
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Geminal bis-borane formation by borane Lewis acid induced cyclopropyl rearrangement and its frustrated Lewis pair reaction with carbon dioxide†
Yun-Lin
Liu
,
Gerald
Kehr
,
Constantin G.
Daniliuc
and
Gerhard
Erker
*
Organisch-Chemisches Institut, Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
First published on 16th September 2016
Abstract
Cyclopropylacetylene reacts with two molar equivalents of Piers' borane [HB(C6F5)2] under mild conditions by an addition/rearrangement sequence with cyclopropyl ring opening to give a mixture of two α-B(C6F5)2 substituted tetrahydroboroles. This compound forms an active frustrated Lewis pair with PtBu3 that heterolytically splits dihydrogen and adds carbon dioxide as a geminal chelate bis-boryl component. The respective reactions of the two-fold HB(C6F5)2 addition to Ph-CH2CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH were studied as a geminal Lewis acid reference. Most of the products were characterized by X-ray diffraction.
CH were studied as a geminal Lewis acid reference. Most of the products were characterized by X-ray diffraction.
Introduction
Bis-boranes featuring pairs of strongly Lewis acidic B(C6F5)2 groups should be ideally matching templates for binding of CO2 under frustrated Lewis pair (FLP) conditions. Although such geminal bis-boranes are principally readily available from terminal alkynes by sequential hydroboration reactions with two molar equivalents of HB(C6F5)2, as it has been shown by Piers et al.,1,2 surprisingly little is known about this CO2-trapping reaction. Stephan et al. had used Siebert's unsaturated geminal BCl2 compound 1 (ref. 3) and the corresponding B(C6F5)2 analogue 2, which was derived from 1 by treatment with Zn(C6F5)2, for FLP/CO2 scavenging,4 but the vast majority of FLP/CO2 chemistry used non-chelate Lewis acidic binding motifs5,6 (Scheme 1). | ||
| Scheme 1 Geminal bis-boranes and their FLP reactions with CO2. | ||
We have now investigated the tBu3P/CO2 trapping reaction using a pair of geminal C6F5 containing bis-boranes. Both were obtained by the treatment of the respective terminal acetylene starting materials with two molar equivalents of Piers' borane [HB(C6F5)2]. While we observed the expected normal behaviour upon reacting the alkyne Ph-CH2CH2CCH (5a) with the hydroboration reagent, we observed a rather complex rearrangement behaviour that took place upon the treatment of cyclopropylacetylene (5b) with the HB(C6F5)2 borane. The characterization of the resulting special rearrangement product, its formation and its FLP reaction with CO2 in the presence of a tert-phosphine will be presented and discussed in this account.
Results and discussion
The Ph-CH2CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CH/2HB(C6F5)2 system
CH/2HB(C6F5)2 system
Terminal acetylenes undergo regioselective 1,2-hydroboration with the HB(C6F5)2 reagent to yield the respective substituted vinyl boranes.7 When the reaction is carried out in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of alkyne and [B]H borane, the respective saturated geminal bis-borane is obtained in many cases under kinetic control.1 This typical reaction path was also observed when we treated the alkyne 5a with HB(C6F5)2 in a 1:2 ratio in toluene solution at r.t. (1 hour reaction time). Workup gave the product 6a, which we isolated as a white solid with a 76% yield. The compound was characterized by C,H-elemental analysis and by spectroscopy, and we carried out some characteristic reactions.
:2 molar ratio of alkyne and [B]H borane, the respective saturated geminal bis-borane is obtained in many cases under kinetic control.1 This typical reaction path was also observed when we treated the alkyne 5a with HB(C6F5)2 in a 1:2 ratio in toluene solution at r.t. (1 hour reaction time). Workup gave the product 6a, which we isolated as a white solid with a 76% yield. The compound was characterized by C,H-elemental analysis and by spectroscopy, and we carried out some characteristic reactions.
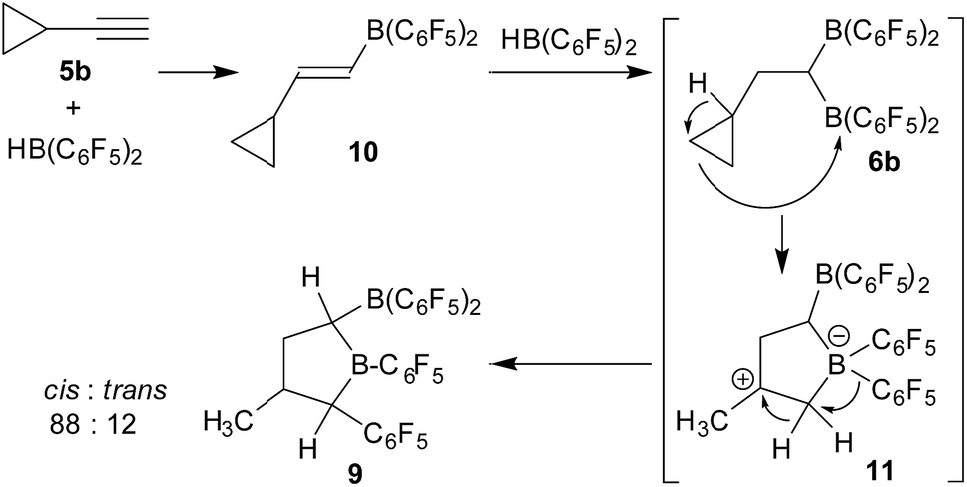
Compound 6a shows a single 11B NMR resonance at δ = 72.1 ppm, which is typical for Lewis acidic planar tricoordinate R–B(C6F5)2 situations.8 Consequently, we observed the three 19F NMR signals of the symmetry-equivalent C6F5 groups at boron. They show a typical large meta/para fluorine NMR chemical shift difference (Δδ19Fm,p = 13.7 ppm). The mixture of compound 6a with the bulky phosphine PtBu3 (1:1) represents a reactive frustrated Lewis pair that is able to heterolytically split dihydrogen9 under mild conditions (r.t., 2.0 bar of H2, overnight in pentane). The product precipitated from the reaction mixture and was isolated as a white solid with an 87% yield. Compound 7 was characterized by X-ray diffraction (single crystals were obtained from pentane/dichloromethane at −35 °C by the diffusion method).
Compound 7 shows a fully extended all anti-periplanar C4-chain featuring the phenyl substituent at one end and the geminal pair of boryl groups at the other. The C1–B1/B2 bonds are almost of the same length and the pair of boron atoms is bridged by the hydride (see Fig. 1). In the crystal there is an independent HPtBu3+ countercation. In solution, compound 7 shows the 31P NMR signal of the [P]H phosphonium cation (δ = 60.2 ppm, 1JPH ∼ 430 Hz). The anion shows the single broad 11B NMR resonance of the symmetry-equivalent pair of B(C6F5)2 groups in the typical tetracoordinated borate range (δ = −18.8 ppm). The C6F5 groups of the boryl groups are diastereotopic. Therefore, we have observed two sets of o,p,m-C6F519F NMR resonances for these units. The bridging [B]–H–[B] hydride gives rise to a broad 1H NMR signal at δ = 2.64 ppm (Scheme 2).
 | ||
| Fig. 1 A projection of the molecular structure of the FLP dihydrogen splitting product 7 (thermal ellipsoids are shown with a 30% probability level). Selected bond lengths (Å) and angles (degrees): B1⋯B2 1.945(5), C1–C2 1.526(4), C1–B1 1.609(5), C1–B2 1.598(5), B1–H01 1.26(4), B2–H01 1.34(3), B2–C1–B1 74.7(2), B2–H01–B1 96.9. | ||
 | ||
| Scheme 2 Frustrated Lewis pair reactions with the geminal bis-borane. 6a. | ||
We then treated the bis(boryl)alkane/phosphine FLP [6a/PtBu3] with carbon dioxide. Exposure of the 6a/PtBu3 mixture in pentane at r.t. to CO2 (2.0 bar) quickly (in 2 hours) resulted in the formation of a white precipitate of compound 8, which was isolated with an 81% yield. Compound 8 is sensitive in solution (CD2Cl2) and decomposed above 0 °C. Single crystals of the FLP/CO2 adduct 8 suitable for characterization by X-ray diffraction were obtained from pentane/dichloromethane at −35 °C by the diffusion method (see Fig. 2). In the crystal, compound 8 shows a gauche/anti-periplanar conformation of the Ph–CH2CH2CH2–CH-chain. The geminal pair of B(C6F5)2 substituents at carbon atom C1 has taken up the CO2 molecule in a rather symmetric way by forming two boron–oxygen bonds of almost the same length, and also the C5–O1/O2 bonds are almost equal in length, indicating a fully delocalized structure for this sub-moiety of compound 8. The resulting six-membered heterocycle features an almost coplanar arrangement of the BOCOB unit with only the carbon atom C1 being localized markedly outside of this plane. The bulky PtBu3 group is found attached at the central carbon atom C5 of this heterocyclic subunit of the overall molecular zwitterionic FLP/CO2 addition product 8.
 | ||
| Fig. 2 A view of the molecular structure of the zwitterionic FLP/CO2 adduct 8 (thermal ellipsoids are shown with a 50% probability level; hydrogen atoms of the PtBu3 group and the C6F5 substituents at boron atoms B1 and B2 are omitted for clarity: for details see the ESI†). Selected bond lengths (Å) and angles (degrees): B1–O1 1.628(4), B2–O2 1.635(4), O1–C5 1.265(4), O2–C5 1.264(4), B2–C1–B1 108.6(2), O2–C5–O1 127.7(3). | ||
In solution we observe the 13C NMR resonance of the scavenged CO2 molecule at δ = 172.7 ppm with a 1JPC coupling constant of 92.6 Hz. Compound 8 shows a typical phosphonium 31P NMR signal at δ = 60.3 ppm, and a single broad 10B NMR resonance at δ = 10.8 ppm. The C6F5 groups at the pair of boron atoms are pairwise diastereotopic, giving rise to two equal intensity pairs of o,p,m19F NMR features, with a rather small chemical shift difference Δδ19Fm,p around 5.5 ppm, as is typical for borate type structures based on the B(C6F5)2 subunit.
The cyclopropylacetylene/2HB(C6F5)2 system: rearrangement to tetrahydroborole derivatives
We next reacted cyclopropylacetylene (5b) with two molar equivalents of Piers' borane [HB(C6F5)2] (toluene, r.t., 1 hour). In this case, we did not obtain the simple cyclopropyl-CH2CH[B(C6F5)2]2 product (6b), but found that a rearrangement had occurred. In situ NMR spectroscopy revealed the formation of a ca. 7:1 mixture of the α-boryl-tetrahydroborole products cis-9 and trans-9. We isolated the compound cis-9 in an almost pure condition (96:4) after workup as a pale yellow solid with a 67% yield (see Scheme 3).
 | ||
| Scheme 3 Formation of the tetrahydroborole derivative 9 (with unsystematical atom numbering scheme as used in Fig. 3). | ||
Compound cis-9 was characterized by X-ray diffraction using single crystals that were grown from a pentane solution of the compound at −35 °C (see Fig. 3, left). The X-ray crystal structure analysis has shown that a five-membered saturated tetrahydroborole framework had been formed in the reaction, bearing a B(C6F5)2 substituent at the α-position C1, a methyl substituent at C3 and a C6F5 substituent at C4. The pair of substituents in cis-9 at carbon atoms C1 and C4 are cis-oriented; both are in a trans-arrangement with the methyl substituent at carbon atom C3. The plane of the C6F5 group at C4 is oriented markedly away from the mean heterocyclic core [dihedral angle θ B1–C4–C21–C22 −119.6(1)°], whereas the C6F5 group at the adjacent boron atom B1 is rotated slightly in the opposite direction [θ C1–B1–C11–C12 49.0(7)°, C4–B1–C11–C12 −136.2(5)°]. Both of the boron atoms B1 and B2 show trigonal planar coordination geometries (∑B1ccc 359.8°, ∑B2ccc 359.9°), which should render these both as strongly Lewis acidic centres. Consequently, we have monitored a pair of 11B NMR features for compound cis-9 in solution (d6-benzene) in the typical range of Lewis acidic B(C6F5)R signals (δ = 78.0 ppm, 67.6 ppm) and a similar appearance of the 19F NMR spectrum was observed for the B(C6F5)2/B(C6F5) units [Δδ19Fm,p = 13.9 ppm, 16.0 ppm] (for details, see the ESI†).
 | ||
| Fig. 3 A view of the molecular structures of the α-boryl-tetrahydroborole cis-9 [left, thermal ellipsoids are shown with a 30% probability level; selected bond lengths (Å) and angles (degrees): B1–C1 1.582(7), C1–B2 1.561(7), B1–C4 1.576(7), B2–C1–B1 120.3(4), C11–B1–C4 125.5(4)] and the trans-9 epimer (right, thermal ellipsoids are shown with a 50% probability level; the separate synthesis of trans-9 is described below). Selected bond lengths (Å) and angles (degrees): B1–C1 1.588(2), C1–B2 1.543(2), B1–C4 1.579(2), B2–C1–B1 114.0(1), C11–B1–C4 124.3(1), ∑B1ccc 360.0, ∑B2ccc 359.8. | ||
We tried to find a mechanistic rationale for the formation of the boryl tetrahydroborole product 9 in the reaction of cyclopropylacetylene (5b) with two HB(C6F5)2 equivalents. It is known that cyclopropanes are often readily opened to the respective olefin isomers upon exposure to boron Lewis acids.10 Therefore, we briefly checked whether the opened isomer of 5b, 2-methyl-1-buten-3-yne, might be involved in this reaction. However, this was not the case. Its reaction with two equivalents of HB(C6F5)2 took a different course (for details, see the ESI†).
Therefore, we assumed a reaction pathway as outlined in Scheme 4. It is known that 5b undergoes a single hydroboration with Piers' borane to give 10, so we assume it to be the initial intermediate.11 With a second HB(C6F5)2 equivalent this can then undergo the subsequent hydroboration reaction to give the geminal bis-boryl substituted compound 6b. In the in situ NMR experiment we observed an intermediate which is likely 6b (for details, see the ESI†). This is not stable under our typical reaction conditions but undergoes Lewis acid induced cyclopropyl ring opening, potentially leading to 11 which is subsequently stabilized by a sequence of hydride/C6F5 1,2-shifts to result in the observed product 9. We must stress that we so far have no information about the alleged intermediates on the way and we cannot convincingly explain, let alone predict, the preferred stereochemical outcome, aside from the assumption that the formation of the observed cis-9 product is following a pathway of least steric hindrance on the way (Scheme 5).
 | ||
| Scheme 4 Rearrangement reaction leading to 9. | ||
 | ||
| Scheme 5 FLP reactions of compound cis-9. | ||
The geminal bis-boryl compound contains a pair of Lewis acidic boron atoms and, consequently, it may serve as a chelate boron Lewis acid component in FLP chemistry. The isolated cis-9 in conjunction with the phosphorus Lewis base PtBu3 served as an active dihydrogen splitting reagent. Thus, treatment of a 1:1 mixture of cis-9 and PtBu3 with dihydrogen (2.0 bar) in pentane solution overnight produced the dihydrogen splitting product cis-12 as a precipitate. The salt cis-12 was isolated as a white solid with a 71% yield. We obtained single crystals of compound cis-12 from pentane/dichloromethane by a diffusion method which were suitable for characterization by X-ray diffraction (see Fig. 4). In the crystal, we see the typical r-1-boryl, t-3-methyl, c-4-C6F5 arrangement12 of the substituents on the tetrahydroborole framework. There is now a hydride bridging between the two boron atoms.13 Consequently, both the boron atoms B1 and B2 have attained distorted tetrahedral coordination geometries (∑B1ccc = 345.3°, ∑B2ccc = 349.8°), and we found the HPtBu3+ cation in the crystal.
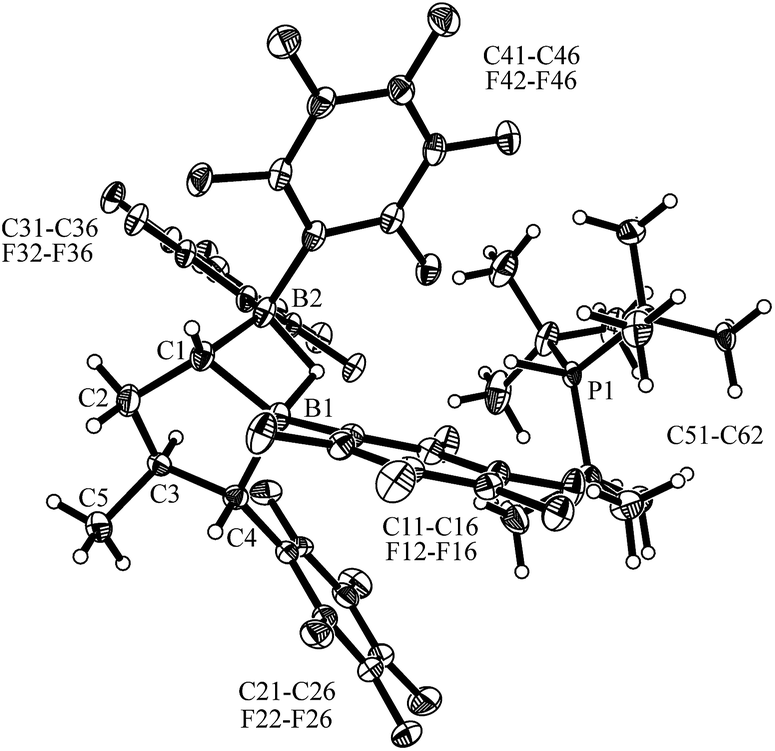 | ||
| Fig. 4 Molecular structure of the dihydrogen splitting product cis-12 (thermal ellipsoids are shown with a 30% probability level). Selected bond lengths (Å) and angles (degrees): B1⋯B2 1.924(6), B2–C1 1.584(5), B2–H1, 1.33(3), C1–B1, 1.603(5), B1–C4 1.653(5), B1–H1 1.35(3), B2–C1–B1 74.3(2), B1–H1–B2 91.7. | ||
The bulk isolated product cis-12 (in CD2Cl2) contained ca. 15–20% contamination of the isomer trans-12 since we had started from a not completely pure starting material (for details, see the ESI;† the characterization of the independently synthesised isomer trans-12 will be described below). Compound cis-12 shows a pair of 11B NMR signals in the typical borate chemical shift range (δ = −14.5 ppm, −19.7 ppm). It shows a 31P NMR phosphonium doublet at δ = 60.6 ppm with 1JPH ∼ 428 Hz. We also exposed the cis-9/PtBu3 FLP (again contaminated with a small amount of trans-9) to carbon dioxide (2.0 bar, r.t., overnight) in pentane solution. Under the typical conditions, the zwitterionic FLP/CO2 addition product precipitated and was recovered by filtration to give cis-13 as a white solid with a 73% yield. The NMR analysis (in THF-d8) again showed the presence of a second isomer (trans-13, see below, ca. 3%).
Single crystals of cis-13 suitable for X-ray crystal structure analysis were obtained from pentane/dichloromethane at −35 °C by the diffusion method (see Fig. 5). The compound contains a central heterocyclic six-membered ring that was formed by double chelate coordination of the geminal bis-boryl acceptor with the oxygen atoms of the phosphine activated carbon dioxide molecule. The structure of this subunit is largely delocalized with similar bond lengths in the B1–O1/B2–O2 pair as well as the C6–O1/O2 pair of carbon–oxygen bonds. Carbon atom C6 has the PtBu3 group attached to it. This chelate heterocycle is interlocked with the five-membered tetrahydroborole framework, which has the boron atom B1 incorporated in it. This section of the molecule shows the same characteristic stereochemical features as we had found for its precursor cis-9. The hydrogen atoms at C1/C4 and the methyl substituent at carbon atom C3 are all in a cis-arrangement on this five-membered ring.
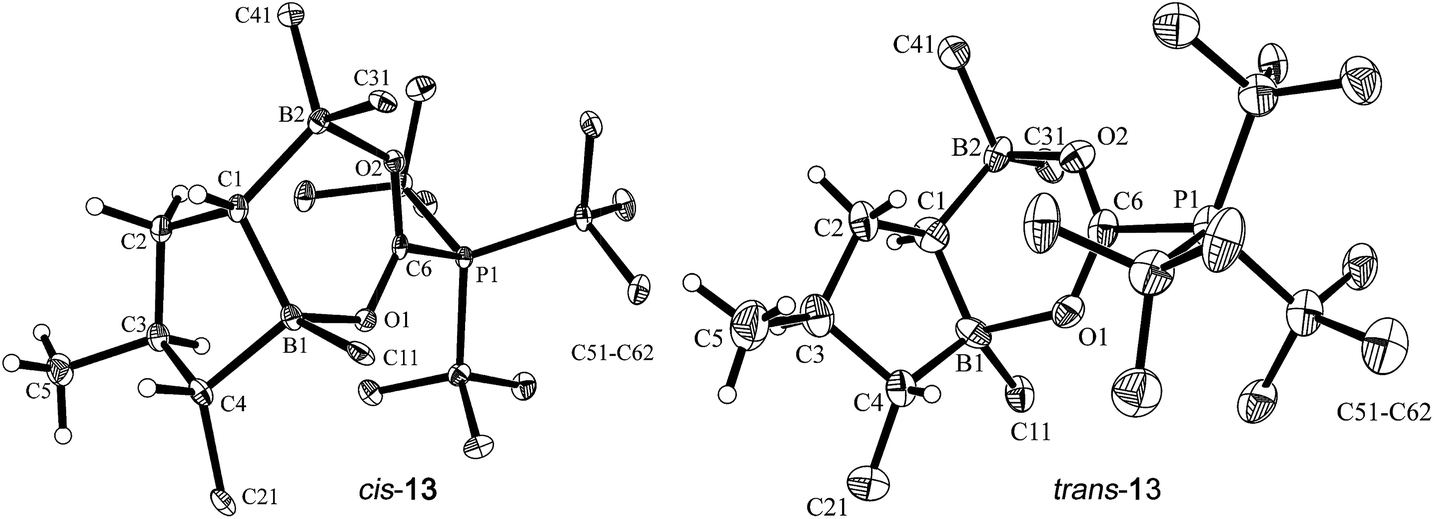 | ||
| Fig. 5 Projection of the molecular structures of the FLP/CO2 addition product cis-13 [left, thermal ellipsoids are shown with a 50% probability level; hydrogen atoms of the PtBu3 group and the C6F5 substituents at boron atoms B1, B2, and at carbon atom C4 are omitted for clarity: for details see the ESI;† selected bond lengths (Å) and angles (degrees): P1–C6 1.905(2), B1–O1 1.657(2), B2–O2 1.636(2), O2–C6 1.258(2), O1–C6 1.255(2), O1–C6–O2 128.0(2), B2–C1–B1 110.9(2)] and trans-13 [right, the independent synthesis of trans-13 is described below; thermal ellipsoids are shown with a 30% probability level; hydrogen atoms of the PtBu3 group and the C6F5 substituents at boron atoms B1, B2, and at carbon atom C4 are omitted for clarity: for details see the ESI;† selected bond lengths (Å) and angles (degrees): P1–C6 1.913(10), B1–O1 1.717(13), B2–O2 1.634(13), O2–C6 1.248(12), O1–C6 1.271(12), O1–C6–O2 128.7(9), B2–C1–B1 118.1(9)]. | ||
The boryl tetrahydroborole system 9 contains three independent carbon chirality centres. Therefore, there is the possibility of forming four diastereoisomers. So far our rearrangement reaction was rather stereoselective and produced the major product cis-9 with the relative stereoselectivity r-1, t-3, c-4 plus a small amount of a minor isomer which probably represents one of the other three diastereoisomers, but whose relative stereochemistry we did not know. We have now prepared and characterized the isomer “trans-9” (of relative r-1, c-3, t-4 stereochemistry) by a selective isomerization process at the saturated central heterocyclic framework.
For that purpose, we treated the substituted tetrahydroborole product cis-9 [r-1, t-3, c-4] with a catalytic amount (20 mol%) of the persistent nitroxide radical TEMPO (pentane, r.t., 4 days).11,14 This reaction apparently proceeded with reversible H-atom abstraction at the activated C1 position of the heterocycle and we isolated the trans-9 epimer [r-1, c-3, t-4] as a colourless solid with a 74% yield. This compound was characterized by C,H-elemental analysis, by NMR spectroscopy (11B: δ = 79.6 ppm, 72.9 ppm, for details see the ESI†) and by X-ray diffraction. Single crystals suitable for the X-ray crystal structure analysis of compound trans-9 were obtained from a pentane/dichloromethane mixture at −35 °C (see Fig. 3, right). It shows the typical five-membered tetrahydroborole framework with the B(C6F5)2 and C6F5 substituents at carbon atoms C1 and C4 now in a trans relationship. The methyl group at C3 has remained trans oriented to the C6F5 group at C4.
Compound trans-9 also formed an active frustrated Lewis pair with PtBu3. The system heterolytically cleaved dihydrogen at near to ambient conditions (pentane, r.t., 2.0 bar H2, overnight), and we isolated the hydridoborate/phosphonium salt with a 62% yield. It shows typical 11B NMR signals at δ = −13.3 ppm and −17.1 ppm and a 31P NMR feature at δ = 60.7 ppm (1JPH ∼ 428 Hz). Compound trans-12 was characterized by X-ray diffraction (single crystals were obtained from pentane/dichloromethane at r.t. by the diffusion method). The X-ray crystal structure analysis (see Fig. 6) showed the presence of the hydride bridged pair of boron atoms inside the anion and the separate HPtBu3+ cation. The framework of compound trans-12 features the expected trans-orientation of the B(H)(C6F5)2/C6F5 pair of substituents at the ring carbon atoms C1/C4 and the vicinal trans-orientation of the C3–CH3 group with the C4–C6F5 substituent. The system has consequently conserved the relative stereochemistry of the starting material trans-9. Compound trans-12 shows a relative stereochemistry of r-1, c-3, t-4 (see Scheme 6 and Fig. 6).
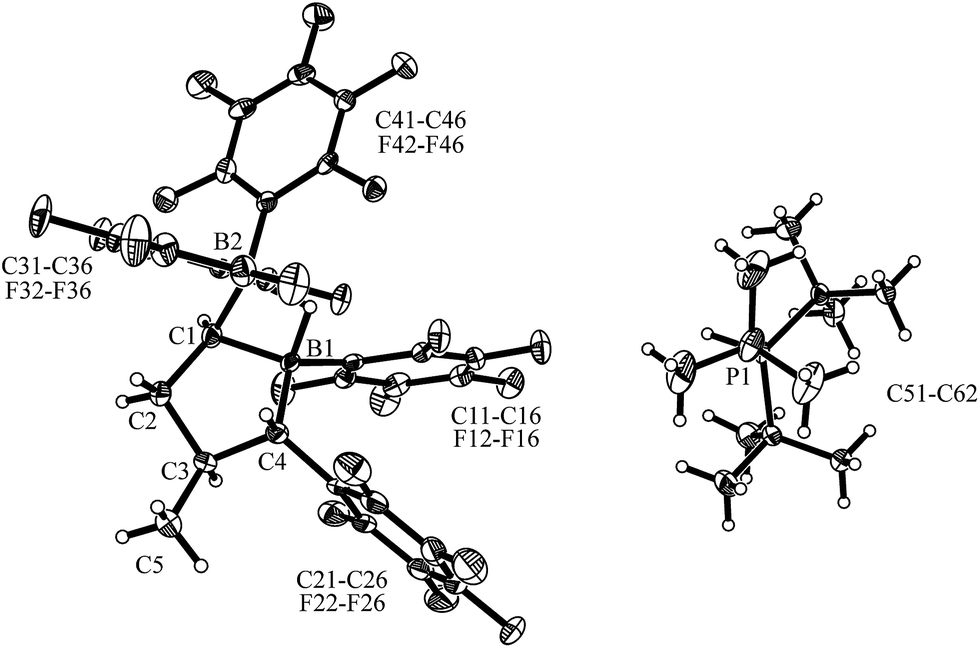 | ||
| Fig. 6 A view of the molecular structure of the hydridoborate/phosphonium salt trans-12 (thermal ellipsoids are shown with a 50% probability level). Selected bond lengths (Å) and angles (degrees): B1⋯B2 1.909(5), B2–C1 1.584(5), B1–C1 1.596(4), B1–C4 1.636(5), B2–H1 1.31(3), B1–H1 1.30(4), B2–C1–B1 73.8(2), B1–H1–B2 93.8. | ||
 | ||
| Scheme 6 Isomerization of compound cis-9 and the FLP reactions of trans-9. | ||
Compound trans-9 also reacts with carbon dioxide in the presence of PtBu3. Exposing a mixture of trans-9 and tris(tert-butyl)phosphine in pentane solution overnight at r.t. to a CO2 atmosphere gave the FLP/CO2 adduct trans-13 as a white precipitate with a 76% yield. The compound turned out to be only sparingly soluble in many solvents. However it could be characterized by X-ray diffraction using single crystals that were directly obtained from the reaction mixture of trans-9/PtBu3 with CO2. The structure (see Fig. 5, right) confirmed the stereochemical assignment of the backbone of the compounds of this trans-series: in compound trans-13 the boryl substituent at carbon atom C1 is in a trans relationship with the C6F5 substituent at the distal ring carbon atom C4, and the latter is oriented trans relative to the methyl group at C3. Consequently, the relative positions of the three substituents at the central tetrahydroborole framework in compound trans-13 are r-1-boryl, c-3-methyl, t-4-C6F5 configured. The CO2 oxygen atoms are found to be bonded to the pair of boron Lewis acid sites and the phosphorus atom is coordinated to the CO2 carbon atom. The CO2 bonding to the geminal bis(borane) acceptor is slightly unsymmetrical with the B1–O1 bond in the central position being markedly longer than the lateral B2–O2 contact and also the P1–C6 linkage is rather long (see Fig. 5). Compound trans-13 was just sufficiently soluble in d8-THF to allow the recording of most of its NMR features. The actual sample used was ca. 90% pure, and it contained a minor compound of unknown composition. Compound trans-13 shows a 31P NMR resonance at δ = 57.4 ppm. The 13C NMR signal of the CO2 derived moiety occurs at δ = 173.4 ppm (1JPC = 90.0 Hz) and compound trans-13 features a total of six o- (two overlapping), four p- and four m-C6F519F NMR signals in d8-THF at 233 K (for further details see the ESI†).
Conclusions
We have shown in this study that the reaction of cyclopropylacetylene with two molar equivalents of Piers' borane [HB(C6F5)2] takes an unusual course. We assume that initially the usual two-fold hydroboration reaction of the terminal alkyne takes place with the anti-Markovnikov orientation generating the respective geminal bis-boryl compound. This is apparently not stable under the applied mild reaction conditions, but undergoes an intramolecular rearrangement process initiated by cyclopropyl ring opening by the adjacent strong borane Lewis acid. This initiates a series of 1,2-migration reactions involving the migration of one C6F5 group from boron to carbon which eventually yields the α-boryl tetrahydroborole system 9. This is obtained with a rather high diastereoselectivity from this rearrangement process. The major compound cis-9 is an active FLP dihydrogen cleavage reagent in the presence of the bulky PtBu3 Lewis base. The cis-9/PtBu3 FLP also sequesters CO2 cleanly in a chelate fashion, similar to the here studied more Lewis acidic geminal R-CH[B(C6F5)2]2 reference systems, despite the loss of one electron withdrawing C6F5 substituent at a boron atom. This probably indicates the favourable influence of the geminal bis-boryl situation for both chelate hydride and chelate CO2 binding.Experimental section
Preparation of compound 6a
A solution of 4-phenyl-1-butyne (5a, 65.0 mg, 0.50 mmol) in toluene (1.0 mL) was added to a suspension of bis(pentafluorophenyl)borane (345 mg, 1.00 mmol) and toluene (3.0 mL). The reaction mixture was stirred at room temperature for 1 hour and then the suspension was filtered by cannula filtration. The volatiles of the obtained filtrate were removed in vacuo to give a colorless oil. Subsequently pentane (4.0 mL) was added and the mixture was stored at ca. −35 °C overnight. The formed white powder was isolated by filtration, washed with pentane (2 × 1 mL) and dried in vacuo to give compound 6a (312 mg, 0.38 mmol, 76%) as a white solid. Anal. calc. for C34H12B2F20: C, 49.68%; H, 1.47%. Found: C, 49.40%; H, 1.40%. For the NMR data see the ESI.†Preparation of compound 7
A solution of compound 6a (82.2 mg, 0.10 mmol) and tri-tert-butylphosphine (20.5 mg, 0.10 mmol) in pentane (3.0 mL) was exposed to a hydrogen atmosphere (2.0 bar) at room temperature and stirred overnight. The resulting white precipitate was collected by cannula filtration and washed with pentane (3 × 2 mL). After the removal of all volatiles in vacuo, compound 7 was obtained (88.6 mg, 0.087 mmol, 87%) as a white solid. Anal. calc. for C46H41B2F20P: C, 53.83%; H, 4.03%. Found: C, 53.81%; H, 4.01%. Single crystals suitable for the X-ray crystal structure analysis were obtained by the slow diffusion of pentane into a solution of compound 7 in dichloromethane at −35 °C.Preparation of compound 8
A solution of compound 6a (123.3 mg, 0.15 mmol) and tri-tert-butylphosphine (30.3 mg, 0.15 mmol) in pentane (5.0 mL) was exposed to CO2 (2.0 bar) at room temperature and then stirred for 2 hours. The resulting white precipitate was isolated by cannula filtration and washed with pentane (3 × 1 mL). After drying the solid in vacuo, compound 8 (129.4 mg, 0.12 mmol, 81%) was obtained as a white powder. Anal. calc. for C47H39B2F20O2P: C, 52.84%; H, 3.68%. Found: C, 53.21%; H, 3.91%. Single crystals of compound 8 suitable for the X-ray crystal structure analysis were obtained by the slow diffusion of pentane into a solution of the white powder in dichloromethane at −35 °C.Preparation of compound cis-9
A solution of compound 5b (33.0 mg, 0.50 mmol) in toluene (1.0 mL) was added to a suspension of bis(pentafluorophenyl)borane (345 mg, 1.00 mmol) and toluene (3.0 mL). After stirring the reaction mixture at room temperature for 1 hour, the solution was separated from the resulting suspension by cannula filtration. Then all volatiles of the filtrate were removed in vacuo to give a yellow oil, which was dissolved in pentane (2.5 mL) and stored at −35 °C overnight. The precipitated pale yellow solid was isolated by filtration and washed with cold pentane (2 × 0.5 mL). The removal of all volatiles in vacuo gave a pale yellow solid (253 mg, 0.34 mmol, 67%). Anal. calc. for C29H8B2F20: C, 45.95 %; H, 1.06%. Found: C, 45.74%; H, 1.07%. Crystals of compound cis-9 suitable for the X-ray crystal structure analysis were obtained from a solution of the yellow solid in pentane at −35 °C.Preparation of compound trans-9
TEMPO (16.6 mg, 0.11 mmol) was added to a solution of compound cis-9 (400 mg, 0.53 mmol) in pentane (15 mL). After stirring the reaction mixture at r.t. for 4 days, the resulting suspension was concentrated to about 2.0 mL, and stored in the fridge (−35 °C) overnight. The precipitated white powder was isolated via cannula filtration, and washed with cold pentane (2 × 1.0 mL). The removal of all volatiles under reduced pressure gave product trans-9 (296 mg, 0.39 mmol, 74%) as a white solid. Anal. calc. for C29H8B2F20: C, 45.95%; H, 1.06%. Found: C, 45.45%; H, 0.95%. Crystals suitable for the X-ray crystal structure analysis were obtained from a solution of compound trans-9 in pentane (1.5 mL) and CH2Cl2 (0.5 mL) at −35 °C.Preparation of compound cis-12
A solution of compound cis-9 (cis/trans≈ 96/4, vide supra) (113.7 mg, 0.15 mmol) and tri-tert-butylphosphine (30.3 mg, 0.15 mmol) in pentane (5.0 mL) was exposed to dihydrogen (2.0 bar) at room temperature and then stirred overnight. The formed white precipitate was collected by cannula filtration and washed with pentane (3 × 1 mL). After the removal of all volatiles in vacuo, a white solid was obtained (101.4 mg, 0.11 mmol, 71%). Anal. calc. for C41H37B2F20P: C, 51.17%; H, 3.88%. Found: C, 50.96%; H, 3.76%. Single crystals of compound cis-12 suitable for the X-ray crystal structure analysis were obtained by the slow diffusion of n-pentane into a solution of the white solid in dichloromethane at room temperature.Preparation of compound trans-12
A solution of compound trans-9 (75.8 mg, 0.10 mmol) and tri-tert-butylphosphine (20.2 mg, 0.10 mmol) in pentane (4.0 mL) was exposed to a dihydrogen atmosphere (2.0 bar) at room temperature and stirred overnight. The formed white precipitate was collected by cannula filtration and washed with n-pentane (2 × 1 mL). The removal of all volatiles in vacuo gave compound trans-12 (59.2 mg, 0.062 mmol, 62%) as a white solid. Anal. calc. for C41H37B2F20P: C, 51.17%; H, 3.88%. Found: C, 51.09%; H, 3.67%. Single crystals suitable for X-ray crystal structure analysis were obtained by the slow diffusion of pentane into a solution of compound trans-12 in dichloromethane at room temperature.Preparation of compound cis-13
A solution of compound cis-9 (cis/trans≈ 96/4, vide supra) (113.7 mg, 0.15 mmol) and tri-tert-butylphosphine (30.3 mg, 0.15 mmol) in pentane (5.0 mL) was exposed to a CO2 atmosphere (2.0 bar) and then stirred overnight at room temperature. The formed white precipitate was collected by cannula filtration and washed with pentane (3 × 1 mL). After the removal of all volatiles in vacuo, a white solid was obtained (108.3 mg, 0.11 mmol, 73%). Anal. calc. for C42H35B2F20O2P: C, 50.23%; H, 3.51%. Found: C, 50.07%; H, 3.36%. Single crystals of compound cis-13 suitable for the X-ray crystal structure analysis were obtained by the slow diffusion of n-pentane into a solution of the obtained white solid in dichloromethane at −35 °C.Preparation of compound trans-13
A solution of compound trans-9 (75.8 mg, 0.10 mmol) and tri-tert-butylphosphine (20.2 mg, 0.10 mmol) in pentane (5.0 mL) was exposed to CO2 (2.0 bar) at room temperature and then stirred overnight. The formed white precipitate was collected by cannula filtration and washed with pentane (3 × 1 mL). After the removal of all volatiles in vacuo, compound trans-13 (76.2 mg, 0.076 mmol, 76%) was obtained as a white solid. Anal. calc. for C42H35B2F20O2P: C, 50.23%; H, 3.51%. Found: C, 49.96%; H, 3.27%. Single crystals of compound trans-13 suitable for the X-ray crystal structure analysis were obtained directly from a reaction solution of compound trans-9 (37.9 mg) and tri-tert-butylphosphine (10.1 mg) and dichloromethane (1.0 mL) in a CO2 atmosphere (2.0 bar) at room temperature.Acknowledgements
Financial support from the Alexander von Humboldt-Stiftung (stipend to Y. L. L.) is gratefully acknowledged.Notes and references
- D. J. Parks, R. E. von H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809 CrossRef CAS.
- (a) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS. See also: (b) J.-H. Lamm, J. Horstmann, J. H. Nissen, J.-H. Weddeling, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2014, 4294 CrossRef CAS.
- W. Siebert, M. Hildenbrand, P. Hornbach, G. Karger and H. Pritzkow, Z. Naturforsch., B: J. Chem. Sci., 1989, 44, 1179 CAS.
- X. Zhao and D. W. Stephan, Chem. Commun., 2011, 47, 1833 RSC.
- Selected examples of CO2 binding using non-chelate borane Lewis acids: (a) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643 CrossRef PubMed; (b) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796 CrossRef PubMed; (c) I. Peuser, R. C. Neu, X. Zhao, M. Ulrich, B. Schirmer, J. A. Tannert, G. Kehr, R. Fröhlich, S. Grimme, G. Erker and D. W. Stephan, Chem.–Eur. J., 2011, 17, 9640 CrossRef CAS PubMed; (d) C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925 CrossRef CAS PubMed; (e) J. Boudreau, M. A. Courtemanche and F. G. Fontaine, Chem. Commun., 2011, 47, 11131 RSC; (f) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826 CrossRef CAS PubMed; (g) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463 CrossRef CAS PubMed; (h) M. Harhausen, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2012, 31, 2801 CrossRef CAS; (i) F. Bertini, V. Lyaskovskyy, B. J. J. Timmer, F. J. J. de Kanter, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201 CrossRef CAS PubMed; (j) X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 6465 CrossRef CAS PubMed; (k) S. Frömel, G. Kehr, R. Fröhlich, C. G. Daniliuc and G. Erker, Dalton Trans., 2013, 42, 14531 RSC; (l) M. J. Sgro and D. W. Stephan, Chem. Commun., 2013, 49, 2610 RSC; (m) D. Voicu, M. Abolhasani, R. Choueiri, G. Lestari, C. Seiler, G. Ménard, J. Greener, A. Guenther, D. W. Stephan and E. Kumacheva, J. Am. Chem. Soc., 2014, 136, 3875 CrossRef CAS PubMed.
- Selected examples of FLP mediated CO2 reduction using non-chelate Lewis acids: (a) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839 CrossRef CAS PubMed; (b) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660 CrossRef CAS PubMed; (c) G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2011, 50, 8396 CrossRef PubMed; (d) K. Takeuchi and D. W. Stephan, Chem. Commun., 2012, 48, 11304 RSC; (e) M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343 CrossRef CAS PubMed; (f) R. Dobrovetsky and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 2516 CrossRef CAS PubMed; (g) M. A. Courtemanche, M. A. Legare, L. Maron and F. G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326 CrossRef CAS PubMed; (h) T. Wang and D. W. Stephan, Chem.–Eur. J., 2014, 20, 3036 CrossRef CAS PubMed; (i) T. Wang and D. W. Stephan, Chem. Commun., 2014, 50, 7007 RSC; (j) R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513 CrossRef CAS; (k) M. A. Courtemanche, A. P. Pulis, É. Rochette, M. A. Légaré, D. W. Stephan and F. G. Fontaine, Chem. Commun., 2015, 51, 9797 RSC.
- For reviews, see: (a) H. C. Brown and P. V. Ramachandran, Pure Appl. Chem., 1991, 63, 307 CrossRef CAS; (b) H. C. Brown and P. V. Ramachandran, J. Organomet. Chem., 1995, 500, 1 CrossRef CAS; (c) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957 CrossRef CAS; (d) R. Barbeyron, E. Benedetti, J. Cossy, J.-J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431 CrossRef CAS.
- D. L. Bryce, R. E. Wasylishen and M. Gee, J. Phys. Chem. A, 2001, 105, 3633 CrossRef CAS.
- For reviews, see: (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed. For leading examples, see: (c) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed; (d) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS PubMed; (e) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- (a) A. Feldmann, A. Iida, R. Fröhlich, S. Yamaguchi, G. Kehr and G. Erker, Organometallics, 2012, 31, 2445 CrossRef CAS; (b) M. M. Hansmann, R. L. Melen, M. Rudolph, F. Rominger, H. Wadepohl, D. W. Stephan and A. S. K. Hashmi, J. Am. Chem. Soc., 2015, 137, 15469 CrossRef CAS PubMed; (c) S. Tamke, Z.-W. Qu, N. A. Sitte, U. Flörke, S. Grimme and J. Paradies, Angew. Chem., Int. Ed., 2016, 55, 4336 CrossRef CAS PubMed; (d) See for a comparison: R. A. Adler Yanez, G. Kehr, C. G. Daniliuc, B. Schirmer and G. Erker, Dalton Trans., 2014, 43, 10794 RSC.
- F. Türkyilmaz, G. Kehr, J. Li, C. G. Daniliuc, M. Tesch, A. Studer and G. Erker, Angew. Chem., Int. Ed., 2016, 55, 1470 CrossRef PubMed.
- The stereochemical nomenclature is used according to IUPAC, see J. Org. Chem., 1970, 35, 2849 Search PubMed.
- (a) P. Moquist, G.-Q. Chen, C. Mück-Lichtenfeld, K. Bussmann, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Sci., 2015, 6, 816 RSC; (b) S. Kohrt, S. Dachwitz, C. G. Daniliuc, G. Kehr and G. Erker, Dalton Trans., 2015, 44, 21032 RSC; (c) J. Yu, G. Kehr, C. G. Daniliuc and G. Erker, Chem. Commun., 2016, 52, 1393 RSC.
- For reviews, see: (a) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; (c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and analytical details are given. CCDC 1487577–1487584. For ESI and crystallographic data in CIF or other electronic formats see DOI: 10.1039/c6sc03468c |
| This journal is © The Royal Society of Chemistry 2017 |