergence in the thioester oxy-Michael cyclization for the formation of 4-hydroxy-2,6-cis- or 2,6-trans-substituted tetrahydropyran rings under different conditions was investigated both computationally and experimentally. Synthetic studies showed that the 4-hydroxyl group was essential for stereodivergence. When the 4-hydroxyl group was present, TBAF-mediated conditions gave the 2,6-trans-tetrahydropyran and trifluoroacetic acid-mediated conditions gave the 2,6-cis-tetrahydropyran. This stereodivergence vanished when the hydroxyl group was removed or protected. Computational studies revealed that: (i) the trifluoroacetic acid catalysed formation of 2,6-cis-tetrahydropyrans was mediated by a trifluoroacetate-hydroxonium bridge and proceeded via a chair-like transition state; (ii) the TBAF-mediated formation of 2,6-trans-tetrahydropyrans proceeded via a boat-like transition state, where the 4-hydroxyl group formed a crucial hydrogen bond to the cyclizing alkoxide; (iii) both reactions are under kinetic control. The utility of this stereodivergent approach for the formation of 4-hydroxy-2,6-substituted tetrahydropyran rings has been demonstrated by the total syntheses of the anti-osteoporotic natural products diospongin A and B.
Introduction
2,6-Disubstituted tetrahydropyran (THP) rings form key structural motifs in many potent biologically active natural products,1 including the phorboxazoles,2 lasonolide A,3 the diospongins4 and psymberin.5 These biological activities and complex molecular frameworks have prompted a large amount of work aimed at increasing the efficiency of the syntheses of THP rings,6 which, problematically, are often formed as mixtures of 2,6-cis- and 2,6-trans-diastereomers. One fundamental strategy regularly used for their formation is the oxy-Michael cyclization onto an α,β-unsaturated carbonyl group, which often leads to the formation of both possible diastereomeric THPs. Here, we report a stereodivergent oxy-Michael reaction which can lead to the diastereoselective formation of either the 2,6-cis- or the 2,6-trans-THP in up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity (Scheme 1). We have also conducted computational and experimental studies which elucidate the origin of this stereodivergence and show the importance of a H-bond between the 4-hydroxyl group and the cyclizing alkoxide in the oxy-Michael cyclization. These studies allow us to propose a general set of guidelines for future syntheses of 2,6-disubstituted THPs.
:1 diastereoselectivity (Scheme 1). We have also conducted computational and experimental studies which elucidate the origin of this stereodivergence and show the importance of a H-bond between the 4-hydroxyl group and the cyclizing alkoxide in the oxy-Michael cyclization. These studies allow us to propose a general set of guidelines for future syntheses of 2,6-disubstituted THPs.
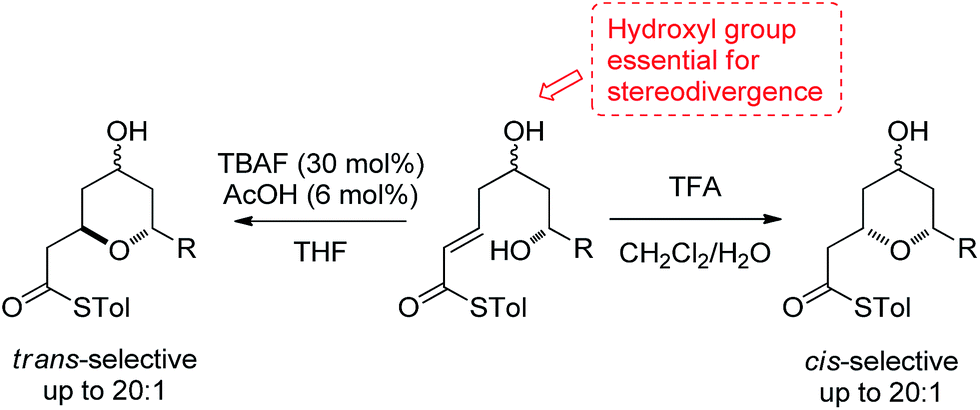 | ||
| Scheme 1 The stereodivergent thioester oxy-Michael cyclization. | ||
Previous studies have investigated oxy-Michael cyclizations in order to gain an understanding of the factors governing the stereoselectivity of the cyclization. This has led to the general opinion that the formation of the 2,6-cis-THP may be favored by performing an oxy-Michael reaction onto an α,β-unsaturated ester under thermodynamic conditions, while the 2,6-trans-THP may be favored by performing the same reaction under kinetically controlled conditions. In practice, the situation is not so straightforward. While 2,6-trans-THPs tend to be formed in good yields with moderate to good diastereoselectivities,11 the higher temperatures and longer reaction times required for the formation of the 2,6-cis-THP ring often result in lower diastereoselectivities and yields.12 The origin of this moderate 2,6-trans-selectivity is generally accepted as arising from better orbital overlap in the transition state of the kinetic cyclization leading to the 2,6-trans-THP compared to the 2,6-cis-THP.8 Although there is no generally accepted mechanism for the acid-mediated cyclization, it has been proposed that the formation of the 2,6-cis-THP is favored due to greater stereoelectronic stabilization of the transition state from both the FMO coefficients of the allylic cation and orbital overlap with the oxygen lone pair, compared to the transition state leading to the 2,6-trans-THP.9
In our studies on the synthesis of the C20–C32 pentasubstituted tetrahydropyran core of the phorboxazoles we encountered an occurrence of stereodivergence13 while utilizing thioesters as electophiles in an oxy-Michael cyclization.14 In this case, stereodivergence occurred when the conditions for the deprotection of a TBS-ether were changed. Deprotection of 1 with AcOH buffered TBAF led to the formation of the 2,6-trans-THP 2 with no trace of 3 being detected. However, deprotection with TFA resulted in the formation of the 2,6-cis-THP product 3 in >13:1 selectivity (Scheme 2). It is worth noting that when the conventional oxoester 4 was submitted to these conditions the 2,6-trans-THP 5 resulted from treatment with TBAF buffered with AcOH in THF; no trace of the cis-diastereomer was seen. However, only decomposition occurred when 4 was treated with TFA.13 Intrigued by these results, especially by the dramatic change in diastereoselectivity seen in the thioester substrate, we resolved to carry out synthetic and computational studies to elucidate the mechanistic origins of this stereodivergence and to establish more general synthetic guidelines for the diastereoselective synthesis of 2,6-disubstituted THPs. The results of these studies are reported in this paper.
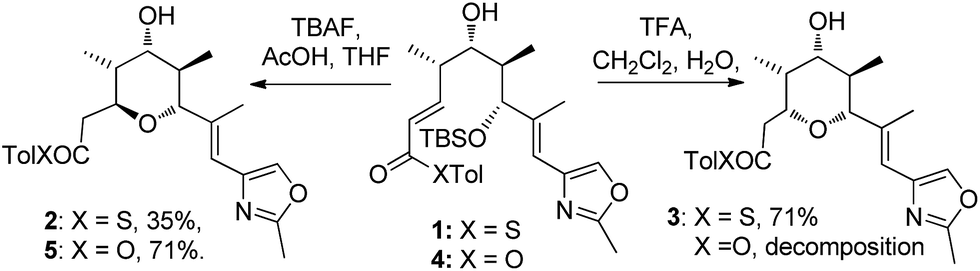 | ||
| Scheme 2 Stereodivergence in the thioester oxy-Michael cyclization to form the C20–C32 fragment of the phorboxazoles. | ||
Results and discussion
Synthetic investigation into the generality of stereodivergence
We initially decided to probe whether the stereodivergence was specific to 1 or whether it was a more general phenomenon. To this end we synthesized cyclization substrates 6a–c, which had the same relative configuration as 1 and 9a–c, which had the opposite relative configuration.15 Each substrate was submitted to both the buffered TBAF and the TFA mediated conditions (Tables 1 and 2).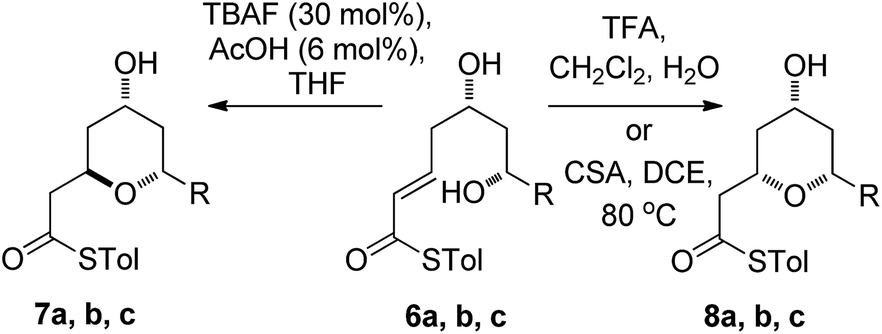
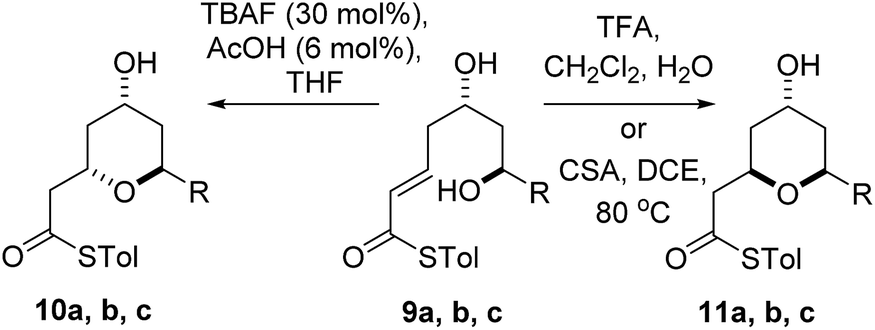
Substrates 6a, b, c, which contain the 4-hydroxyl group were submitted to both the TBAF-mediated and the Brønsted acid promoted cyclization conditions (Table 1). In this case, TBAF mediated reactions smoothly generated 2,6-trans-THP products 7a–c in good yields and with excellent selectivity (with the exception of 6c), while Brønsted acid promoted conditions gave the 2,6-cis-THP products 8a–c with excellent selectivity and in good yields. In the case of the phenyl substituted compound 6b, the TFA conditions led to decomposition although the CSA conditions led to 2,6-cis-THP product being isolated in 74% yield.
Diastereomeric diol substrates 9a, b, c were studied next (Table 2). Once again, TBAF-mediated cyclizations generated the 2,6-trans-THP predominantly. As before, Brønsted acid promoted conditions gave the 2,6-cis-THP products 11a–c with excellent selectivity and in good yields. In the case of 9b (R = Ph), CSA promoted conditions had to be used to avoid decomposition under TFA conditions. Thus it would appear that the stereodivergence seen in the cyclization of 1 is not limited to that particular system.
In order to ascertain if the reactions were under thermodynamic control 2,6-cis-substrate 11b was submitted to the TBAF conditions and found to be unchanged after several hours and 2,6-trans-substrate 10b was submitted to the Brønsted acid conditions and was also re-isolated unchanged. These results imply that both the TBAF and TFA-mediated reactions are not under thermodynamic control.
Computational studies on the stereodivergence
With stereodivergent behavior being exhibited by all the substrates investigated, we decided to conduct DFT studies in order to elucidate the origin of this behaviour. DFT investigations were conducted on both the buffered TBAF-mediated reaction which produced the 2,6-trans-THPs 2, 7a–c and 10a–c and the TFA-mediated reactions which produced 2,6-cis-THPs 3, 8a–c and 11a–c. Conformation searches were conducted at the molecular mechanics level and using MacroModel and MMFF force field.16 DFT geometries were optimized and energies calculated using the B3LYP density functional,17 and split-valence polarized 6-31G*+ basis set with diffuse functions.18 Geometries were first optimized in gas-phase and afterwards in the solvent indicated using PBF solvent model.19Fluoride-mediated cyclization
As all of the TBAF-mediated reactions were trans-selective we initially chose to model the cyclization of 6a to 7a (Scheme 3). We rationalized that the active molecule is the alkoxide 12, which then attacks the conjugate double bond to form either the enolate of the 2,6-trans-THP 13 or of 2,6-cis-THP 14. The enolates can be formed as either E or Z-isomers. Since the interconversion would likely be slow, the enolate geometry should be determined by the thioester conformation (s-cis or s-trans) in the transition state.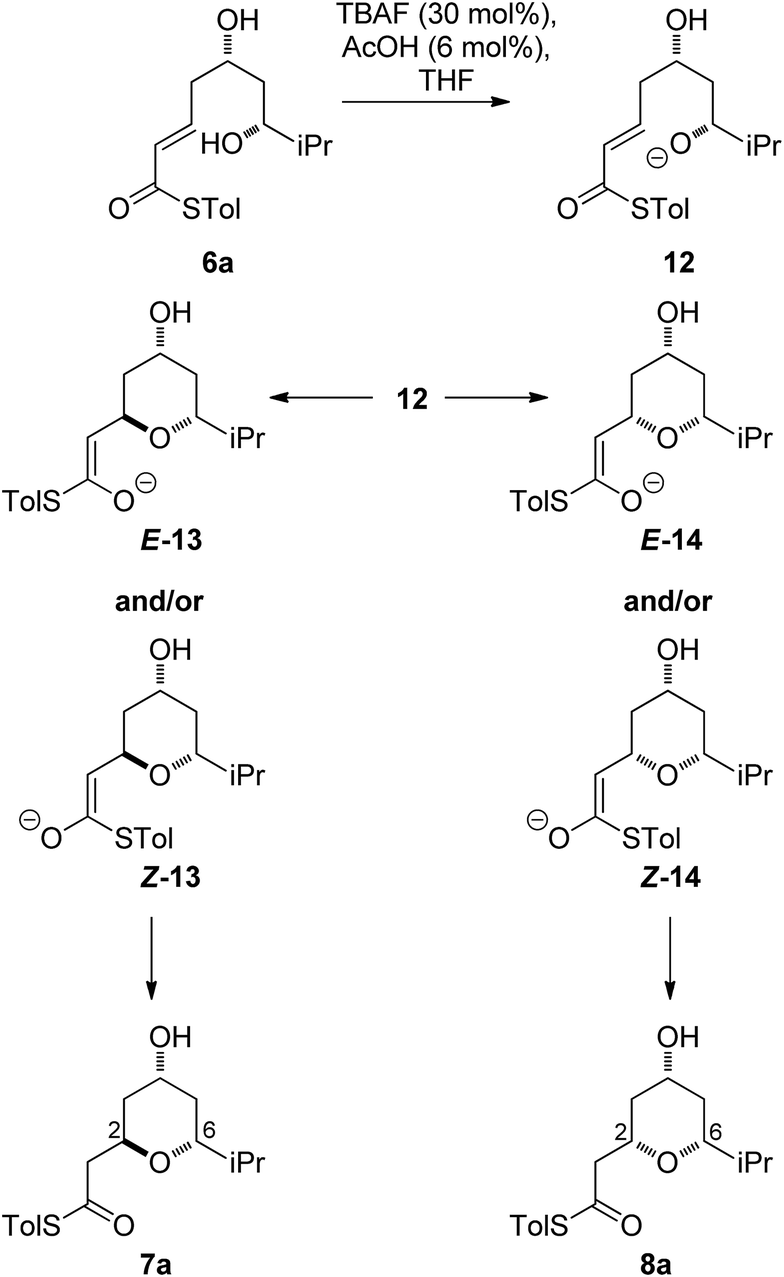 | ||
| Scheme 3 Mechanistic considerations for the TBAF cyclization. | ||
With this in mind, a thorough search for the transition states leading to the four possible enolates E-13, Z-13, E-14, Z-14 was conducted. Several transition states leading to each of the enolates were found, lowest energy of which are shown in Fig. 1. Notably, the conformations of the lowest energy TSs were boat-like instead of the more common chair-like conformation. A strong intramolecular hydrogen bond between the 4-hydroxyl and the alkoxide stabilizes this conformation and makes it more favourable.20,21
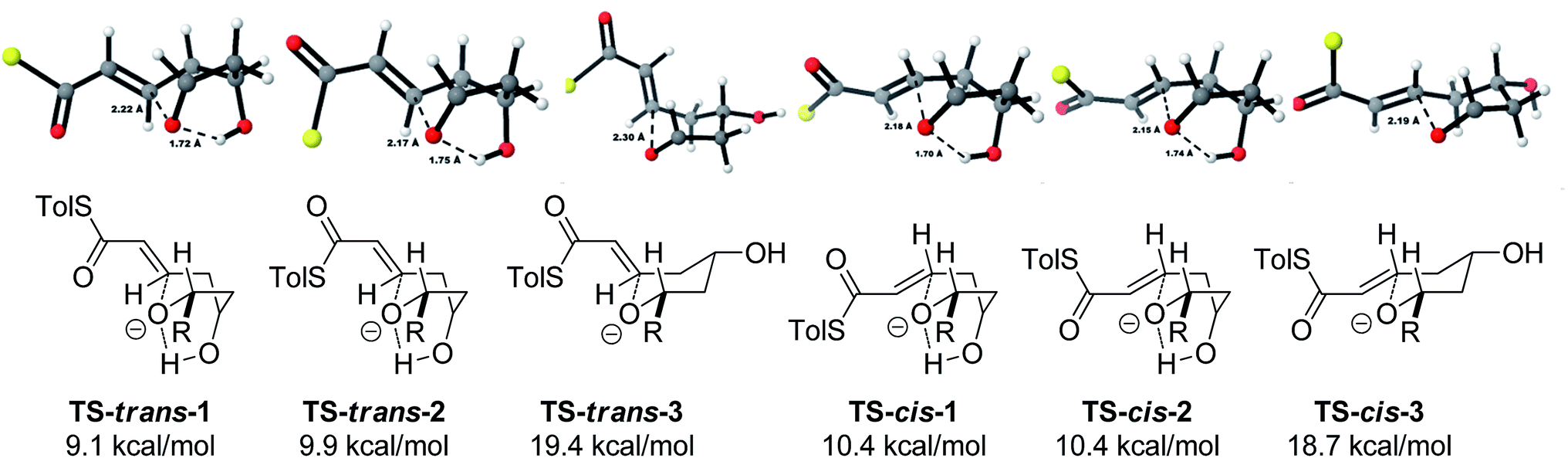 | ||
| Fig. 1 Transition states for the TBAF mediated cyclization. Activation enthalpies calculated in THF implicit solvent model and shown relative to the ground state conformation of alkoxide 12. Tolyl and iPr groups omitted for clarity. | ||
Alternative chair-like transition states leading to both products were also found and are shown in Fig. 1. These, however, are significantly higher in energy, and therefore not significant.
E-transition states are lower than the corresponding Z-transition states, but the differences are not large. Similarly, the Z-thioenolates Z-13 and Z-14 are higher in energy than E-thioenolates because of the increased steric interactions. Once the product is formed, the boat conformation is no longer favourable and the THP thioenolates relax to the chair conformations, all of which were calculated to be lower in energy by 2–4 kcal mol−1.
While normal ester enolates are much more basic than alkoxides, thioester enolate pKa is much lower and comparable to alkoxides.22 It is therefore not surprising that the E-thioenolates and the 4-alkoxides were found to be quite similar in energy. For the 2,6-trans-substrate the reaction end point before reprotonation would be the E-enolate, with the alkoxide being 1.5 kcal mol−1 higher in energy. For the 2,6-cis-substrate the 4-alkoxide is favoured over the E-thioenolate by 1.6 kcal mol−1. The overall thermodynamic product of the reaction should be the 2,6-cis-4-alkoxide, which is 0.5 kcal mol−1 lower in energy than the 2,6-trans-E-thioenolate. This adds further support to kinetic control in this reaction, because the major observed product is the 2,6-trans-THP.
Using all of this information a reaction energy profile was constructed (Fig. 2). The barrier for the forward reaction is 9.1 kcal mol−1 for the 2,6-trans-product and 10.4 kcal mol−1 for the 2,6-cis-product. Both are very low and are consistent with the observed speed of the reaction, which is usually complete in less than 10 minutes at or below room temperature. For the reverse reaction, the total energy barrier is 14.4 kcal mol−1, making it several orders of magnitude slower. This clearly shows that the reaction is under kinetic control, which matches experimental observations. Therefore the activation energies for the diastereomeric pathways are also determining the diastereoselectivity of the reaction. The 2,6-trans-boat transition state is 1.3 kcal mol−1 lower than the 2,6-cis-boat transition state, matching the observed diastereoselectivity well. One possible reason for this energy difference is the semi-eclipsed interaction of the β and γ-hydrogen atoms of the α,β-unsaturated thioester. This is present in the TS-cis-1 (dihedral angle 37°), but not in the TS-trans-1 (dihedral angle 161°). Another contributing factor would be the increased steric clash in the TS-cis-1 from a pseudo-1,3-diaxial interaction between the protons at the 2- and 6- positions in the forming ring. This interaction is absent in the trans-transition states, but could be particularly pronounced in the cis-transition states because the protons are pointing slightly towards each other to allow the alkoxide attack from a favourable trajectory.
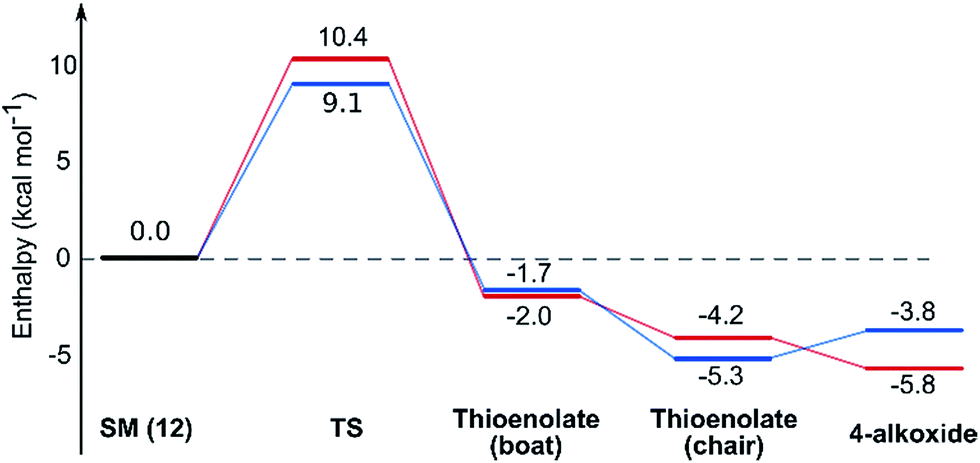 | ||
| Fig. 2 Energy diagram for the TBAF mediated lowest energy pathways to the 2,6-trans7a (blue) and 2,6-cis8a (red). Enthalpies calculated in THF implicit solvent model and are relative to the ground state conformation of alkoxide 12. | ||
Acid-mediated cyclization
With an explanation in hand for the trans-selectivity of the buffered TBAF-mediated reaction we turned our attention to the TFA-mediated cis-selective cyclization reaction (Scheme 4). As shown by Fuwa and ourselves in interconversion experiments, the process is likely to be under kinetic control.13,14 Therefore a simple thermodynamic preference for the 2,6-cis-diastereomers is not an adequate explanation for the observed stereoselectivity.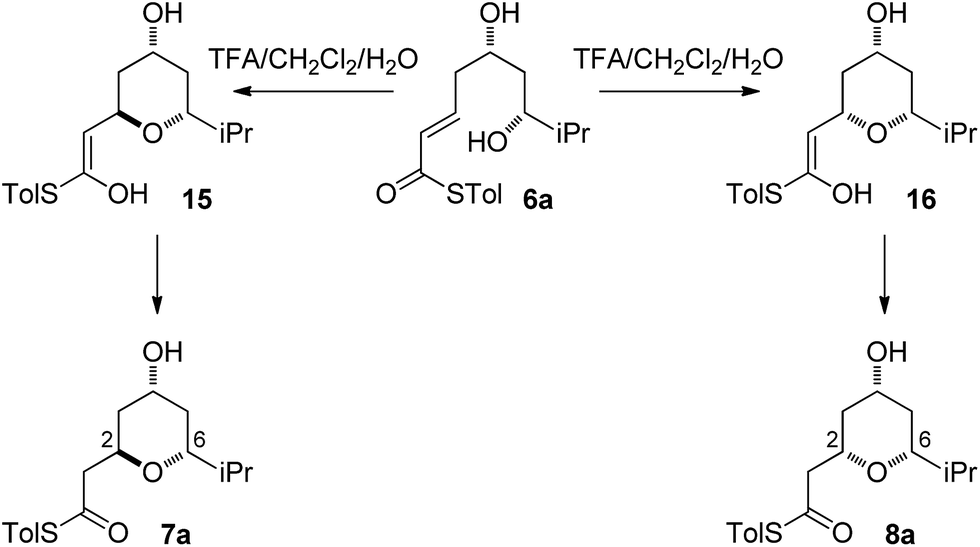 | ||
| Scheme 4 Mechanistic considerations for the acid-mediated cyclization. | ||
Currently there is no generally accepted mechanism for the acid mediated oxy-Michael cyclization. Based on studies by Houk,8a Fuwa proposed an allylic cation type mechanism23 (Scheme 5), although no further experimental evidence to support this proposal has been reported.
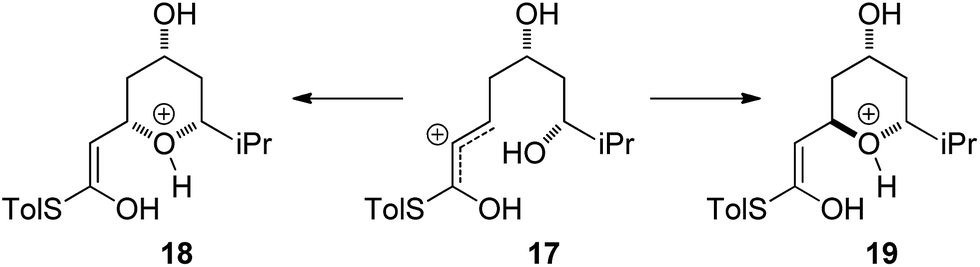 | ||
| Scheme 5 Allylic cation mechanism. | ||
Firstly, the viability of the allylic carbocation mechanism was tested by DFT calculations. When the lowest energy conformations of the cyclized intermediates 18 and 19 were submitted to geometry optimization at the DFT level, the THP rings opened back up during the process. This implies that there is no energy barrier for the opening of the ring and that the protonated cyclized intermediates 18 and 19 are unstable. Therefore it is highly unlikely that a simple protonation is the mechanism for the acid catalysis in this reaction. This mechanism would also provide little room to explain the different levels of diastereoselectivity achieved by the use of different acids as reported by Fuwa.14
Other potential modes of activation were then explored (Fig. 3). Among the mechanisms identified were two where TFA acts as proton shuttle, protonating the thioester and deprotonating the alcohol almost simultaneously. Another potential mechanism requires two different molecules of TFA, one of which acts as an acid and protonates the thioester and the other one acts as a base and deprotonates the alcohol nucleophile during the attack. Of these three mechanisms the 1,3-TFA proton shuttle mechanism was calculated to have the smallest activation energy, and the rest of the computational study focused on investigating it.
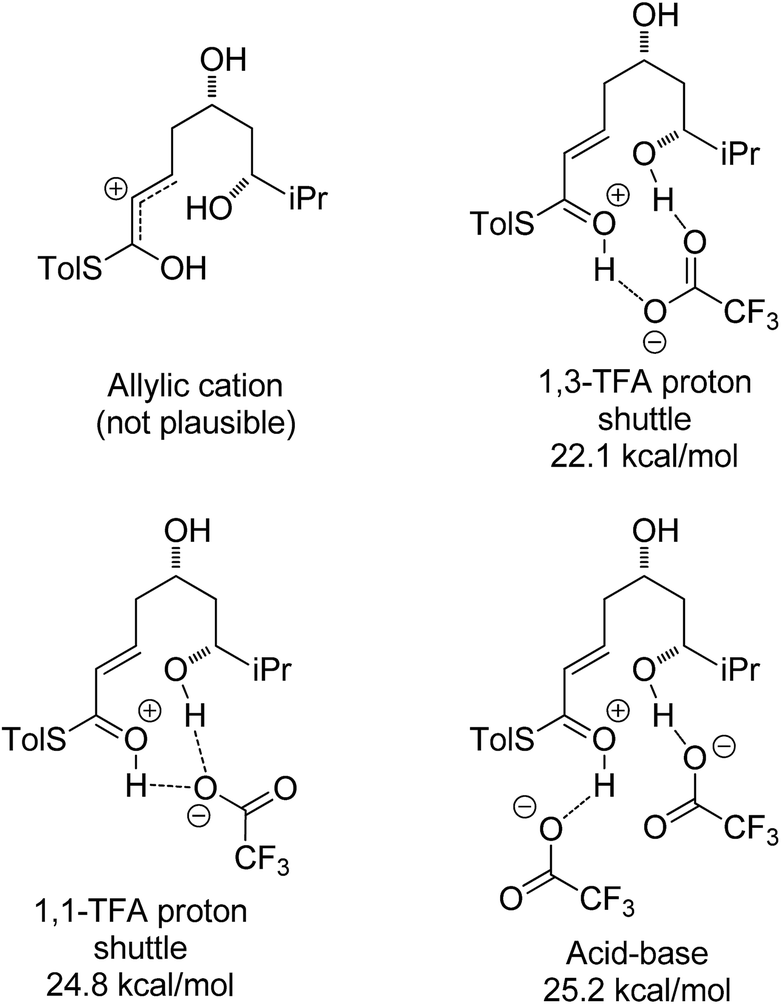 | ||
| Fig. 3 Possible activation modes in TFA mediated cyclization. Enthalpies shown are the activation enthalpies calculated in gas phase relative to the corresponding starting material-TFA complex ground state conformation. | ||
A thorough search for transition states leading to the two diastereomeric enols 15 and 16 revealed several chair- and boat-like transition states, of which the lowest energy ones are shown in Fig. 4. Because the TFA proton shuttle imposes a distance constraint between the alcohol and the thioester carbonyl group, only the transition states leading to the E-enols 15 and 16 are possible. The activation enthalpies were calculated to be 19.3 kcal mol−1 for the 2,6-cis-chair-like transition state and 21.7 kcal mol−1 for the 2,6-trans-chair-like transition state. TS-cis-chair is 1.9 kcal mol−1 lower in energy, consistent with the observed diastereoselectivity. The higher energy of the trans-transition state appears to be caused by an increased pseudo-1,3-diaxial steric clash between the 6-proton and the 2-thioester substituent. This interaction is not present in the cis-transition state. In all of the transition states, there are two hydrogen bonds between the TFA anion and the alcohol, and between the TFA anion and the protonated thioester. This allows the TFA to act as a proton shuttle and to simultaneously improve the electrophilicity of the thioester and the nucleophilicity of the alcohol.
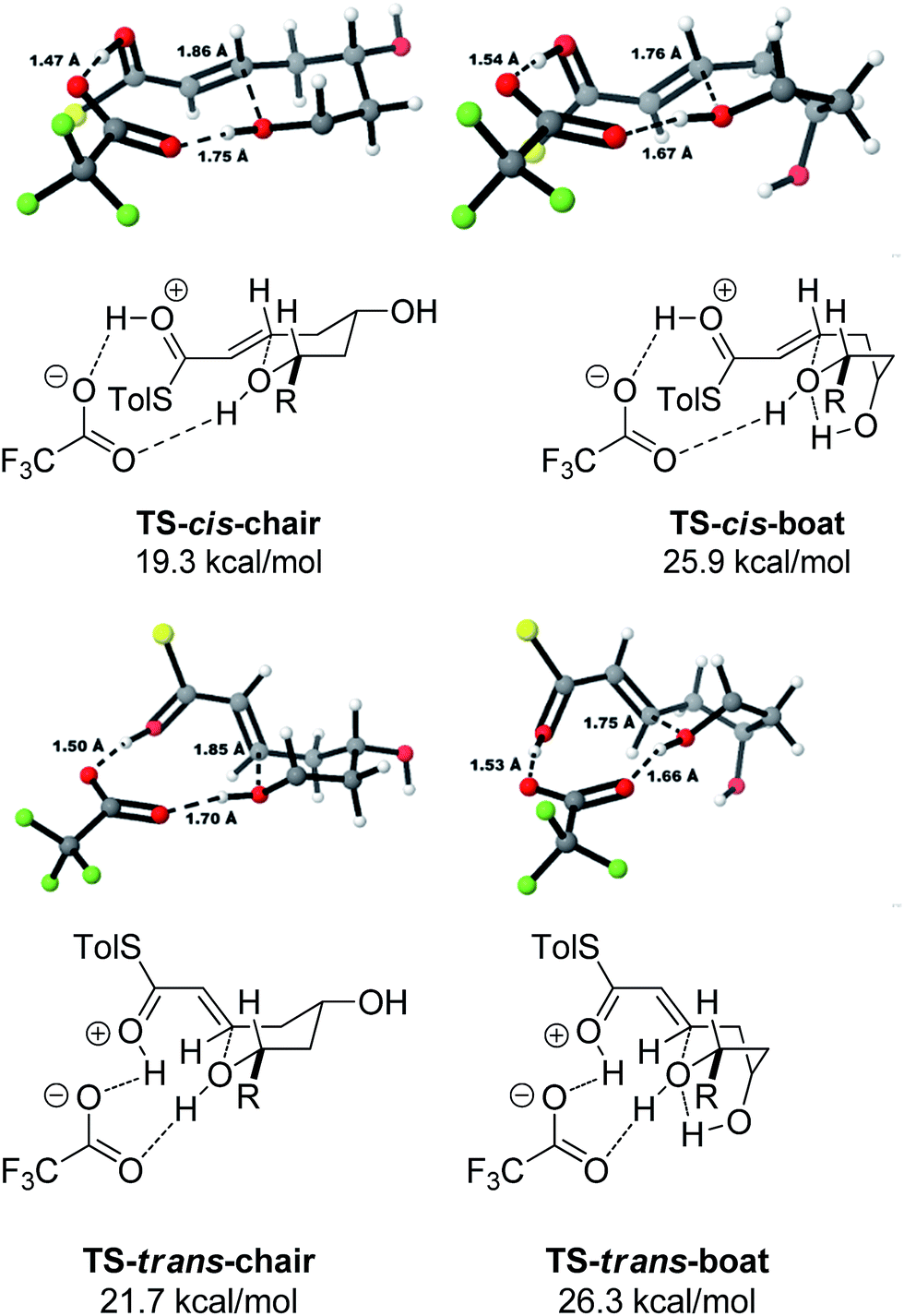 | ||
| Fig. 4 Transition states for the TFA mediated cyclization. Activation enthalpies calculated in DCM implicit solvent model and shown relative to the ground state conformation of diol 7a complex with TFA. Tolyl and iPr groups omitted for clarity. | ||
In contrast with the TBAF case, the 4-hydroxyl group is not directly involved in the stabilization of the transition state, and the boat-like transition states are both some 5 kcal mol−1 higher in energy than the corresponding chair-like transition states. Calculations also confirmed kinetic control in this reaction, as the activation energy of the reverse reaction is 8.4 kcal mol−1 higher than the forward reaction (Fig. 5).
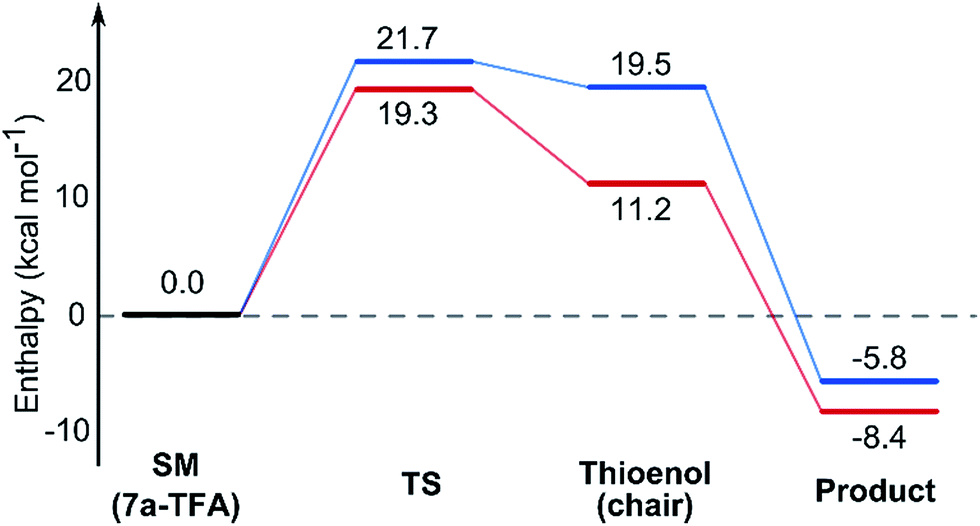 | ||
| Fig. 5 Energy diagram for the TBAF mediated lowest energy pathways to the 2,6-cis9a (red) and 2,6-trans8a (blue). Enthalpies calculated in DCM implicit solvent model and are relative to the ground state conformation of 7a complex with TFA. | ||
Synthetic investigation of the role of the 4–OH group
As the computational studies had implicated the 4-hydroxyl as the source of the stereodivergence, it was decided to test this hypothesis on a number of substrates. Substrates chosen for study were 20a, 20b and 20c, which did not have the 4-hydroxyl group present and a substrate where the hydroxyl group was protected as a methyl ether 22.When TFA was used the cyclization of 20a, b, c proceeded smoothly to form the 2,6-cis-THP products 21a, b, c in moderate to good yields and with reasonable diastereoselectivities, however, the products of the TBAF mediated cyclization reactions were also the 2,6-cis-THP 21a, b, c and were formed with even higher diastereoselectivity than under the TFA-mediated reaction conditions (Table 3).
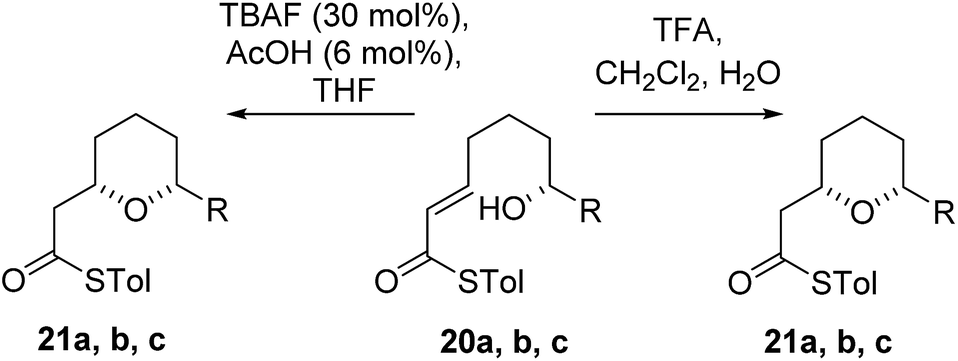
The final substrate studied was where the 4-hydroxyl group was capped as a methyl ether 22 (Scheme 6). Cyclization precursor 22 was subjected to both sets of cyclization conditions. While the TFA-mediated conditions yielded the cis-diastereomer 23 cleanly, the TBAF-mediated conditions proceeded very slowly, with substantial hydrolysis of the thioester. However, despite this 1H NMR analysis of the crude reaction mixture showed that only the cis-product 23 had been formed, confirming our hypothesis and previous results that a hydrogen-bond donor in the 4-position is required for the formation of the 2,6-trans-diastereomer under buffered TBAF-mediated conditions. The results of these synthetic studies are in complete agreement with the predictions of the computational studies. The computational studies showed that chair-like transition states in the TBAF-mediated reaction would provide the reverse diastereoselectivity with the 2,6-cis-chair TS being 1.5 kcal mol−1 lower in energy than the 2,6-trans-chair TS. This explains why the 2,6-trans selectivity is not observed in the 4-dehydroxy substrates 20a–c and the methyl ether 22, because in those cases there is no way of stabilizing the boat-like TS and the chair-like transition state would be more favoured, giving the 2,6-cis products.
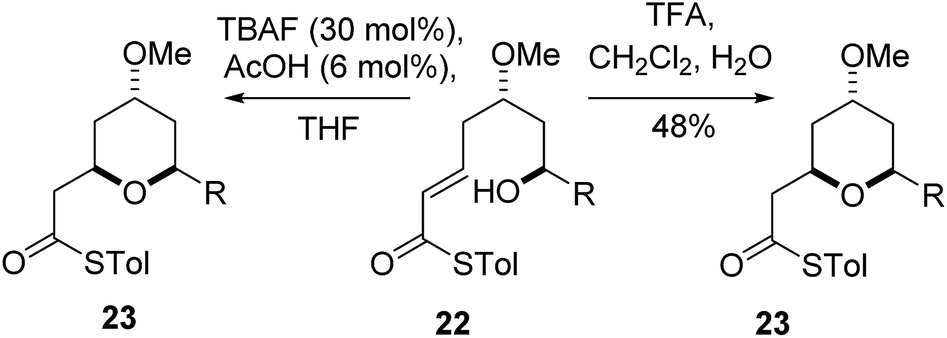 | ||
| Scheme 6 Cyclization of methyl ether 23. | ||
Differences in the reactivity of thioester 1 and oxoester 4
With explanations for the origins of the stereodivergence in the cyclization reactions, the last remaining question for the computational study was the dramatic difference in the reactivity of thioester 1 and oxoester 4 in this oxy-Michael cyclization. Only the 2,6-cis-pathways were investigated as we were primarily interested in the reactivity of the substrate: thioester 1 cyclised to the 2,6-cis-THP whereas oxoester 4 decomposed. We identified a similar transition state for the oxoester as in the case of thioester, however, the energy profile of the reaction showed a much higher transition state energy for oxoester. This difference of 7.6 kcal mol−1 compared to the thioester would make the oxoester cyclization much slower. The slower cyclization would allow for competing decomposition reactions to dominate. One possible decomposition pathway is that the potentially acid sensitive styrenyl alcohol's ionization competes with conjugate addition under the Brønsted acid conditions. The transition state geometries of the thioester and oxoester cyclizations are very similar and therefore it appears very unlikely that steric effects would be the cause for the dramatic difference. An alternative cause would be the differences in the electronic structure; that the oxoester 4 is a less efficient electrophile than the thioester 1, which would manifest itself in the LUMOs of the substrates.Two very similar low lying conformations of the thioester-TFA and the oxoester-TFA complex were compared (Fig. 6). The electron density distribution is very similar for both substrates but the energy for the thioester-TFA complex LUMO is −1.43 eV and −1.06 eV for the oxoester-TFA complex. While this difference is relatively small, it is significant and shows that the sulfur atom makes the LUMO more accessible for nucleophiles.
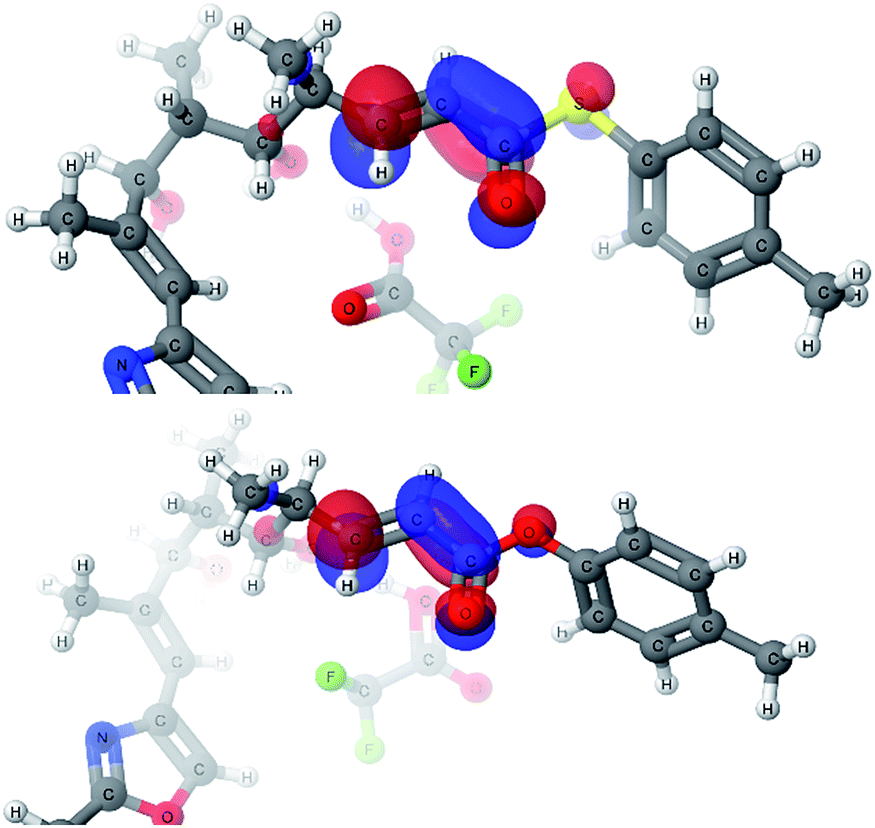 | ||
| Fig. 6 LUMOs of the thioester-TFA complex (top) and oxoester-TFA complex (bottom). | ||
A possible explanation for the difference in the LUMO energies between thioesters and oxoesters might be that sulfur lone pair (in sp3 orbital made up from a contribution of the 3p orbitals) has a smaller overlap with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O π* orbital of the ester than the oxygen lone pair (in sp3 orbital made up from a contribution of the 2p orbitals). This would have the effect of making the thioester electrophile more enone-like and more electrophilic in comparison with the α,β-unsaturated oxoester,24 this reactivity profile has recently been reported in an intramolecular oxy-Michael cyclization onto an enone versus an α,β-unsaturated oxoester.10a
O π* orbital of the ester than the oxygen lone pair (in sp3 orbital made up from a contribution of the 2p orbitals). This would have the effect of making the thioester electrophile more enone-like and more electrophilic in comparison with the α,β-unsaturated oxoester,24 this reactivity profile has recently been reported in an intramolecular oxy-Michael cyclization onto an enone versus an α,β-unsaturated oxoester.10a
Stereodivergent synthesis of diospongins A and B
We realized that compounds 10b and 11b, the products of the stereodivergent oxy-Michael reactions of 9b, were possible precursors to the natural products diospongins B and A respectively. Diospongins A and B are members of the biaryl heptanoid class of natural products and have been reported to exhibit anti-osteoporotic activity.4 There have been several syntheses of these molecules,25 but none to date have exploited the potential for a stereodivergent synthesis from a common precursor, and so we sought to showcase the stereodivergent oxy-Michael reaction with a synthesis of both these natural products. In theory both diospongin A and B could be accessed in one step from these precursors by a Liebeskind–Srogl type coupling reaction of the thioester with phenyl boronic acid under Pd-catalysis.26,14a Indeed this reaction was successfully employed in the synthesis of diospongin A 25, from 11b in 75% yield (Scheme 7). However, application of the same conditions to 10b resulted only in re-isolation of starting material. Despite the investigation of several different solvents, temperatures and catalyst loadings and ligands we were unable to get 10b to react. Application of the alternative Liebeskind organostannane conditions27 also resulted in no reaction taking place. We are unable to explain this lack of reactivity for the 2,6-trans-diastereomer 10b at this time. We therefore had to adopt an alternative strategy for the conversion of the thioester to the desired phenyl ketone. Tetrahydropyran-4-ol 10b was instead treated with PhLi at −78 °C and warmed to RT overnight, which did give diospongin B 24 in 52% yield (Scheme 7). Spectroscopic data of both diospongins were identical to those reported previously in the literature.25 This marks the first syntheses of these diastereomeric natural products using a stereodivergent process from the same common precursor.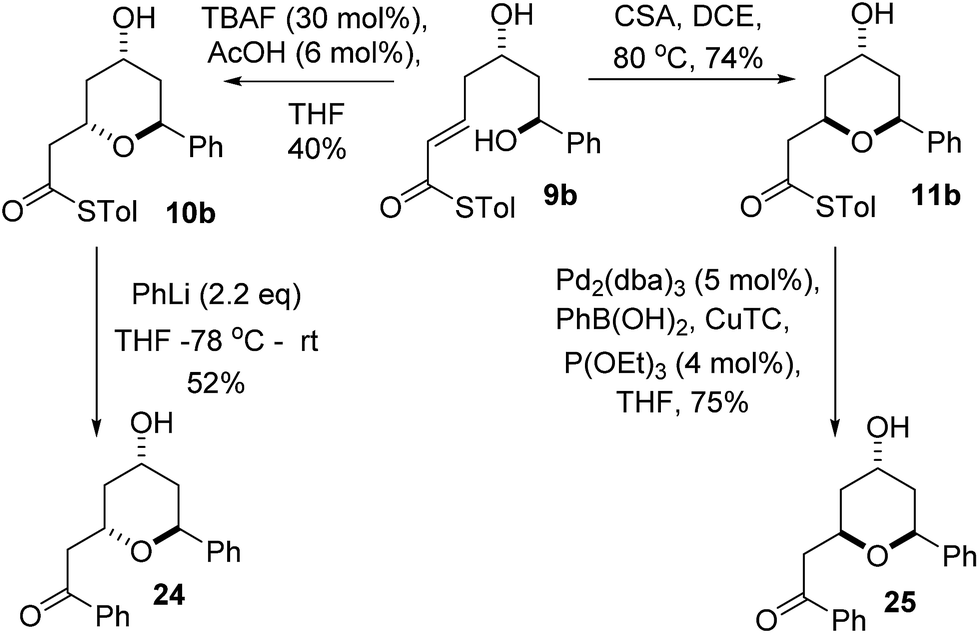 | ||
| Scheme 7 Stereodivergent synthesis of diospongins A and B. | ||
Conclusions
A stereodivergence was observed in the oxy-Michael reaction of α,β-unsaturated thioesters to form THP rings. These THP rings are present in a large number of structurally complex and biologically active natural products. Computational investigations of this stereodivergence indicated that it resulted from the participation of the 4-hydroxyl group in a hydrogen bond with the cyclising alkoxide which enforced a boat-like transition state in the buffered TBAF mediated reaction which led to the 2,6-trans-THP product. The Brønsted acid mediated reaction had no such interaction and proceeded through a chair-like transition state to generate the 2,6-cis-THPs. These computational studies suggested that both reactions were under kinetic control. Synthetic studies confirmed these computational predictions. When the 4-hydroxyl group was present the substrates exhibited stereodivergent reaction pathways under the reaction conditions. However, when the 4-hydroxyl group was removed or protected no stereodivergence was seen. These results allow us to suggest guidelines for the future diastereoselective synthesis of 2,6-disubstituted THPs.• Use α,β-unsaturated thioesters: they are more reactive in cyclizations than α,β-unsaturated oxoesters.
• For the formation of 2,6-cis-THPs use a Brønsted acid promoted cyclization.
• For the formation of 2,6-trans-THPs, a 4-hydroxyl group is essential in a buffered TBAF promoted cyclization.
The utility of these guidelines and the stereodivergent oxy-Michael reaction was further demonstrated by the stereodivergent synthesis of the anti-osteoporotic natural products diospongin A and B from a common precursor.
In summary, we have used a combined computational and experimental approach to develop a robust and simple procedure for the synthesis of 4-hydroxy-2,6-cis- and 4-hydroxy-2,6-trans-THP rings and elucidated the mechanism of this stereodivergence. We believe this knowledge will be extremely useful for those seeking to synthesize functionalized THP rings in high yields and with high selectivities in the context of natural product synthesis.
Acknowledgements
We acknowledge the University of York, Department of Chemistry's Wild Fund (K. E. and Y.-T. H) and the EU's ERASMUS + program (U. K.) for financial support.Notes and references
- (a) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC; (b) K. W. Armbrust, M. G. Beaver and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 6941 CrossRef CAS PubMed; (c) B. A. Loy, A. B. Lesser, D. Staveness, K. L. Billingsley, L. Cegelski and P. A. Wender, J. Am. Chem. Soc., 2015, 137, 3678 CrossRef CAS PubMed; (d) B. M. Trost, C. E. Stivala, K. L. Hull, A. Huang and D. R. Fandrick, J. Am. Chem. Soc., 2014, 136, 88 CrossRef CAS PubMed; (e) H. Kraus, A. Français, M. O'Brien, J. Frost, A. Diéguez-Vázquez, A. Polara, icordi, R. Horan, D.-S. Hau, T. Tsunoda and S. V. Ley, Chem. Sci., 2013, 4, 1989 RSC.
- P. A. Searle and T. F. Molinski, J. Am. Chem. Soc., 1995, 117, 8126 CrossRef CAS.
- P. A. Horton, F. E. Koehn, R. E. Longley and O. J. McConnell, J. Am. Chem. Soc., 1994, 116, 6015 CrossRef CAS.
- J. Yin, K. Kouda, Y. Tezuka, Q. Le Tran, T. Miyahara, Y. Chen and S. Kadota, Planta Med., 2004, 70, 54 CrossRef CAS PubMed.
- F. A. Cichewicz and P. Crews, Org. Lett., 2004, 6, 1951 CrossRef PubMed.
- (a) M. A. Perry, S. D. Rychnovsy and N. Sizemore, Synthesis of Saturated Oxygen Heterocycles I, Topics in Heterocyclic Chemistry 35, ed. J. Cossy, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 43–95 Search PubMed; (b) R. J. Beattie, T. W. Hornsby, G. Craig, M. C. Galan and C. L. Willis, Chem. Sci., 2016, 7, 2743 RSC; (c) H. Faustino, I. Varela, J. Mascareñas and F. López, Chem. Sci., 2015, 6, 2903 RSC; (d) Y. Xie and P. E. Floreancig, Chem. Sci., 2011, 2, 2423 RSC.
- (a) For a review see: C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988 RSC; (b) For an example in the synthesis of aspergillide A and B see: M. Kanematsu, M. Yoshida and K. Shishido, Angew. Chem., Int. Ed., 2011, 50, 2618 CrossRef CAS PubMed.
- (a) M. G. Banwell, C. T. Bui, H. T. T. Pham and G. W. Simpson, J. Chem. Soc., Perkin Trans. 1, 1996, 967 RSC; (b) J. M. Betancourt, V. S. Martín, J. M. Padrón, J. M. Palazón, M. A. Ramírez and M. A. Soler, J. Org. Chem., 1997, 62, 4570 CrossRef; (c) M. A. Ramírez, J. M. Padrón, J. M. Palazón and V. S. Martín, J. Org. Chem., 1997, 62, 4584 CrossRef; (d) M. G. Banwell, B. D. Bissett, C. T. Bui, H. T. T. Pham and G. W. Simpson, Aust. J. Chem., 1998, 51, 9 CrossRef CAS; (e) C. Schneider and A. Schuffenhauer, Eur. J. Org. Chem., 2000, 73 CrossRef CAS; (f) K. N. Houk, M. N. Paddon-Row, N. G. Rondan, Y.-D. Wu, F. K. Brown, D. C. Spellmeyer, J. T. Metz, Y. Li and R. J. Loncharich, Science, 1986, 231, 1108 CAS.
- (a) K. N. Houk and R. W. Strozier, J. Am. Chem. Soc., 1973, 95, 4094 CrossRef CAS; (b) J. L. Jensen and D. J. Carré, J. Org. Chem., 1974, 39, 2103 CrossRef CAS; (c) J. L. Jensen and A. T. Thibeault, J. Org. Chem., 1977, 42, 2168 CrossRef CAS.
- (a) For computational and synthetic studies on an intramolecular system see: T. P. A. Hari, B. I. Wilke, J. A. Davey and C. N. Boddy, J. Org. Chem., 2016, 81, 415 CrossRef CAS PubMed; (b) In a spiroketal system see: Y. Y. Khomutnyk, A. J. Arguelles, G. A. Winschel, Z. Sun, P. M. Zimmerman and P. Nagorny, J. Am. Chem. Soc., 2016, 138, 444 CrossRef CAS PubMed.
- (a) K. Lee, H. Kim and J. Hong, Org. Lett., 2011, 13, 2722 CrossRef CAS PubMed; (b) S. R. Byeon, H. Park, H. Kim and J. Hong, Org. Lett., 2011, 13, 5816 CrossRef CAS PubMed; (c) W. J. Buffham, N. A. Swain, S. L. Kostiuk, T. P. Goncalves and D. C. Harrowven, Eur. J. Org. Chem., 2012, 1217 CrossRef CAS; (d) S. Athe, B. Chandrasekhar, S. Roy, T. K. Pradhan and S. Ghosh, J. Org. Chem., 2012, 77, 9840 CrossRef CAS PubMed.
- (a) M. Ball, B. J. Bradshaw, R. Dumeunier, T. J. Gregson, S. MacCormick, H. Omori and E. J. Thomas, Tetrahedron Lett., 2006, 47, 2223 CrossRef CAS; (b) I. Paterson and L. E. Keown, Tetrahedron Lett., 1997, 38, 5727 CrossRef CAS; (c) C. S. Lee and C. J. Forsyth, Tetrahedron Lett., 1996, 37, 6449 CrossRef CAS.
- P. A. Clarke and K. Ermanis, Org. Lett., 2012, 14, 5550 CrossRef CAS PubMed.
- (a) H. Fuwa, K. Noto and M. Sasaki, Org. Lett., 2011, 13, 1820 CrossRef CAS PubMed; (b) H. Fuwa, N. Ichinokawa, K. Noto and M. Sasaki, J. Org. Chem., 2012, 77, 2588 CrossRef CAS PubMed.
- See ESI† for details.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (b) P. M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499 CrossRef CAS.
- (a) D. J. Tannor, B. Marten, R. Murphy, R. A. Friesner, D. Sitkoff, A. Nicholls, M. Ringnalda, W. A. Goddard III and B. Honig, J. Am. Chem. Soc., 1994, 116, 11875 CrossRef CAS; (b) B. Marten, K. Kim, C. Cortis, R. A. Friesner, R. B. Murphy, M. N. Ringnalda, D. Sitkoff and B. Honig, J. Phys. Chem., 1996, 100, 11775 CrossRef CAS.
- M. Pellicena, K. Krämer, P. Romea and F. Urpí, Org. Lett., 2011, 13, 5350 CrossRef CAS PubMed.
- I. Paterson and G. W. Haslett, Org. Lett., 2013, 15, 1338 CrossRef CAS PubMed.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1991, 56, 4218 CrossRef CAS.
- H. Fuwa, Heterocycles, 2012, 85, 1255 CrossRef CAS.
- Thioesters have previously been shown to exhibit characteristics similar to ketones: A. M. M. El-Asar, C. P. Nash and L. L. Ingraham, Biochemistry, 1982, 21, 1972 CrossRef.
- (a) K. B. Sawant and M. P. Jennings, J. Org. Chem., 2006, 71, 7911 CrossRef CAS PubMed; (b) S. Chandrasekhar, T. Shyamsunder, S. J. Prakash, A. Prabhakar and B. Jagadeesh, Tetrahedron Lett., 2006, 47, 47 CrossRef CAS; (c) C. Bressy, F. Allais and J. Cossy, Synlett, 2006, 3455 CAS; (d) R. W. Bates and P. Song, Tetrahedron, 2007, 63, 4497 CrossRef CAS; (e) M.-A. Hiebel, B. Pelotier and O. Piva, Tetrahedron, 2007, 63, 7874 CrossRef CAS; (f) N. Kawai, S. M. Hande and J. Uenishi, Tetrahedron, 2007, 63, 9049 CrossRef CAS; (g) J. S. Yadav, B. Padmavani, B. V. S. Reddy, C. Venugopal and A. B. Rao, Synlett, 2007, 2045 CrossRef CAS; (h) H. Wang, B. J. Shuhler and M. Xian, Synlett, 2008, 2651 Search PubMed; (i) G. Sabitha, P. Padmaja and J. S. Yadav, Helv. Chim. Acta, 2008, 91, 2235 CrossRef CAS; (j) K. Lee, K. H. Kim and J. Hong, Org. Lett., 2009, 11, 5202 CrossRef CAS PubMed; (k) G. Kumaraswamy, G. Ramakrishna, P. Naresh, B. Jagadeesh and B. Sridhar, J. Org. Chem., 2009, 74, 8468 CrossRef CAS PubMed; (l) J. D. More, Synthesis, 2010, 2419 CrossRef CAS; (m) R. N. Kumar and H. M. Meshram, Tetrahedron Lett., 2011, 52, 1003 CrossRef CAS; (n) T.-L. Ho, B. Tang, G. Ma and P. Xu, J. Chin. Chem. Soc., 2012, 59, 455 CrossRef CAS; (o) E. Stefan, A. P. Nalin and R. E. Taylor, Tetrahedron, 2013, 69, 7706 CrossRef CAS PubMed; (p) H. Yao, J. Ren and R. Tong, Chem. Commun., 2013, 49, 193 RSC; (q) S. B. Meruva, R. Mekala, A. Raghunadh, K. Raghavendra Rao, V. H. Dahanukar., T. V. Pratap, U. K. Syam Kumar and P. K. Dubey, Tetrahedron Lett., 2014, 55, 4739 CrossRef CAS; (r) T. Rybak and D. G. Hall, Org. Lett., 2015, 17, 4156 CrossRef CAS PubMed; (s) P. A. Clarke, N. M. Nasir, P. B. Sellars, A. M. Peter, C. A. Lawson and J. L. Burroughs, Org. Biomol. Chem., 2016, 14, 6840 RSC.
- L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260 CrossRef CAS.
- R. Wittenberg, J. Srogl, M. Egi and L. S. Liebeskind, Org. Lett., 2003, 5, 3033 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data and details of the computational studies. See DOI: 10.1039/c6sc03478k |
| This journal is © The Royal Society of Chemistry 2017 |