
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon dioxide binding at a Ni/Fe center: synthesis and characterization of Ni(η1-CO2-κC) and Ni-μ-CO2-κC:κ2O,O′-Fe†
Changho
Yoo
and
Yunho
Lee
*
Department of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: yunholee@kaist.ac.kr; Fax: +82 42 350 2810; Tel: +82 42 350 2814
First published on 30th August 2016
Abstract
The degree of CO2 activation can be tuned by incorporating a distinct electronic coordination environment at the nickel center. A mononuclear nickel carboxylate species (Ni–CO2, 3) and a dinuclear nickel–iron carboxylate species (Ni–CO2–Fe, 5) were prepared. The structure of 3 reveals a rare η1-κC binding mode of CO2, while that of 5 shows bridging CO2 binding (μ2-κC:κ2O,O′) between the nickel and iron, presented as the first example of a nickel-μ-CO2-iron species. The structural analyses of 3 and 5 based on XRD and DFT data reveal a higher degree of CO2 activation in 5, imparted by the additional interaction with an iron ion.
Introduction
Activation of carbon dioxide is currently receiving much attention due to its relevance to environmental and energy related issues.1 In the area of transition metal catalyzed reactions, one of the main challenges is selective reduction of CO2 to a product such as formate, carbon monoxide, methanol or methane.2 In a 2-electron process, the binding mode of the CO2 may determine the eventual product formation, e.g. formate vs. carbon monoxide.3 When the initial metal–oxygen interaction occurs to form a metal CO2 adduct M-η1-CO2-κO, subsequent hydride transfer via CO2 addition to a M–H bond generates a metal-formate species. Alternatively, the metal–carbon bond formation can produce a metallacarboxylate species (M-η1-CO2-κC), followed by C–O bond cleavage to generate CO. In the latter case, an additional Lewis acid interaction can stabilize the negative charges at the oxygen atoms of the bound CO2.4,5 Therefore, CO2 activation with a bimetallic system can be one way to guide the selectivity of the CO2 catalyst and is receiving much attention.5,6 In fact, an excellent example of a bimetallic center utilized in an efficient catalytic conversion of CO2 can be found in the active site of carbon monoxide dehydrogenase (CODH).7 According to recent studies, CO2 coordination at a heterobimetallic nickel–iron active site can be found in CODH’s intermediate species, which possesses a Ni-μ-CO2-Fe moiety, Scheme 1.8 Although X-ray analysis provides a structural snapshot of the CO2 reduction sequence, the role of the unique iron ion is currently not well-understood.7 Thus, acquiring an understanding of iron assisted CO2-nickel coordination is of fundamental interest and is crucial for gaining mechanistic insight into this and other enzymatic reactions. | ||
| Scheme 1 The active site of carbon monoxide dehydrogenase (CODH, left), and 4- and 5-coordinate nickel CO2 adducts (right). | ||
In organonickel chemistry, there are few mononuclear Ni-η2-CO2 adducts possessing both M–C and M–O bonds.9 In 1975, Aresta and co-workers reported the first structurally characterized nickel–CO2 adduct (PCy3)2Ni(η2-CO2), Scheme 1.9a An analogous complex, (dtbpe)Ni(η2-CO2) (dtbpe = 1,2-bis(di-tert-butylphosphino)ethane) was recently reported by Hillhouse and co-workers.9d More recently, our group reported a similar but unique five-coordinate nickel–CO2 adduct (PPMeP)Ni(η2-CO2) (PPMeP = PMe(2-PiPr2-C6H4)2).9f According to its structural analysis, the five coordinate nickel CO2 species supported by three neutral P donors has a weak Ni–O bond available for electrophilic attack.9f Additionally, by utilizing an anionic tridentate PNP ligand (PNP− = N[2-PiPr2-4-Me-C6H3]2−), our group reported the nickel hydroxycarbonyl species (PNP)NiCOOH (1), (PNP)NiCOONa (2) and {(PNP)Ni}2-μ-CO2-κ2C,O (4), the first examples of Ni–CO2 complexes that reveal a Ni–CO2-κC binding mode, Scheme 2.10 The carboxylate group in these species is stabilized by a Lewis acid such as a proton, sodium or another nickel ion. Our interest then moved to comparing (PPMeP)Ni–CO2 and (PNP)Ni–CO2 to evaluate their fundamental differences in CO2 activation. The different geometries favored with a PPMeP or PNP ligand affect the identity of the nickel–CO2 moiety, which can be Ni(II)–CO22− or Ni(0)–CO2 or an open-shell Ni(I)–CO2˙−, vide infra. Furthermore, by isolating the native nickel–CO2 species, we can further study the effect of the second iron ion. Although several nickel carboxylate species are already known, iron has never been introduced synthetically into a Ni–CO2 moiety.
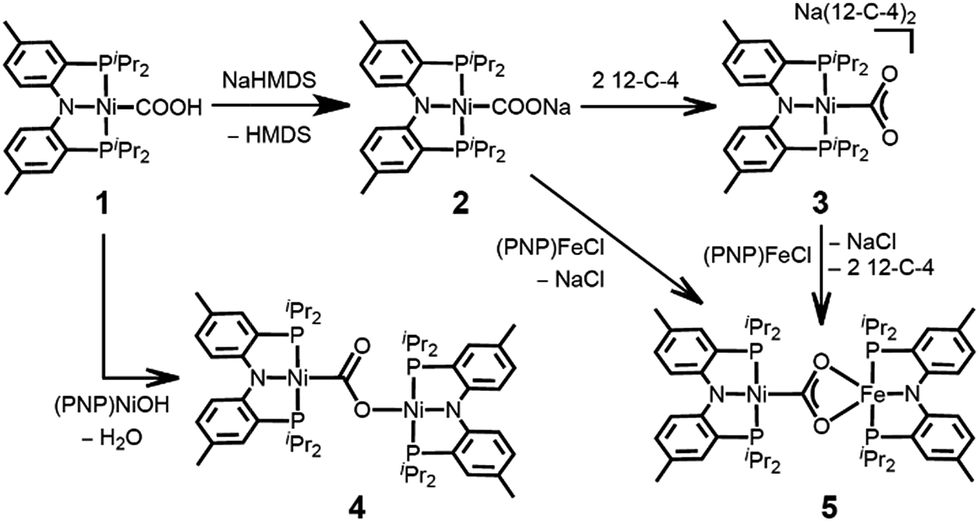 | ||
| Scheme 2 Preparation of mononuclear- and dinuclear-CO2 adducts. | ||
Here, we present a nickel carboxylate species {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3), in which the nickel–CO2 moiety does not have any Lewis acid interactions. We also prepared a dinuclear nickel–iron carboxylate species (PNP)Ni-μ-CO2-κC:κ2O,O′-Fe(PNP) (5), reminiscent of the NiFe-binuclear active site of CODH. This is an unprecedented example of a nickel–iron hetero-bimetallic complex possessing a bridging CO2 ligand. The levels of CO2 activation in compounds 3 and 5 are compared with other Ni–CO2 adducts and the Ni-μ-CO2-Fe moiety found in CODH.
Results and discussion
Synthesis and characterization of the Ni-η1-CO2-κC complex
The coordination of a hydroxycarbonyl moiety via a Ni–C bond was previously realized at a divalent nickel center supported by a PNP ligand.10 Following deprotonation of (PNP)NiCOOH (1), its anionic congener (PNP)NiCOONa (2) was also prepared and recently reported by our group.10 The X-ray structure reveals that two molecules of 2 are oriented to form a pair with ionic interactions with two sodium ions in the crystal lattice, Fig. 1.11 The corresponding CO2 ligand coordinates to the nickel center in a μ3-κ1C:κ2O,O′:κ1O′ mode with a Ni–C1 bond distance of 1.882(1) Å. There are additional bonds of the CO2 moiety to sodium ions with Na–O bond distances of 2.352(1), 2.217(1) and 2.459(1).11 To obtain a sodium-free adduct, 2 equiv. of 12-crown-4 was added to a solution of 2, resulting in the formation of {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3). The crystal structure of 3 revealed the successful generation of a mononuclear nickel adduct possessing an η1-κC coordinated carbon dioxide species with a Ni–C bond distance of 1.911(2) Å, as shown in Fig. 1 and Table 1. The oxidation state of the nickel ion in 3 can be assigned as 2+ based on its similar structural features to previously known nickel(II) species such as (PNP)NiCOOH (1) and {(PNP)Ni}2-μ-CO2-κ2C,O (4), vide infra. The geometry of 3 is square planar (τ4 = 0.1212) with a similar but slightly elongated Ni–C bond distance in comparison to those of 1 and 4 (dNi–C = 1.866(2) and 1.888(2) Å, respectively, Table 1). This is probably due to a lower degree of π back-bonding between the nickel and CO2. In fact, π back-donation from the nickel center to a CO2 ligand in such nickel carboxylate species is indicated by shorter Ni–C distances (1.858–1.911 Å) than those of the nickel alkyl species (PNP)NiR (R = Me, Et, nPr) (1.963–2.004 Å).13 The molecular orbitals generated from DFT calculations also show the presence of π back-donation from the Ni dxz to the CO2 π* orbital (see ESI†). Due to the absence of Lewis acid interactions in 3, a lower π-accepting ability of the CO2 ligand is expected. Its structural data also revealed that the plane of the CO2 ligand is perpendicular to that of the square planar (PNP)Ni moiety. One of the oxygen atoms (dNi1–O2 = 2.614(1) Å) is slightly closer to the nickel center than the other (dNi1–O1 = 2.776(1) Å), Table 2. These Ni–O distances are much longer than those for other known Ni-η2-CO2 adducts (1.9–2.2 Å, Table 2), suggesting that neither of the oxygen atoms are bound.9 The DFT analysis also supports minimal interaction between the nickel and oxygen atoms (Wiberg index = 0.1358 for Ni1–O2 and 0.1679 for Ni1–O1, see Table 2). The two C–O bond distances are nearly identical (dC1–O1 = 1.247(2) Å, dC1–O2 = 1.248(2) Å, Table 1) and slightly shorter than in the analogous carboxylate complexes 2 and 4 (Table 1), due to the absence of a Lewis acid, Na or Ni. According to the DFT analysis, the HOMO of 3 possesses contributions from both a nickel dx2−y2 orbital and a CO2 π* orbital, see Fig. 2. Due to additional electron density from CO22− being shifted to the nickel, the CO2 moiety is slightly oxidized compared to the sp2 hybridized carboxylate ligands found in 2 and 4. The larger O–C–O angle (128.4(2)°) of 3 compared to others (124.0(1) and 123.7(2)°) also supports this electronic feature, vide infra. Although an η1-κC CO2 coordination mode has been proposed for many CO2 reduction strategies,2,3 the only example of a crystallographically identified metal η1-κC CO2 complex is a rhodium CO2 adduct, Rh(CO2)(Cl)(diars)2 (diars = o-phenylene-bis(dimethylarsine)), reported by the Herskovitz group.14 According to their C–O bond distances (1.20(2) and 1.25(2) Å) and O–C–O angle (126(2)°), the CO2 moiety in 3 shares similar structural and electronic features. Thus, compound 3 is a unique example possessing η1-κC CO2 binding, since such a binding mode is unknown for 1st row transition metals and is rare in structurally characterized metal–CO2 adducts. | ||
| Fig. 1 Displacement ellipsoid (50%) representations for (a) (PNP)NiCOONa (2) in a dimeric assembly with co-crystallized THF molecules,11 and (b) {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3). A co-crystallized 12-crown-4 molecule and hydrogen atoms are omitted for clarity. | ||
| 1 10 | 2 11 | 3 | 4 10 | 5 | CODH8b | |
|---|---|---|---|---|---|---|
| a M = Na. b M = Ni. c M = Fe. | ||||||
| d Ni–C (Å) | 1.866(2) | 1.882(1) | 1.911(2) | 1.888(2) | 1.858(1) | 1.805(31) |
| d M–O (Å) | — | 2.352(1)a | — | 1.897(2)b | 2.204(1)c | 2.030(18)c |
| 2.217(1)a | 2.066(1)c | |||||
| 2.459(1)a | ||||||
| d C–O (Å) | 1.269(3) | 1.260(1) | 1.247(2) | 1.240(3) | 1.269(2) | 1.298(30) |
| 1.313(3) | 1.271(1) | 1.248(2) | 1.296(3) | 1.289(2) | 1.316(30) | |
| ΔdC–O (Å) | 0.044 | 0.011 | 0.001 | 0.056 | 0.020 | 0.018 |
| ∠O–C–O (°) | 119.6(2) | 124.0(1) | 128.4(2) | 123.7(2) | 116.5(1) | 117.2(26) |
| Ni(PCy3)2(η2-CO2)9a | (dtbpe)Ni(η2-CO2)9d | (PPMeP)Ni(η2-CO2)9f | 3 | 5 | |
|---|---|---|---|---|---|
| a Wiberg bond indices were calculated using single-point calculations, for which geometries were obtained from the XRD data. | |||||
| Structural parameters | |||||
| d Ni–C (Å) | 1.84(2) | 1.868(2) | 1.904(1) | 1.911(2) | 1.858(1) |
| d Ni–O (Å) | 1.99(2) | 1.904(2) | 2.191(1) | 2.614(1) | 2.718(1) |
| 2.776(1) | 2.792(1) | ||||
| d C–O (Å) | 1.17(2) | 1.200(3) | 1.218(2) | 1.248(2) | 1.269(2) |
| 1.22(2) | 1.266(3) | 1.252(2) | 1.247(2) | 1.289(2) | |
| ∠O–C–O (°) | 133 | 138.0(2) | 135.1(1) | 128.4(2) | 116.5(1) |
| ν CO2 (cm−1) | 1740 | 1724 | 1682 | 1620 | 1510 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Wiberg bond indices | |||||
| Ni–C | — | 0.5766 | 0.5286 | 0.6143 | 0.6277 |
| Ni–O | — | 0.4300 | 0.3117 | 0.1679 | 0.0798 |
| 0.1358 | 0.0644 | ||||
| C–O | — | 1.6927 | 1.6384 | 1.5112 | 1.3993 |
| 1.4080 | 1.4701 | 1.4949 | 1.2933 | ||
 | ||
| Fig. 2 Combination of the Ni dx2−y2 and CO2 π* orbitals provides the DFT calculated HOMO of {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3). | ||
Synthesis and characterization of the heterobimetallic nickel–iron CO2 complex
To gain a better understanding of the role of the second metal ion in the CODH active site, we prepared a heterobimetallic nickel–iron carboxylate species possessing a Ni–CO2–Fe fragment by addition of {(PNP)Fe}+ to a Ni-η1-CO2-κC species. To a yellow solution of {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3) in toluene, a purple solution of (PNP)FeCl was added. The immediate formation of a new orange species (PNP)Ni-μ-CO2-κC:κ2O,O′-Fe(PNP) (5) was confirmed, using the 1H NMR spectrum, from the absence of peaks for 3 and (PNP)FeCl and the presence of new paramagnetically shifted signals. The same product was also prepared by substitution of the sodium ion of (PNP)NiCOONa (2) with (PNP)FeCl. The solid-state structure of 5 clearly revealed a dinuclear nickel–iron complex with a bridging CO2 ligand in the μ2-κC:κ2O,O′ mode (Fig. 3). The Ni and Fe ions are separated by a distance of 4.3690(3) Å. The two C–O bond distances are 1.269(2) and 1.289(2) Å, revealing that a significant elongation has occurred due to the iron interaction compared to 3 (Table 1). The bond distances between the iron and both oxygen atoms are 2.204(1) and 2.066(1) Å. The nickel center possesses a square planar geometry (τ4 = 0.1012). The geometry around the iron is distorted square pyramidal (τ = 0.13,15Fig. 3). The O1–C1–O2 angle (116.5°) reflects the sp2 hybridization of the carboxylate ligand in 5. In fact, recent crystallographic data of CODH at atomic resolution (dmin = 1.03 Å) revealed that the bound CO2 molecule (∠O–C–O = 117.2(26)°) is a carboxylate anion (CO22−).8b,8c Regarding the similarity between these angles, the carboxylate moiety in 5 might be close to CO22−. The asymmetric vibration for CO2 observed at 1510 cm−1, which is similar to that observed for the dinickel carboxylate species (4) at 1518 cm−1, also indicates a reduced state of CO2. The effective magnetic moment of 5 was determined using the Evans' method (μeff = 4.95 μB in C6D6), which indicated an S = 2 spin state.16 According to DFT calculations, most of the spin density is located on the iron center (see ESI†). For CODH, the unique iron, Feu, was assigned as a high spin iron(II) (ferrous component II, FCII) using Mössbauer spectroscopy,17 and a low-spin nickel(II) was demonstrated using X-ray absorption spectroscopy (XAS).18 The current structural and spectroscopic analyses suggest that 5 might share a similar electronic structure to that found in CODH. Gibson classified the μ2-κC:κ2O,O′ binding modes of CO2 into two types according to the difference between the two C–O distances.19 Due to the two similar C–O distances of the CO2 moiety, compound 5 (ΔdC–O = 0.020 Å) can be assigned as a class I complex.20 In the dinickel CO2 species (4), the CO2 molecule is coordinated in a μ2-κC:κO mode with the absence of a Ni2–O2 interaction (dNi2–O2 = 3.14(7)) and two different C–O bond distances (ΔdC–O = 0.056 Å). In CODH, CO2 is coordinated in a μ2-κC:κO fashion between the nickel and iron ions, but the two C–O bond distances are quite comparable (ΔdC–O = 0.018 Å), akin to the μ2-κC:κ2O,O′ mode. This might be due to hydrogen bonding with the protein matrix, since both the CO2 oxygens are hydrogen bonded to His93 and Lys563, respectively.8 | ||
| Fig. 3 Displacement ellipsoid (50%) representation for (PNP)Ni-μ-CO2-κC:κ2O,O′-Fe(PNP) (5). Hydrogen atoms are omitted for clarity. | ||
Compound 5 is the first example of a dinuclear nickel–iron–CO2 complex. While dinuclear CO2 complexes mostly employ 2nd and 3rd row transition metals,19 several bimetallic iron carboxylates (Fe–CO2–M, M = Ti, Zr, Sn, Re) have been reported.5a,5c–e,21 However, such complexes typically possess an Fe–C bond rather than an Fe–O bond with CO2. There have been numerous examples of nickel–iron bimetallic complexes reported for synthetic model studies of NiFe hydrogenase,22 but a bimetallic complex possessing a Ni-μ-CO2-Fe moiety closely related to CODH chemistry is not known. The Holm group constructed a series of [NiFe3S4] cubanes as structural model complexes for the [NiFe4S4] core in CODH.23 However, the installation of an iron species corresponding to the Feu in CODH and reactions involving CO and CO2 have not yet been investigated. More recently, the Holm group also reported a bimetallic complex containing nickel and iron supported by a binucleating macrocycle.24 With respect to CODH chemistry, bridging hydoroxo, cyanido and formato species have been generated, however, a Ni-μ-CO2-Fe fragment had not yet been isolated.
Activation of CO2 in 3 and 5
Previously known 4-coordinate Ni–CO2 complexes possessing an η2-CO2 binding mode have a formally zero-valent nickel center, Scheme 1.9 This suggests limited CO2 activation in such species. Interestingly, the 5-coordinate nickel CO2 adduct (PPMeP)Ni(CO2) also has a similar level of activation of CO2 based on the C–O bond distances and the O–C–O angle, Table 2. Regarding CO2 binding and activation, {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3) is a unique example. It is striking that a neutral pincer-type ligand PPMeP (PPMeP = PMe[2-PiPr2-C6H4]2) favors 5-coordinate η2-CO2 coordination at a single nickel center, while 3 remains as a 4-coordinate species with η1-CO2 coordination.9f Although the total number of Ni d-electrons and CO2 π*-electrons in both 3 and (PPMeP)Ni(CO2) is the same, (PPMeP)Ni(CO2) can be considered as formally Ni(0)–(CO2) while 3 can be better described as a Ni(II)–(CO22−) species. The asymmetric CO2 stretching frequency for 3 is significantly shifted to a lower vibration, 1620 cm−1, compared to those of the Ni-η2-CO2 complexes (Table 2), which is evidence of a reduced CO2 moiety in 3.9 This may be due to the influence of the trans atom: an anionic amide nitrogen vs. a neutral phosphorus atom. The anionic nitrogen in 3 electrostatically favors a divalent nickel center, while the neutral π-acidic P atom in the PPMeP ligand favors a Ni(0) center. In fact, the PNP ligand typically stabilizes a square planar geometry while the PPMeP ligand favors a pseudo-tetrahedral geometry. Thus, 3 prefers to accommodate a divalent nickel center while (PPMeP)Ni(CO2) prefers Ni(0). However, the reduction state of the CO2 moiety in 3 is a little ambiguous according to the O–C–O angle. The O–C–O angle in 3 of 128.4(2)° is larger than those of an ideal sp2 hybridized carbon (120°) and the other nickel(II) carboxylate species 1, 2 and 4 (119.6(2)°, 124.0(1)° and 123.7(2)°, respectively, Table 1). The O–C–O angle of a CO2 radical anion (CO2˙−) is suggested to be 133°,25 which is fairly similar to those of the previously reported Ni-η2-CO2 complexes, Table 2. Thus, the geometry of the CO2 moiety in 3 may be thought of as being between a CO2 radical anion and a carboxylate.Upon addition of iron to compound 3, the CO2 is further reduced to carboxylate (CO22−). The C–O bond distances and O–C–O angle in 5 clearly show a 2-electron reduced state of the CO2 moiety, Table 2. This was also indicated by the asymmetric CO2 vibration observed at 1510 cm−1, which is significantly lower than those of other CO2 species and 3. The Wiberg bond indices nicely agree with the bond distances, Table 2. These analyses of a series of nickel–CO2 compounds demonstrate how the degree of CO2 activation can be tuned by incorporating a distinct electronic coordination environment at the metal center, and may have parallels to the efficient CO2 conversion found in CODH.
In order to study further activation of the bound CO2via C–O bond cleavage, protonation of 3 and 5 was attempted. Our group previously reported that reversible C–O bond cleavage/formation occurs with a nickel hydroxycarbonyl species (1).10 From reaction of 3 with 1 equiv. of HBF4·Et2O, a nickel hydroxycarbonyl species (1) was produced and isolated with a 74% yield. A similar reaction of 3 with 2 equiv. of HBF4·Et2O resulted in the formation of a carbonyl species {(PNP)NiCO}{BF4} in 75% yield, revealing that two sequential protonations can occur with compound 3 possessing a Ni-η1-CO2-κC moiety, which are key steps in the transformation of CO2 to CO. Although protonation of compound 5 seems to produce 1 and {(PNP)NiCO}{BF4}, unfortunately, their yields were not clear due to thermal decomposition of 5 and the generation of multiple products. Demetallation of the iron seems to be one of the decomposition processes.
Conclusions
In conclusion, the generation of unprecedented nickel–carbon dioxide adducts possessing a Ni–C bond accommodated by a (PNP)Ni scaffold was accomplished. A mononuclear CO2 adduct {Na(12-C-4)2}{(PNP)Ni-η1-CO2-κC} (3) and a dinuclear nickel–iron carboxylate species (PNP)Ni-μ-CO2-κC:κ2O,O′-Fe(PNP) (5) were synthesized successfully. While the solid state structure of 3 revealed a rare η1-κC binding mode, compound 5 was structurally characterized to reveal a unique class I type μ2-κC:κ2O,O′ binding mode. This heterobimetallic CO2 adduct is the first example of a nickel–iron carboxylate species, of which the structural and electronic features are reminiscent of those of the Ni-μ-CO2-Fe fragment found in the C-cluster of CODH. Comparison of the η1-CO2-κC species 3 and dinuclear Ni-μ-CO2-Fe species 5 with previously reported Ni–CO2 adducts suggested that the CO2 ligand can be stabilized and activated by interaction with the second metal. Protonation of 3 produces a nickel carbonyl species {(PNP)NiCO}{BF4} via C–O bond cleavage, while the reactivity of 5 is limited. Further studies on incorporating a stable iron species and the subsequent reactivity toward protonation are currently underway.Acknowledgements
This work was supported by the C1 Gas Refinery Program (NRF-2015M3D3A1A01064880) through the National Research Foundation of Korea (NRF-2015R1A2A2A01004197), and KAIST and the Aramco Overseas Company. This work was also supported by the Supercomputer Center/Korea Institute of Science and Technology (KSC-2015-S1-0005).Notes and references
- (a) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (b) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (c) M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC; (b) C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392 RSC; (c) E. Fujita, Coord. Chem. Rev., 1999, 185–186, 373 CrossRef CAS.
- (a) J. Song, E. L. Klein, F. Neese and S. Ye, Inorg. Chem., 2014, 53, 7500 CrossRef CAS PubMed; (b) M. R. Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974 CrossRef PubMed; (c) E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC.
- (a) M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917 CrossRef CAS PubMed; (b) S. J. K. Forrest, J. Clifton, N. Fey, P. G. Pringle, H. A. Sparkes and D. F. Wass, Angew. Chem., Int. Ed., 2015, 54, 2223 CrossRef CAS PubMed.
- (a) M. Hirano, M. Akita, K. Tani, K. Kumagai, N. C. Kasuga, A. Fukuoka and S. Komiya, Organometallics, 1997, 16, 4206 CrossRef CAS; (b) K. E. Litz, K. Henderson, R. W. Gourley and M. M. B. Holl, Organometallics, 1995, 14, 5008 CrossRef CAS; (c) J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21 CrossRef CAS; (d) J. R. Pinkes and A. R. Cutler, Inorg. Chem., 1994, 33, 759 CrossRef CAS; (e) J. C. Vites, B. D. Steffey, M. E. Giuseppetti-Dery and A. R. Cutler, Organometallics, 1991, 10, 2827 CrossRef CAS; (f) E. G. Lundquist, J. C. Huffman, K. Folting, B. E. Mann and K. G. Caulton, Inorg. Chem., 1990, 29, 128 CrossRef CAS; (g) E. G. Lundquist, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1986, 108, 8309 CrossRef CAS; (h) S. Gambarotta, F. Arena, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1982, 104, 5082 CrossRef CAS; (i) G. Fachinetti, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1978, 100, 7405 CrossRef CAS.
- (a) C. W. Machan and C. P. Kubiak, Dalton Trans., 2016 10.1039/C6DT01956K , in press; (b) S. Bagherzadeh and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 10898 CrossRef CAS PubMed; (c) O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716 CrossRef CAS PubMed; (d) J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582 CrossRef CAS PubMed; (e) B. D. Steffey, C. J. Curtis and D. L. DuBois, Organometallics, 1995, 14, 4937 CrossRef CAS.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149 CrossRef CAS PubMed.
- (a) J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461 CrossRef CAS PubMed; (b) J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560 CrossRef CAS PubMed; (c) M. W. Ribbe, Angew. Chem., Int. Ed., 2015, 54, 8337 CrossRef CAS PubMed.
- (a) M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636 RSC; (b) M. Aresta and C. F. Nobile, J. Chem. Soc., Dalton Trans., 1977, 708 RSC; (c) A. Döhring, P. W. Jolly, C. Krüger and M. J. Romão, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 484 Search PubMed; (d) J. S. Anderson, V. M. Iluc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203 CrossRef CAS PubMed; (e) R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Dalton Trans., 2013, 42, 1461 RSC; (f) Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458 RSC.
- C. Yoo, J. Kim and Y. Lee, Organometallics, 2013, 32, 7195 CrossRef CAS.
- C. Yoo and Y. Lee, Inorg. Chem. Front., 2016, 3, 849 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- C. Yoo, S. Oh, J. Kim and Y. Lee, Chem. Sci., 2014, 5, 3853 RSC.
- J. C. Calabrese, T. Herskovitz and J. B. Kinney, J. Am. Chem. Soc., 1983, 105, 5914 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003 RSC; (b) S. K. Sur, J. Magn. Reson., 1989, 82, 169 CAS.
- Z. Hu, N. J. Spangler, M. E. Anderson, J. Xia, P. W. Ludden, P. A. Lindahl and E. Münck, J. Am. Chem. Soc., 1996, 118, 830 CrossRef CAS.
- W. Gu, J. Seravalli, S. W. Ragsdale and S. P. Cramer, Biochemistry, 2004, 43, 9029 CrossRef CAS PubMed.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS PubMed.
- Classification of the μ2-κC:κ2O,O′ binding modes of CO2 for a bimetallic center (M and M′): while the CO2 coordinates via a M–C bond, class I complexes have the two oxygen atoms of the CO2 ligand symmetrically bonded to M′ and class II complexes possess two asymmetric M′–O bonds.
- (a) D. H. Gibson, J. F. Richardson and T.-S. Ong, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 259 CrossRef; (b) D. H. Gibson, M. Ye and J. F. Richardson, J. Am. Chem. Soc., 1992, 114, 9716 CrossRef CAS; (c) D. H. Gibson, J. F. Richardson and O. P. Mbadike, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 784 CrossRef; (d) D. H. Gibson, M. Ye, J. F. Richardson and M. S. Mashuta, Organometallics, 1994, 13, 4559 CrossRef CAS; (e) D. H. Gibson, M. Ye, B. A. Sleadd, J. M. Mehta, O. P. Mbadike, J. F. Richardson and M. S. Mashuta, Organometallics, 1995, 14, 1242 CrossRef CAS; (f) M. Lutz, M. Haukka, T. A. Pakkanen and L. H. Gade, Organometallics, 2001, 20, 2631 CrossRef CAS.
- (a) S. Kaur-Ghumaan and M. Stein, Dalton Trans., 2014, 43, 9392 RSC; (b) A. C. Marr, D. J. E. Spencer and M. Schröder, Coord. Chem. Rev., 2001, 219–221, 1055 CrossRef CAS.
- (a) S. Ciurli, P. K. Ross, M. J. Scott, S.-B. Yu and R. H. Holm, J. Am. Chem. Soc., 1992, 114, 5415 CrossRef CAS; (b) R. Panda, C. P. Berlinguette, Y. Zhang and R. H. Holm, J. Am. Chem. Soc., 2005, 127, 11092 CrossRef CAS PubMed; (c) J. Sun, C. Tessier and R. H. Holm, Inorg. Chem., 2007, 46, 2691 CrossRef CAS PubMed.
- D. Huang and R. H. Holm, J. Am. Chem. Soc., 2010, 132, 4693 CrossRef CAS PubMed.
- (a) D. W. Ovenall and D. H. Whiffen, Mol. Phys., 1961, 4, 135 CrossRef CAS; (b) J. W. Rabalais, J. M. McDonald, V. Scherr and S. P. McGlynn, Chem. Rev., 1971, 71, 73 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for 3 and 5. CCDC 1492006 and 1492007. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03450k |
| This journal is © The Royal Society of Chemistry 2017 |