
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of the physicochemical properties of [Pt(dien)(nucleobase)]2+ for HIVNCp7 targeting†
S. D.
Tsotsoros
a,
P. B.
Lutz
bc,
A. G.
Daniel
a,
E. J.
Peterson
ad,
R. E. F.
de Paiva
 a,
E.
Rivera
a,
Y.
Qu
a,
C. A.
Bayse
*c and
N. P.
Farrell
*ad
a,
E.
Rivera
a,
Y.
Qu
a,
C. A.
Bayse
*c and
N. P.
Farrell
*ad
aDepartment of Chemistry, Virginia Commonwealth University, 1001 W. Main Street, Richmond, VA 23284-2006, USA. E-mail: npfarrell@vcu.edu
bDepartment of Science, Technology and Mathematics, Regent University, Virginia Beach, Virginia 23464, USA
cDepartment of Chemistry and Biochemistry, Old Dominion University, Norfolk, VA 23529, USA. E-mail: CBayse@odu.edu
dMassey Cancer Center, Virginia Commonwealth University, Richmond, VA, USA
First published on 6th October 2016
Abstract
Physicochemical properties of coordination compounds can be exploited for molecular recognition of biomolecules. The inherent π–π stacking properties of [Pt(chelate)(N-donor)]2+ ([PtN4]) complexes were modulated by systematic variation of the chelate (diethylenetriamine and substituted derivatives) and N-donor (nucleobase or nucleoside) in the formally substitution-inert PtN4 coordination sphere. Approaches to target the HIV nucleocapsid protein HIVNCp7 are summarized building on (i) assessment of stacking interactions with simple tryptophan or tryptophan derivatives to (ii) the tryptophan-containing C-terminal zinc finger and (iii) to the full two-zinc finger peptide and its interactions with RNA and DNA. The xanthosine nucleoside was identified as having significantly enhanced stacking capability over guanosine. Correlation of the LUMO energies of the modified nucleobases with the DFT π-stacking energies shows that frontier orbital energies of the individual monomers can be used as a first estimate of the π-stacking strength to Trp. Cellular accumulation studies showed no significant correlation with lipophilicity of the compounds, but all compounds had very low cytotoxicity suggesting the potential for antiviral selectivity. The conceptual similarities between nucleobase alkylation and platination validates the design of formally substitution-inert coordination complexes as weak Lewis acid electrophiles for selective peptide targeting.
Introduction
Design of defined coordination compounds in medicine uses the inherent physical and chemical properties of the coordination compound, or metal ion, to achieve specific effects. Properties such as paramagnetism and/or radioactive emission of Gd and Tc, respectively, coupled with appropriate chemical structure produce useful imaging agents whose biological properties can be further modified by suitable chemical design.1 Development of platinum-based anticancer agents has been predicated on the necessity for Pt–DNA bond formation where the conformational distortion subsequently produced by binding of the square planar coordination sphere eventually disrupts nucleic acid function. In the latter case, challenges to the orthodoxy of the necessity for Pt–DNA bond formation has come from the recent demonstrations that formally substitution-inert polynuclear platinum complexes can display significant in vitro and in vivo anti-tumor activity equivalent to cisplatin itself.2 Metallohelicates are a further example of use of “non-covalent” recognition of discrete DNA sequences with consequences for protein recognition.3 Use of formally substitution-inert compounds is attractive as it may allow greater control of the biologically relevant reactions as well as improving pharmacokinetics through elimination of wasteful non-specific biomolecule covalent bond formation.A further example of formally substitution-inert compounds for biological applications is in the use of PtN4 nucleobase compounds to act as Lewis acid electrophiles targeting zinc fingers (ZF), and especially the HIV NCp7 nucleocapsid protein (NCp7).4,5 NCp7 is a small basic zinc finger protein containing two Cys2HisCys zinc coordination motifs and is involved in nearly all stages of the viral life cycle.6,7 NCp7 is of considerable interest as a drug target because it is highly conserved among retroviruses and is intolerant to mutation.6,8 A critical feature of NCp7–DNA/RNA recognition is the stacking of aromatic residues (Trp, Phe) with purine and pyrimidine bases (guanine, cytosine) of the oligonucleotide.9–11 Nucleobase metallation, analogous to protonation or alkylation, enhances their π–π stacking to aromatic amino acids.12,13 Metallation of 9-EtGua in [M(dien)(9-EtGua)]n+ (M = Pt, Pd n = 2; M = Au, n = 3) produces a 2–5-fold increase in the association constant with N-acetyltryptophan (N-AcTrp) compared to the free nucleobase.12–15 The general enhancement also extends to pyrimidines such as 1-mecytosine and even heterocyclic aromatic ligands based on pyridine and thiazole.12,14,16 The application in this manner of an intrinsic biophysical property of a complex such as [Pt(dien)(9-EtGua)]2+ – the π–π stacking of the platinated nucleobase with a tryptophan moiety – is therefore a recognition motif for binding to an appropriate peptide such as NCp7 capable of modulation to inhibit the “intrinsic” nucleic acid interaction. Measured association constants to the C-terminal NCp7 for [Pt(dien)(9-EtGua)]2+ and [Pt(dien)(5′-GMP)] (7.5 and 12.4 × 103 M−1 respectively) compared to that observed for a representative hexanucleotide d(TACGCC) where Ka = 60.03 × 103 M−1.4,17
A second advantage of formally substitution-inert complexes is to infer selectivity toward eventual Pt-bond forming reactions upon target recognition. An inherent problem for any application of zinc finger targeting is the variety of relevant structural motifs and functions.18 Analysis of the protein packing and electrostatic screening in a wide range of zinc fingers led to the attractive postulate that weak organic electrophiles were capable/likely candidates for selective attack on the highly nucleophilic zinc-cysteinates of NCp7.19,20 The concept has been applied for targeting both the HIVNCp7 and also the DNA binding domain of the estrogen receptor (ERDBD).20,21 For coordination compounds the PtN4 chemotype (in this case {PtN3(nucleobase)}) is the Lewis acid equivalent of a weak “organic” electrophile, significantly less reactive than the corresponding MClN3 unit with a substitution-labile M–Cl bond. In agreement, substitution of the nucleobase in [Pt(dien)(9-EtGua)]2+ by the sulfur nucleophile N-AcCys is significantly slower than for the [PtCl(dien)]+ species.22 Incorporation of a Trp into a short 4-amino acid sequence (GAWG versus GAMC) increases the rate of reaction, and perhaps specificity, with [Pt(dien)(9-EtGua)]2+.22 Theoretical calculations on the [Pt(dien)(9-EtGua)]2+–GWMG/GAMG interactions show that the formation of the GWMG species is roughly 5 kcal mol−1 more stable than for the GAMG species (−9.3 and −3.9 kcal mol−1), due to the additional stacking interaction.
The [Pt(dien)(nucleobase)]n+ coordination sphere is a useful template for systematic studies as it allows for modification on both the dien ligand and nucleobase to enhance stacking interactions and reactivity as well as the properties of cellular accumulation and cytotoxicity. In this contribution we examine the modulation of non-covalent interactions on the C-terminal zinc finger (ZF2) and the ‘full” 2-finger peptide (NCp7) and show that ZF2 studies are a good prognosticator for the reactivity of the full peptide with significant enhancement of stacking interactions using xanthosine over guanosine as nucleoside. The complexes are weak inhibitors of the NCp7–DNA/RNA interaction and the inhibition correlates with the observed stacking interaction. We examine the origins of the enhanced effects of xanthosine over guanosine complexes and show that computational approaches are useful in analysis of the strength of the π-stacking interaction as a quick estimation of the ability to inhibit NCp7 and can be used to identify secondary interactions in the binding site to aid in design of effective inhibitors. The studies confirm the potential for modulation of the physicochemical properties of platinum-based compounds to enhance inhibition of a fundamental biological property – the NCp7–RNA(DNA) annealing.
Results and discussion
Characterization and chemical properties of Pt–purine compounds
The target compounds were designed to firstly examine the effect of the planar ligand on the stacking interaction using modified purines and nucleosides. Secondly, steric effects and lipophilicity were incorporated into the “carrier” portion of the molecule by varying the nature of the dien ligand. The structures are shown in Fig. 1. Platination at the purine N7 in all cases is confirmed by the typical downfield shifts of the H(8) protons (see ESI†). Xanthine itself exists in tautomeric equilibrium between the keto and enol forms with consequently a potential array of possible binding sites. The xanthine derivative itself gave multiple H(8) peaks indicative of linkage isomers and was not considered further. The addition of methyl groups to the dien ligand induces considerable steric hindrance about the platinum–nucleobase bond, as observed in the 1H NMR spectra of 1c and 1d. In both cases the H8 signal of the 9-EtGua splits into a doublet due to restricted rotation. Temperature dependence studies of the H8 signal reveal a significant difference between the N,N′-Me2dien compound and the N,N′-Me4dien compound (Fig. 2). For the N,N′-Me2dien ligand the doublet coalesces to a broad peak at 35 °C and then begins to sharpen into a new doublet, with significantly less separation between the peaks. In contrast, little change is observed for the N,N′-Me4dien ligand, even up to 70 °C – implying little interconversion.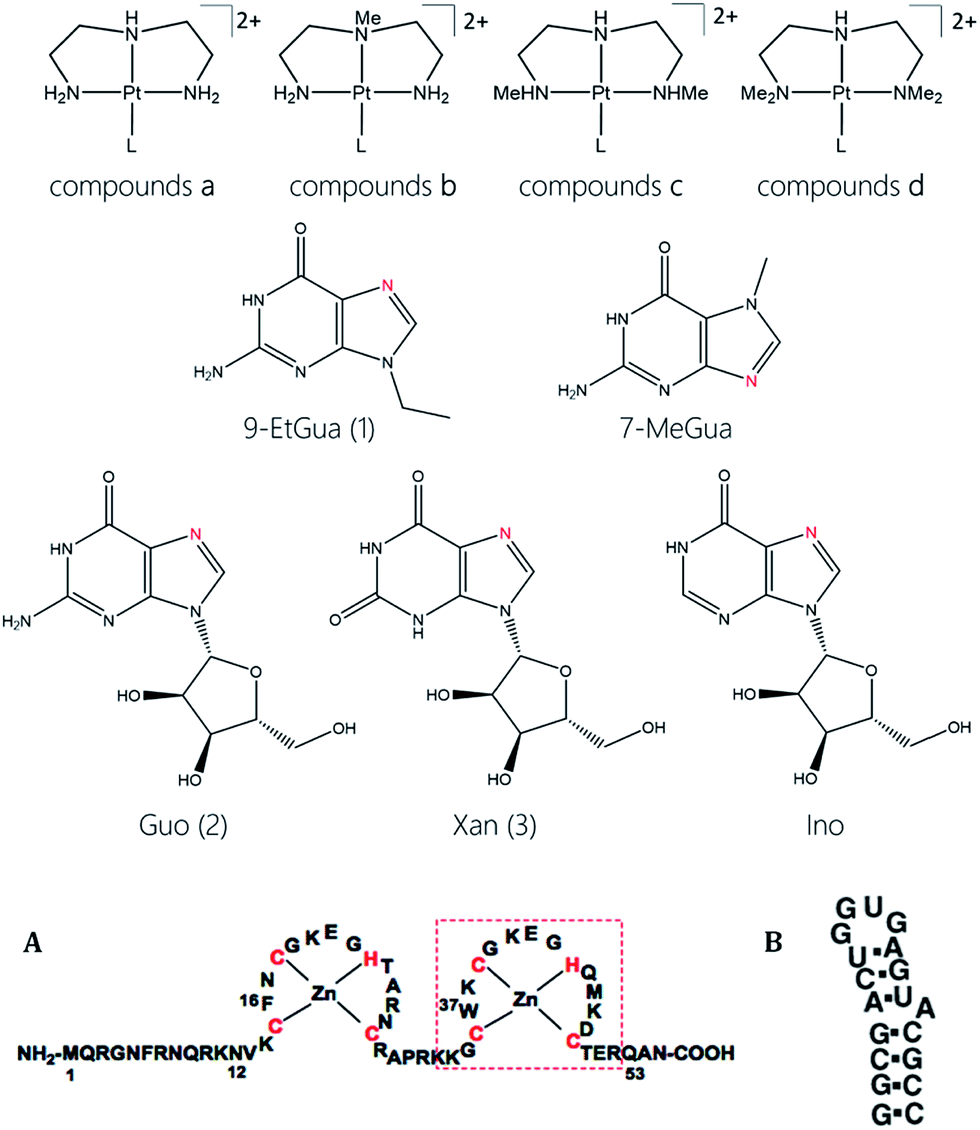 | ||
| Fig. 1 General structure for [Pt(dien)L]2+ compounds, where the dien can be methylated N–Me, N,N′-dimethyl or N,N′-N,N′-tetramethyl and L is a nucleobase or nucleoside, as shown. Structure of the HIVNCp7 nucleocapsid protein showing the two zinc fingers (A). The red dashed line shows the C-terminal finger used in our studies. On right is the SL2RNA sequence used (B). | ||
 | ||
| Fig. 2 Temperature dependence of the 1H NMR splitting of the H8 signal for [Pt(N,N′-Me4dien)(9-EtGua)]2+ (A) and [Pt(N,N′-Me2dien)(9-EtGua)]2+ (B). The small peak in (B) at high T may be due to formation of a minor new species with increasing T. | ||
The 195Pt NMR spectra of both the N,N′-Me4dien and N,N′-Me2dien compounds also show broad signals in agreement with the 1H NMR spectra, Fig. S2.† These peaks show some coalescence but further broaden with increased temperature. The 1H NMR spectra of the xanthosine derivatives also confirm the steric hindrance – in this case the N,N′-Me2dien compound is a broad singlet whereas the more sterically demanding N,N′-Me4dien compound shows two clear singlets at 8.97 and 9.00 ppm. Their corresponding 195Pt NMR spectra again showed broad signals consistent with the presence of rotamers.
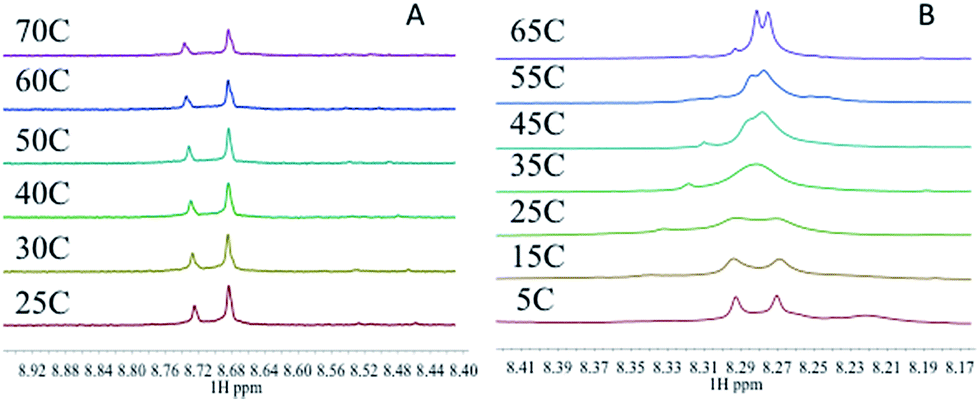
The energies of the rotation barrier, Ebarrier, were calculated following literature methods.23 There is a >2 kcal mol−1 difference between Ebarrier for the two compounds, 15.90 and 17.92 kcal mol−1 for N,N′-Me2dien and N,N′-Me4dien respectively. The energy difference highlights the significant steric hindrance created by the methyl groups. In the case of the N,N′-Me4dien ligand, DFT calculations gave two limiting structures (Scheme 1). In structure I, the 9-EtGua forms a close contact within hydrogen-bonding distances between H(8) and the N,N′-Me4dien ligand (3.06 Å). The C(6)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O atom then has a similar close contact (2.64 Å) to the same NMe2 group of the chelate. The alternative geometry, essentially resulting from rotation of the purine around the platinum square plane, results in the same contacts. Based on the model, structure 1 corresponds to the upfield peak and structure 2 corresponds to the downfield peak of the [Pt(N,N′-Me4dien)(9-EtGua)]2+.
O atom then has a similar close contact (2.64 Å) to the same NMe2 group of the chelate. The alternative geometry, essentially resulting from rotation of the purine around the platinum square plane, results in the same contacts. Based on the model, structure 1 corresponds to the upfield peak and structure 2 corresponds to the downfield peak of the [Pt(N,N′-Me4dien)(9-EtGua)]2+.
 | ||
| Scheme 1 | ||
Biological properties
The modular nature of the zinc finger template lends itself to a systematic targeting approach through studying firstly the fundamental stacking interactions with the simple tryptophan or N-AcTrp followed by extension to the C-terminal finger (containing the critical Trp ligand) and then the full zinc finger protein itself where the stacking interaction is incorporated into the protein and steric effects can be examined. Finally, the effect of the complexes on the NCp7–nucleic acid interaction can be assayed. Cellular properties and selectivity may also be examined to obtain insight into possible selectivity as it is important to understand the biological properties of the platinum–nucleobases with respect to cellular accumulation and cytotoxicity.| L | K a (×103) M−1 | |||
|---|---|---|---|---|
| [Pt(dien)L]2+ | [Pt(N-Medien)L]2+ | [Pt(N,N′-Me2dien)L]2+ | [Pt(N,N′-Me4dien)L]2+ | |
| 7-MeGua | 9.01 ± 0.09 | — | — | — |
| (10.42 ± 0.29) | — | — | — | |
| Ino | 5.63 ± 0.3 | — | — | — |
| (3.20 ± 0.29) | — | — | — | |
| 9-EtGua | 6.88 ± 0.36 | 13.52 ± 0.48 | 7.40 ± 0.93 | 11.78 ± 1.02 |
| (13.10 ± 0.94) | (14.70 ± 1.60) | (12.02 ± 0.37) | (18.30 ± 0.34) | |
| (20 ± 0.08) | ||||
| Guo | 13.10 ± 1.29 | 12.78 ± 0.22 | 6.84 ± 0.67 | 15.19 ± 1.21 |
| (15.80 ± 2.83) | (12.11 ± 1.13) | (11.37 ± 2.01) | (9.16 ± 1.46) | |
| (14 ± 0.05) | ||||
| Xan | 16.13 ± 1.10 | 11.56 ± 0.33 | 11.74 ± 0.53 | (11.30 ± 0.36) |
| (46.64 ± 0.28) | (60.22 ± 2.18) | (35.80 ± 0.76) | (48.67 ± 0.29) | |
| (46.91 ± 0.06) | — | |||
 | ||
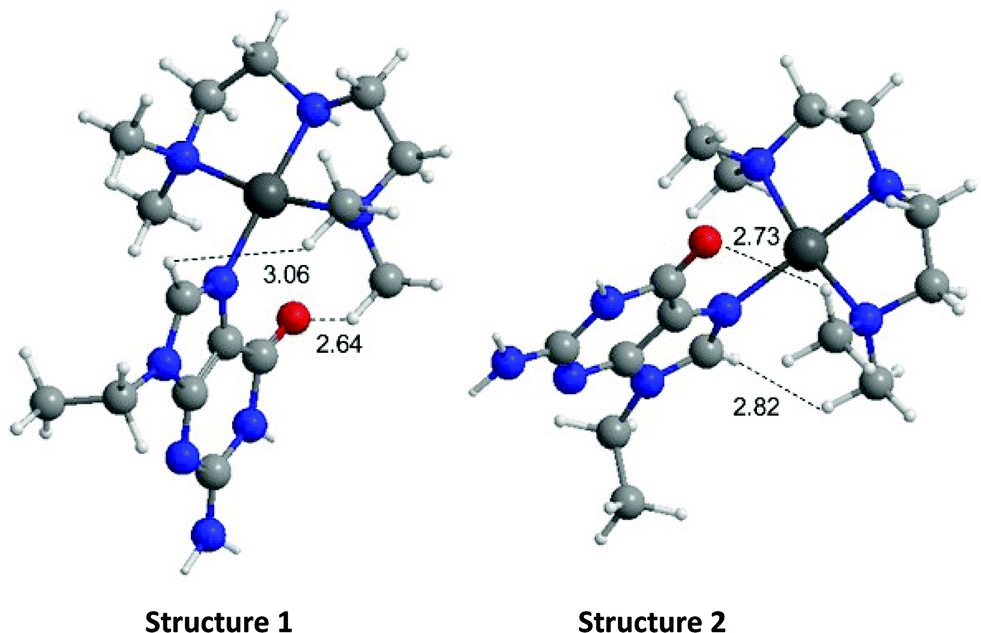
| Fig. 3 (A) Fluorescence quenching of NCp7 in presence of increasing amounts of [Pt(dien)(9-EtGua)]2+ (B) circular dichroism spectrum of “full” NCp7 zinc finger peptide in presence of [Pt(dien)(9-EtGua)]2+. | ||
To analyze the trends in association constants from Table 1, we performed DFT calculations with empirical dispersion corrections to investigate the enhancement of the Gua(Xan)–Trp interaction using “small models” of metalated MeGua and Xan π-stacked with methylindole (MeInd) as a model for Trp. The π-stacking energy, LUMO energies, and charge transfer were examined in order to determine whether the modified Gua with the lowest LUMO energies have the largest charge transfer and the strongest π-stacking interactions. Secondly, we examined a “large model” using the hybrid ONIOM method to determine the effects of π-stacking and hydrogen-bonding within a larger fragment of the C-terminal zinc finger of NCp7.
Small π–π stacked structures
Small models of the π-stacking interaction between MeGua and N-methylindole (MeInd), as a model for Trp, were optimized in the same orientation (A) as found in the NMR solution structure of the NCp7 nucleocapsid complexed with the DNA primer binding site (PDB 2EXF).34 The interaction energies for π-stacking of the dimers MeInd-[Pt(NH3)3(9Me-Gua)]2+, MeInd-[Pt(NH3)3(7-Me-Gua)]2+ and MeInd-[Pt(NH3)3(Xan)]2+ were compared to experimental equilibrium constants for interaction of Trp with the analogous [Pt(dien)(nucleobase)]2+ complexes (Table 1).12,13 The DFT/B97-D optimized geometries of the individual square-planar [Pt(NH3)3(MeGua)]2+ (where MeGua refers to 7/9-MeGua) complexes and their LUMO energies were comparable to previous studies (Table 2).13 The numbering scheme and optimized structures for the [Pt(NH3)3(purine)]2+ (purine = 9-EtGua, 7-MeGua and xanthine) are given in Fig. S3.†| Small structures | LUMO Gua (eV) | LUMO+1 Gua (eV) | ΔE (kcal mol−1) | d(e) | b(e) | d–b(e) | K a (×103) M−1 |
|---|---|---|---|---|---|---|---|
| MeInd-MeGua | −0.87 | −0.82 | −13.69 | 0.048 | 0.052 | −0.004 | 3.50 |
| MeInd-(NH3)3Pt(9-MeGua)2+ | −9.36 | −8.25 | −26.70 | 0.124 | 0.027 | 0.097 | 6.88 |
| MeInd-(NH3)3Pt(7-MeGua)2+ | −9.52 | −8.12 | −31.34 | 9.01 | |||
| MeInd-(NH3)3Pt(Xan)2+ | −9.92 | −8.97 | −30.83 | 0.147 | 0.027 | 0.121 | 16.13 |
| Struct. B MeInd-(NH3)3Pt(9-MeGua)2+ | −9.36 | −8.25 | −27.57 | 0.140 | 0.023 | 0.117 |
In the optimized π-stacked structures, Gua/Xan are not directly eclipsed over MeInd, consistent with general trends in π-stacking interactions (Fig. 4).35 The inter-ring distances for MeInd-9-MeGua (3.2–3.3 Å) were slightly smaller than those found in the crystal structures of Ind–Gua stacked pairs (3.4–3.5 Å).36 Metalation resulted in π-stacking interactions in which the C2 end of the purine is closer to MeInd than the N7 end (i.e., 3.2 and 3.5 Å, respectively, for MeInd-[Pt(NH3)3(9-MeGua)]2+). The tilt angles are similar to that found by Rutledge et al. for an adenine–histidine dimer (10°) and increase as MeInd-MeGua < MeInd-[Pt(NH3)3(9-MeGua)]2+ < MeInd-[Pt(NH3)3Xan]2+.37 An electrostatic interaction between the MeInd π cloud and a hydrogen causes one ammine ligand of the metalated base to extend down into the space between the π-stack, but this interaction is likely to compete with hydrogen bonding with the aqueous solvent. Metalation enhanced the π-stacking energy by 13–18 kcal mol−1 relative to uncomplexed 9-MeGua, consistent with the enhancement of the donor–acceptor interaction through stabilization of the metal-complexed nucleobase LUMO (Fig. 5). Charge decomposition analysis shows that the net electron donation from MeInd to Gua increases by 0.1e upon metallation, comparable to that found for a π-stacked dimer of benzene within a trinuclear Cu(I) triiodide cluster (0.14e).38
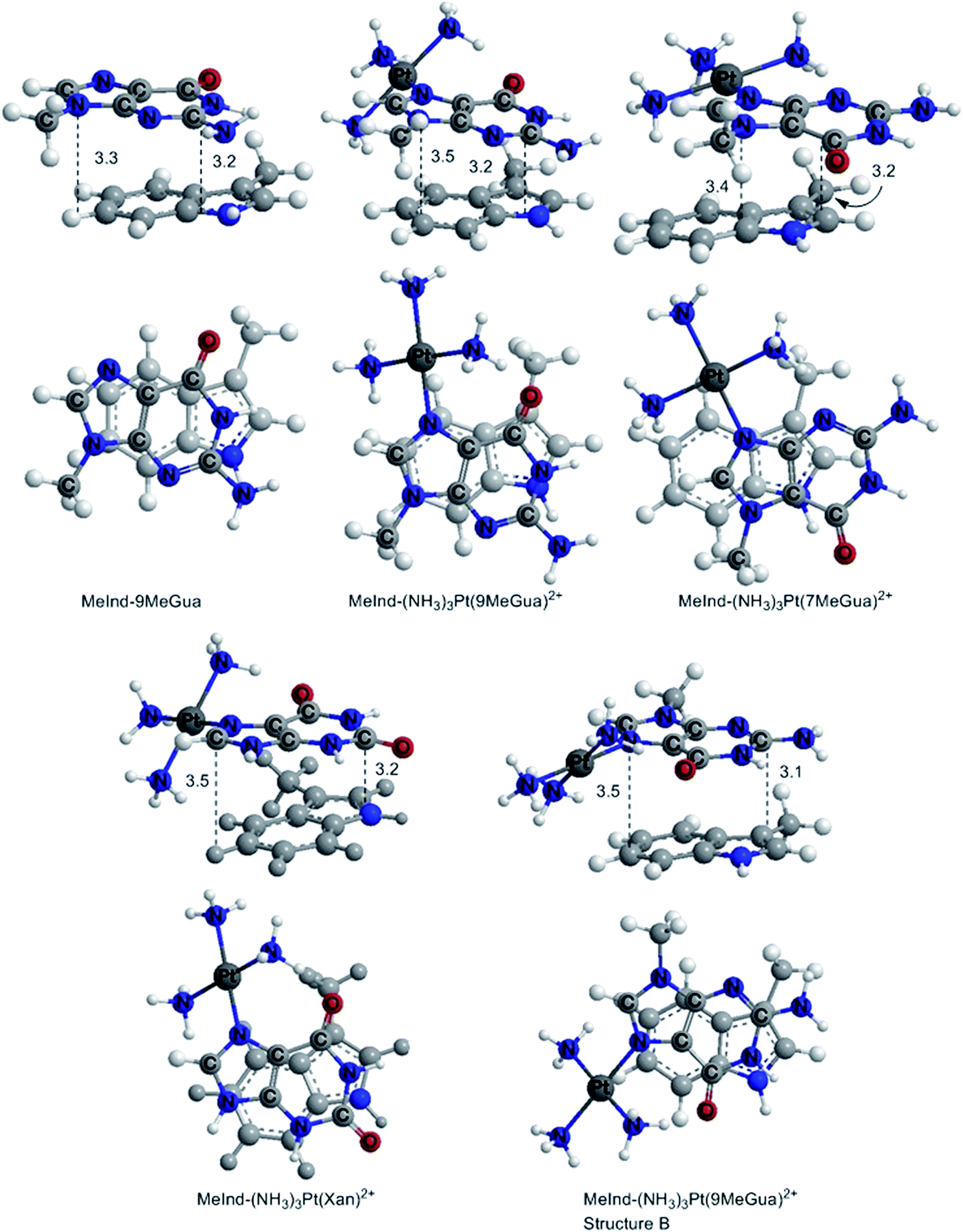 | ||
| Fig. 4 DFT B97-D optimized π-stacked structures of the small models of MeInd with Gua derivatives and Xan. The distances are labeled in Å. | ||
 | ||
| Fig. 5 Correlation of the π-stacking interaction with the nucleobase LUMO energy (eV). | ||
Larger models
Although the analysis of the donor–acceptor HOMO–LUMO gap is useful when one deals with the simple π-stacked structures, factors such as hydrogen bonding and steric effects also contribute to binding to the recognition site. For example, molecular dynamics studies of NCp7 bound to DNA and RNA show hydrogen bonding interactions involving Gua and residues G35, W37 and M46.28 The effect of the surrounding residues on the Trp–Gua interaction was explored using the ONIOM(B97-D:PM6) hybrid quantum-mechanical-semiempirical method on a “large model” of the C-terminal NCp7 zinc finger truncated to residues 32 to 40 and 44 to 49 of the NMR structure of HIV-1 NCp7 complexed with DNA(−) primer binding site (PDB 2EXF), (Fig. 6).34 Lysines 34 and 38 were further truncated to Ala and the side chain of K47 was replaced by an ethyl group. The DFT region, indicated in Fig. 6 with a ball-and-stick representation, was allowed to optimize freely. The remainder of the model was treated with the PM6 semiempirical method and constrained to the NMR structure.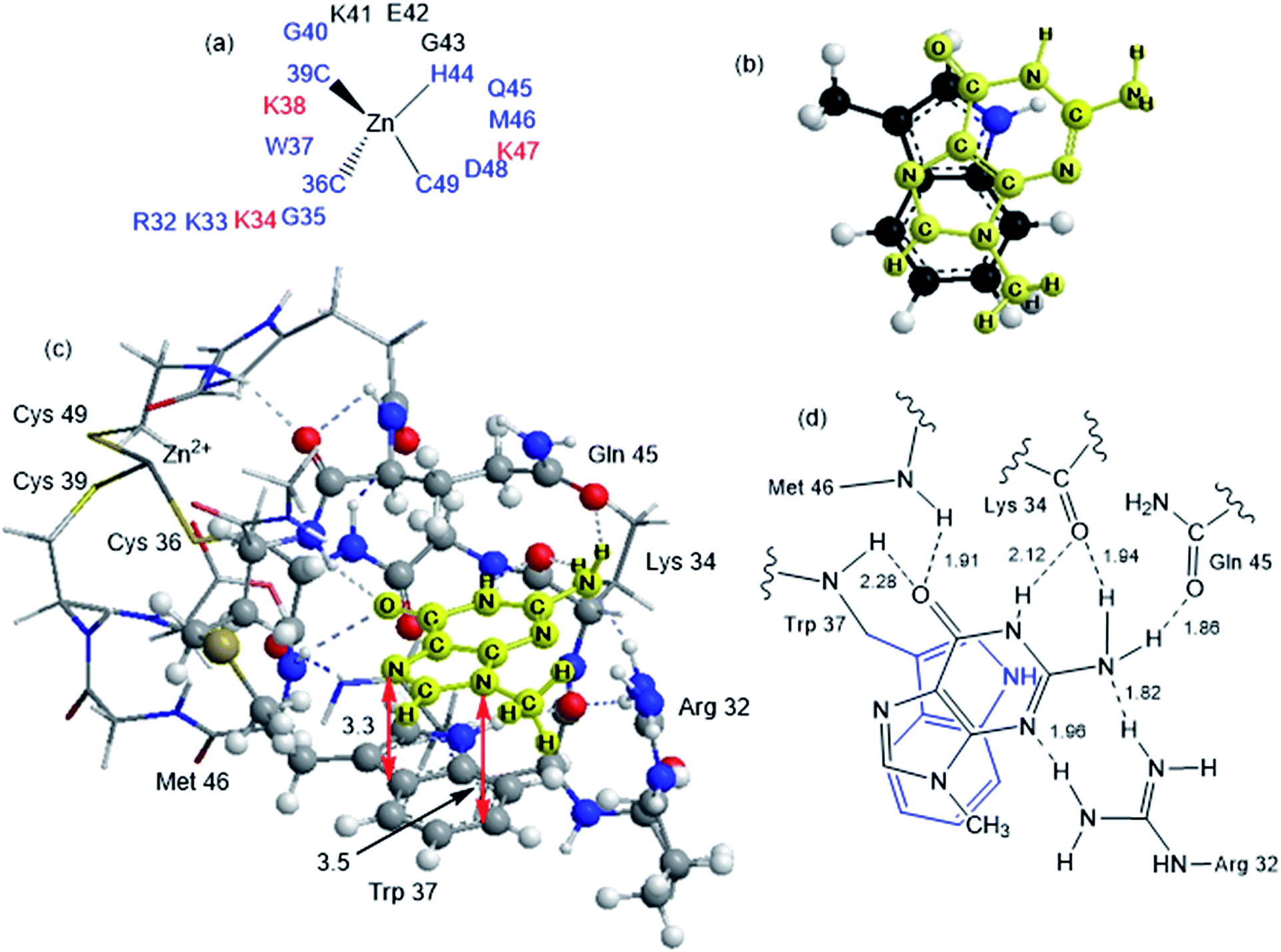 | ||
| Fig. 6 ONIOM(B97-D:PM6)-optimized structure of the truncated model of NCp7 bound with 9-MeGua in the native conformation A. (a) The truncated NCP7 finger 2 used in calculations, blue residues were used without truncation, the red residues were truncated to Ala, K47 was truncated to a modified Ala with ethyl group and black residues were eliminated. (b) Structure A. The truncated optimized model of NCp7 complexed with 9-MeGua with the π interaction shown only. 9-MeGua is shown in yellow. (c) ONIOM optimized native structure. Ball and stick representations correspond to the QM region, stick representation correspond to the SE region. (d) The hydrogen bonding interactions structure A. Bond lengths are given in Å. | ||
Interactions between the protein and DNA phosphate backbone limit Gua to the native conformation A observed in the NMR structure (Fig. 6). Without the phosphate backbone, Gua derivatives could theoretically π-stack with Trp in different orientations. Geometry optimizations of the “large model” were performed with Gua in the native A and three additional conformations (B–D in Fig. S2†). The orientation of MeGua affects both the nature of the π-stacking interaction and the number and type of hydrogen bonding interactions with the protein. In A, π-stacking is supplemented by hydrogen bonding between the Gua carbonyl oxygen and the W37 NH on the backbone as expected from the NMR structure. In the ONIOM model, the Q45 and R32 side chains interact with Gua instead of the absent phosphate backbone. The relative energies of A–D are related to the number and strength of their H-bonding interactions (Fig. S4, Table S2†). Structure B was the most stable conformation followed by the native structure A (+6.2 kcal mol−1), D (11.7 kcal mol−1) and C (+15.7 kcal mol−1). The higher relative energies for C and D can be attributed in part to a smaller number of hydrogen-bonding interactions to the protein fragment. To quantify the effect of the hydrogen bonding, natural bond orbital donor–acceptor energies (ΔEd→a) were calculated for key hydrogen bonding interactions in A–D (Table S2†). A small model of MeInd-[Pt(NH3)3(9-MeGua)]2+ in orientation B was more stable that A, suggesting that B is preferred due to the more favorable electrostatics of the hydrogen bonding interactions in that orientation.
Platinated Gua and Xan were modeled in orientations A and B to determine the effect of hydrogen-bonding and LUMO stabilization on the ability of the Pt(NH3)3 fragment to fit into the binding site in a different conformation (Fig. 7 and Table 3). C and D were omitted because they could not accept the Pt(NH3)3 fragment due to steric constraints. The interaction energy of the metalated Gua/Xan with the NCp7 models was calculated relative to the MeGua bound model by eqn (1):
| NCp7–MeGua + M(NH3)3MeGua/Xan2+ → NCp7–M(NH3)3MeGua/Xan2+ + MeGua | (1) |
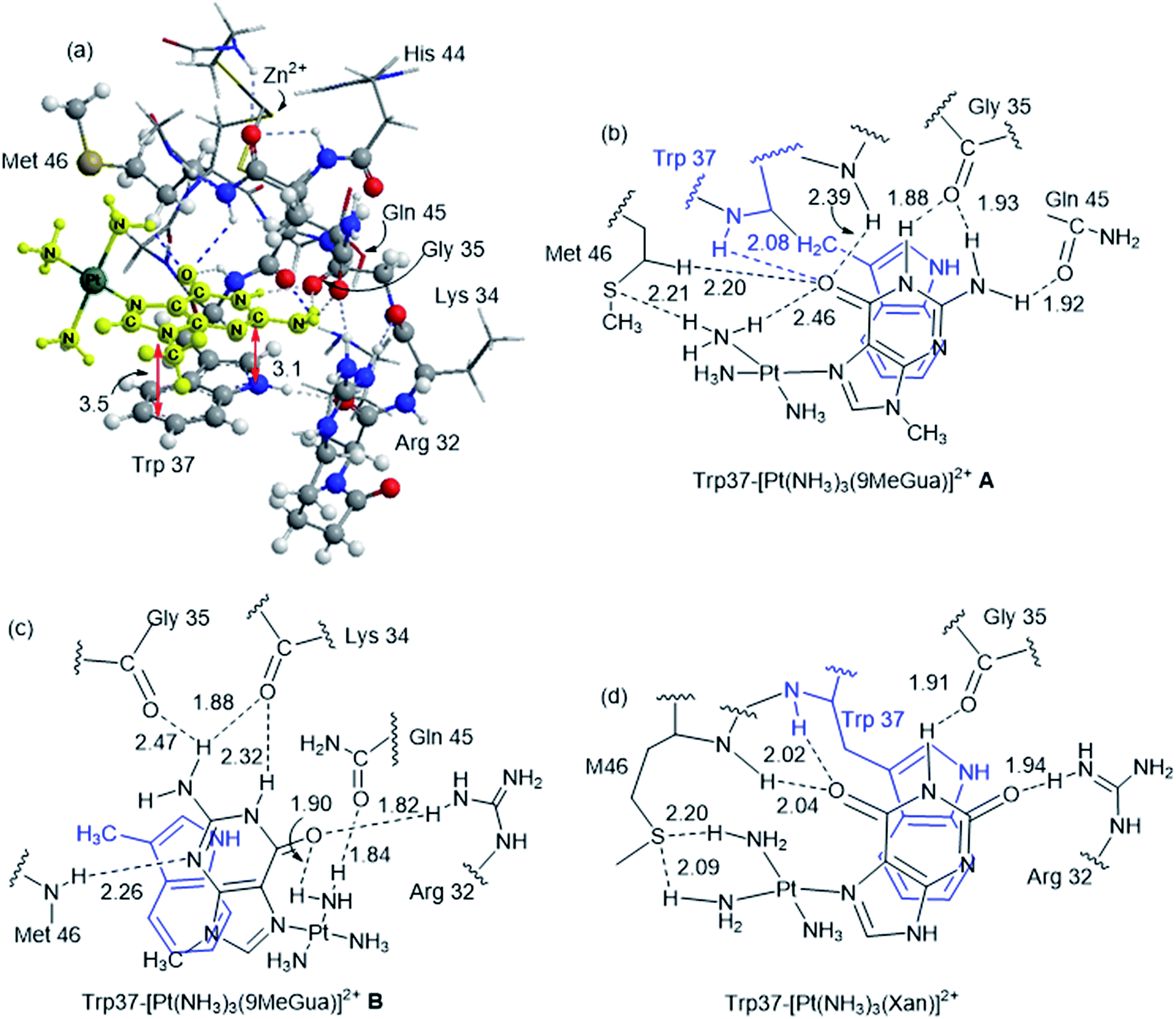 | ||
| Fig. 7 (a) ONIOM(B97-D/PM6)-optimized structure of the large NCp7 model with [Pt(NH3)3(9MeGua)]2+ in native conformation A. Hydrogen bonding interactions and π-stacking conformations for complexes of the large model with [Pt(NH3)3(9-MeGua)]2+ (A (b) and B (c)) and [Pt(NH3)3(Xan)]2+ (d). | ||
| Donor | q D (e) | Acceptor | q A (e) | d, Å | ΔEd→a (kcal mol−1) | WBI |
|---|---|---|---|---|---|---|
| NCp7-[Pt(NH 3 ) 3 (9-MeGua)] 2+ native A | ||||||
| MeGau CO |
−0.712 | W37 BB N–H | 0.407 | 2.08 | 6.44 | 0.025 |
| G35 BB CO |
−0.779 | MeGua N1–H | 0.462 | 1.88 | 12.26 | 0.044 |
| G35 BB CO |
−0.779 | MeGau NH2–Ha | 0.432 | 1.93 | 10.47 | 0.039 |
| Q45 SC CO |
−0.745 | MeGau NH2–Hb | 0.435 | 1.92 | 12.32 | 0.048 |
| M46 SC S | 0.065 | Pt–NH3 N–H | 0.435 | 2.21 | 13.63 | 0.084 |
| MeGau CO |
−0.712 | M46 BB N–H | 0.400 | 2.39 | 1.25 | 0.008 |
| MeGau CO |
−0.712 | M46 SC C–H | 0.211 | 2.20 | 1.59 | 0.005 |
| MeGau CO |
−0.712 | Pt–NH3 N–H | 0.444 | 2.46 | 0.58 | 0.005 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| NCp7-[Pt(NH 3 ) 3 (9-MeGua)] 2+ B | ||||||
| MeGau N3 | −0.627 | M46 SC N–H | 0.411 | 2.26 | 4.63 | 0.018 |
| K34 BB CO |
−0.736 | MeGau NH2–Ha | 0.441 | 1.88 | 14.41 | 0.050 |
| K34 BB CO |
−0.736 | MeGua N1–H | 0.461 | 2.32 | 2.45 | 0.012 |
| G35 BB CO |
−0.694 | MeGau NH2–Hb | 0.436 | 2.47 | 0.94 | 0.004 |
| Q45 SC CO |
−0.750 | Pt–NH3 N–H | 0.466 | 1.84 | 17.69 | 0.053 |
| MeGau CO |
−0.731 | R32 SC N–H | 0.430 | 1.82 | 16.26 | 0.058 |
| MeGau CO |
−0.731 | Pt–NH3 N–H | 0.451 | 1.90 | 10.77 | 0.043 |
|
||||||
| NCp7-[Pt(NH 3 ) 3 (7-MeGua)] 2+ B | ||||||
| MeGau CO |
−0.679 | R32 SC N–H | 0.437 | 1.80 | 17.19 | 0.059 |
| K34 BB CO |
−0.732 | MeGau N1–H | 0.465 | 2.09 | 6.04 | 0.041 |
| K34 BB CO |
−0.732 | MeGau NH2–Ha | 0.436 | 2.21 | 4.44 | 0.020 |
| W37 BB N | −0.727 | MeGau NH2–Hb | 0.426 | 2.35 | 3.65 | 0.016 |
| M46 SC S | 0.056 | Pt–NH3 N–H | 0.421 | 2.22 | 17.23 | 0.082 |
| M46 SC S | 0.056 | Pt–NH3 N–H | 0.424 | 2.39 | 7.75 | 0.051 |
| MeGau N3 | −0.641 | Pt–NH3 N–H | 0.449 | 2.38 | 1.70 | 0.013 |
|
||||||
| NCp7-[Pt(NH 3 ) 3 (Xan)] 2+ | ||||||
| Xan C6O |
−0.687 | M46 SC N–H | 0.419 | 2.04 | 6.34 | 0.023 |
| Xan C6O |
−0.687 | W37 BB N–H | 0.413 | 2.02 | 7.42 | 0.024 |
| G35 BB CO |
−0.762 | Xan N1–H | 0.483 | 1.91 | 12.41 | 0.040 |
| M46 SC S | 0.077 | Pt–NH3 N–H | 0.423 | 2.20 | 13.81 | 0.084 |
| M46 SC S | 0.077 | Pt–NH3 N–H | 0.412 | 2.09 | 16.21 | 0.116 |
| Xan C2O |
−0.653 | R32 SC N–H | 0.442 | 1.94 | 7.80 | 0.023 |
[Pt(NH3)3(9-MeGua)]2+ complexed to the large model in orientation A was more strongly interacting than free 9-MeGua by 35.2 kcal mol−1. The π-stacking interactions shift slightly to accommodate steric and hydrogen-bonding interactions resulting from platination but, similar to the small models, the purine and indole rings have a 9.5° tilt angle Pt(NH3)3 fragment. In both structures, hydrogen bonding between the ammine ligands and the Gua CO influences the interactions with the protein. In A, the interaction of the latter with the peptide backbone at W37 is weakened, but in B, the amide side chain of Gln45 flips so that its CO can interact with the Pt ammine ligands. The lower stability of B (+4.3 kcal mol−1) can be attributed to the electrostatic interactions between the Pt(NH3)3 fragment and the Arg32 side chain. In A, Pt(NH3)3 forms a close interaction with the divalent sulfur of M46 (2.26 Å; ΔEd→a = 13.6 kcal mol−1), which agrees with the NMR structure of the C-terminal NCp7 peptide complexed with the {Pt(dien)-d(TACGCC)} in which the Pt(dien) fragment is in close proximity to the methionine sulfur.17 This result both confirms the validity of the large model and emphasizes the importance of the non-zinc-bonded residues in dictating the overall reaction.
The large models of NCp7-[Pt(NH3)3(Xan)]2+ binding assumed an orientation similar to A to allow for the ammine–Met46 interaction and the more favorable electrostatic interaction between Arg32 and the C2 carbonyl. As a result, the interaction of metallated Xan was ∼10 kcal mol−1 more favorable that of [Pt(NH3)3(9-MeGua)]2+. Hydrogen-bonding interactions were similar to the 9-MeGua analogue, except for a shift in stacking to allow for the C6 carbonyl to interact with backbone amides of W37 and M46 (Fig. 7).
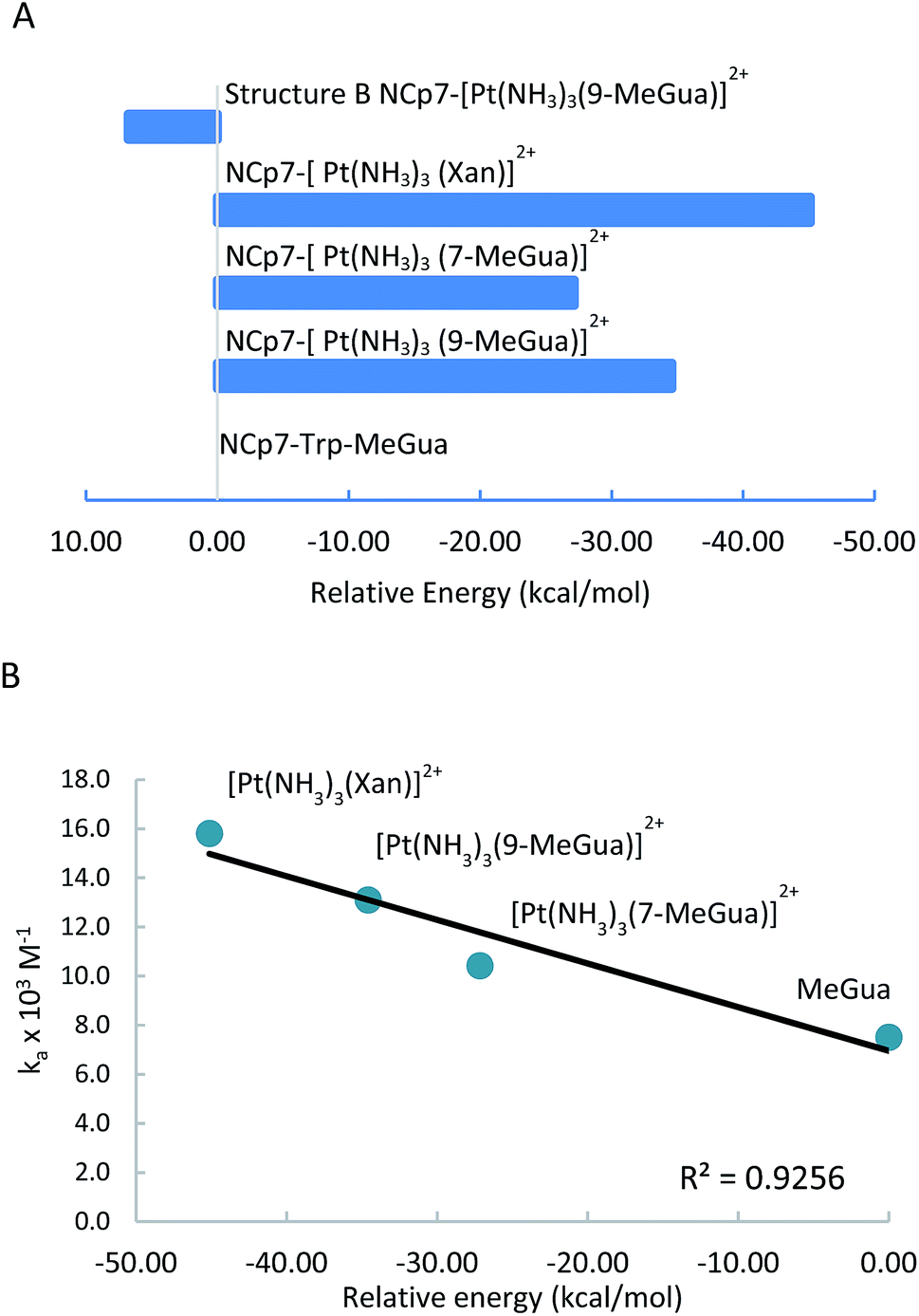 | ||
| Fig. 8 ONIOM(B97-D:PM6) interaction energies for platinated nucleobases with the large NCp7 model calculated relative to the free 9-MeGua complex (A). These energies correlate well with the experimental association constants Ka (Kπ) (B). | ||
Inhibition of the NCp7–RNA(DNA) interaction
Given the trends in the association constants and the good correlations with computational studies the next question to ask is how does the stacking interaction affect the peptide–nucleic acid interaction? As part of the viral packaging process, NCp7 binds to viral RNA, which is comprised of four stem loops (SL). SL2 and SL3 bind strongly to NCp7 and with similar affinity compared to SL1 and SL4.39,40 The ability of the compounds to inhibit or dissociate the complex between NCp7 and SL2 was evaluated by electromobility gel shift assays (EMSAs).41 As previously reported, hairpin SL-2 RNA interacts with the zinc-knuckle motifs present in NCp7.42 The interaction of NCp7 with 32P-SL2 is apparent in Fig. 9A–D as a shift from the faster migrating species (free RNA, lane 1) to the slower migrating species (bound RNA, lane 2). Both [Pt(dien)(9-EtGua)]2+ and [Pt(dien)(xanthosine)]2+ disrupt the interaction of SL-2 with NCp7. The addition order of SL-2 or inhibitors to the reaction mixture with NCp7 did not alter their capacity to disrupt the NCp7–SL2 complex. [Pt(dien)(Xan)]2+ inhibits the complex more effectively when added to preformed NCp7–SL2 (Fig. 9C and D), and requires 4 times less concentration than [Pt(dien)(9-EtGua)]2+ (Fig. 9A and B).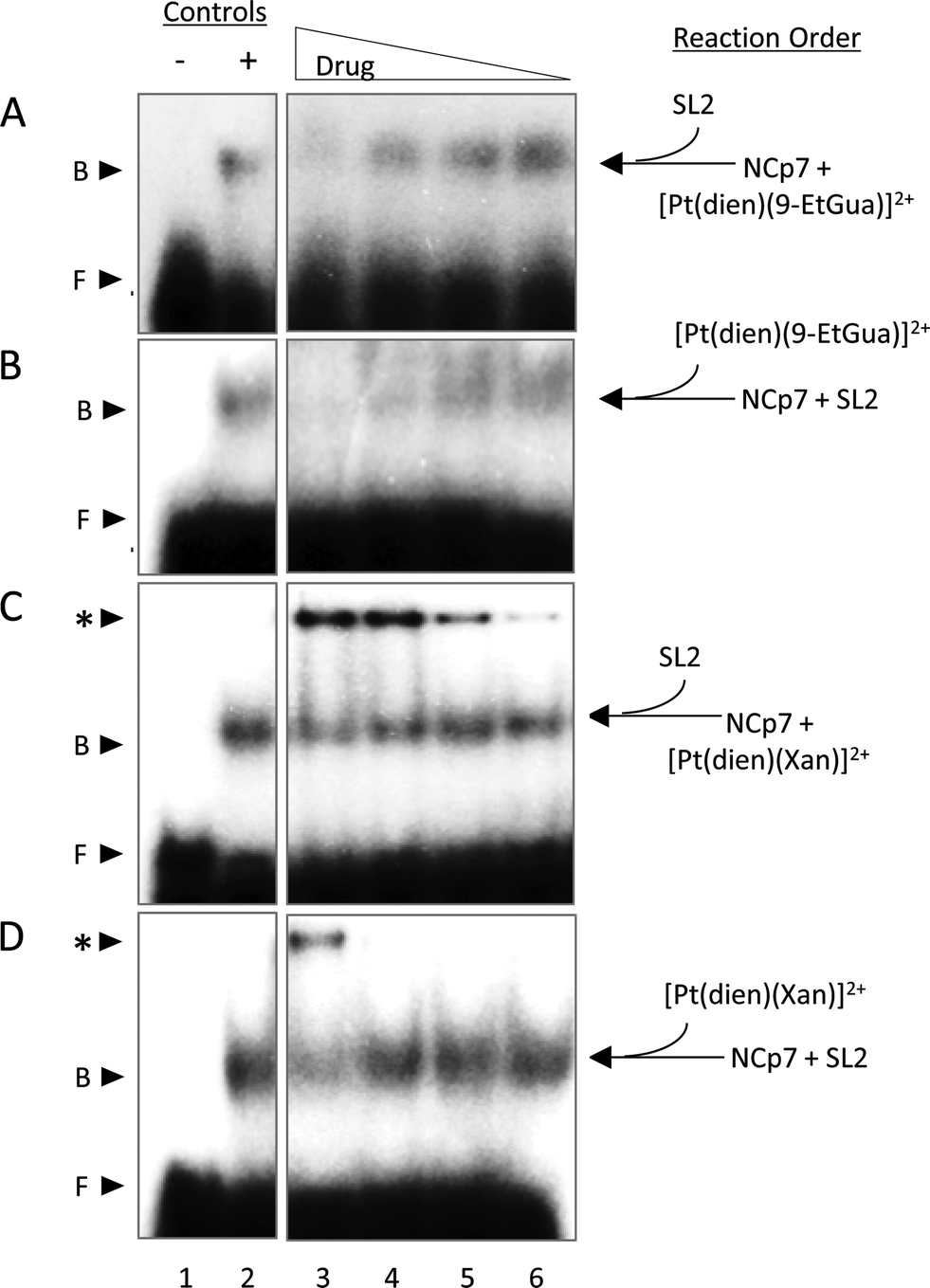 | ||
| Fig. 9 Effects of [Pt(dien)(9-EtGua)]2+ and [Pt(dien)(Xan)]2+ on SL2 RNA-NCp7 protein interaction. (A) [Pt(dien)(9-EtGua)]2+ incubated with NCp7 for 30 min prior to addition of SL2. Lane 1, SL2 RNA; lane 2, SL2 RNA + NCp7; lanes 3–6 NCp7, SL2, and 1000, 500, 250, 125 μM [Pt(dien)(9-EtGua)]2+. (B) NCp7 was incubated with SL2 for 30 min prior to addition of [Pt(dien)(9-EtGua)]. Lane 1, SL2 RNA; lane 2, SL2 RNA + NCp7; lanes 3–6 NCp7, SL2, and 1000, 500, 250, 125 μM [Pt(dien)(9-EtGua)]2+ (C) [Pt(dien)(9-EtGua)]2+ incubated with NCp7 for 30 min prior to addition of SL2. Lane 1, SL2 RNA; lane 2, SL2 RNA + NCp7; lanes 3–6 NCp7, SL2, and 1000, 500, 250, 125 μM [Pt(dien)(Xan)]2+. (D) NCp7 was incubated with SL2 for 30 min prior to addition of [Pt(dien)(Xan)]2+. Lane 1, SL2 RNA; lane 2, SL2 RNA + NCp7; lanes 3–6 NCp7, SL2, and 250, 125, 62.5, and 31.3 μM [Pt(dien)(Xan)]2+. F, free SL-2; B, bound SL-2; *, inhibitor-SL2 aggregate. | ||
The [Pt(dien)(Xan)]2+ reactions with NCp7 and SL2 showed an additional slower migrating band on the gel (Fig. 9C and D). Control experiments identified this species as inhibitor – RNA aggregates, which appear to form at the expense of the pool of free SL2. The study of model DNA quadruplexes formed exclusively by guanine and xanthine showed, for the latter case, that a major interaction responsible for maintaining the helical structure was π-stacking.43 Thus, an inevitable consequence of enhancing stacking is also to enhance RNA affinity. Yet, the affinity of [Pt(dien)(Xan)]2 interactions for DNA or RNA is expected to be relatively low.16 Specific NCp7–SL2 binding would likely out-compete this type of interaction, particularly once the complex is formed. Therefore, it is hypothesized that [Pt(dien)(Xan)]2+ undergoes two concurrent binding events: (1) specific (1:1) high affinity binding with NCp7 and (2) nonspecific and low affinity binding with RNA.
Cellular accumulation and cytotoxicity
The end-product of any biophysical study is to achieve suitable cellular activation. Two parameters which dictate anti-viral specificity are cellular accumulation and cytotoxicity. In the case of the series here, we hypothesized that the addition of methyl groups to the dien ligand would increase lipophilicity, in turn increasing the cellular accumulation. To evaluate this hypothesis, we tested CCRF-CEM and Jurkat cell lines. Both cell lines are CD4+ and HIV susceptible. The methylation of the dien did not result in a significant difference in the cellular accumulation for platinum complexes containing 9-EtGua (1a–d), Gua (2a–d) or Xan (a–d) (Fig. S5†). Three compounds show greater accumulation compared to all others: 1a, 1b, and 2a. From 3 to 6 hours, a negligible increase in accumulation is seen for most compounds, with the exception of the three compounds that see the highest concentration of platinum per cell. The cell uptake trends are the same for the CCRF-CEM and Jurkat cells, which may be indicative of a similar mechanism. To rationalize the trend upon dien methylation, the octanol–water coefficients (logPoct/water) for compounds 1a–1d were calculated, Table S3.† There does not appear to be a trend in the number of methyl groups and logPoct/water or the lipophilicity and cellular accumulation. Based on the cellular accumulation profile, cytotoxicity studies were performed for compounds 1a, 1b and 2a as well as compound 3a (to test the cytotoxicity of a xanthosine derivative). None of the compounds exhibited significant cytotoxic properties with IC50 values over 100 μM in all cases, Table S3B.† This is a desirable property, as the compounds are ultimately designed to inhibit HIV infectivity without affecting normal cellular function. The lack of cytotoxicity exhibited by the compounds reported here is also favorable for anti-viral development.
Conclusions
The work presented highlights a systematic strategy to understand the interaction between platinum–nucleobase compounds and tryptophan and the tryptophan-containing NCp7 as well as some of their basic biological properties. Modulation of an intrinsic physicochemical property – π–π stacking interactions – can be achieved by suitable chemical design. The use of xanthosine greatly increases the association constant with the C-terminal ZF2 over the “parent” guanine-based compounds. Both DFT models of the ‘small” structures and the computational approaches using a “large” model are consistent with the experimental association constants. The correlation of the LUMO energies of the modified nucleobases with the DFT π-stacking energies shows that frontier orbital energies of the individual monomers can be used as a first estimate of the π-stacking strength to Trp. This correlation extends to interaction with the zinc fingers themselves where hydrogen bonding and steric interaction can also determine the final recognition, as shown in more detailed atomistic modeling of the interaction of metallated nucleobases with the large NCp7 model. In this case, the calculations confirm the interaction of the Pt(dien) fragment with Met37 as suggested in the NMR structure of the C-terminal NCp7 peptide with the Pt(dien)-d(TACGCC) hexanucleotide.17 These combined theoretical and experimental results emphasize the importance of the non-zinc-bonded residues in dictating the overall reaction and the potential for targeting interactions identified through computational modeling to enhance the promising proof-of-concept results reported in the inhibition studies.Both components of the NC–nucleic acid chaperone activity have been targeted.44–46 General approaches to inhibit the NCp7–DNA(RNA) interaction through zinc finger targeting have used both covalent and “non-covalent” approaches to zinc ejection.47 The results discussed here show similarities and analogies with these broad approaches which have been used for many organic molecules. Alkylation of nucleobase antagonists has been proposed as a mechanism for enhancing binding to the essential Trp37 of NCp7.48–51 Increased π-stacking between the positively charged methylated base and aromatic amino acid side chains is proposed to be responsible for the preferential recognition for an alkylated base, through lowering of the energy of its lowest unoccupied molecular orbital (LUMO) to make it a better acceptor for electron density from aromatic side chains.50,52 In this sense the formal analogy we have made between alkylation and platination (and metallation in general) to guide design of coordination compounds for medicinal applications is strengthened.
Overall, the biophysical properties displayed by the [Pt(dien)(nucleobase)]2+ are favorable. A study of approximately 2000 small molecules from the NCI Diversity Set suggested a possible fluorescein-based pharmacore with a good correlation between tryptophan quenching and inhibition of NC–nucleic acid binding.53 A second high-throughput screening of small molecules for inhibition of NC-mediated destabilization of the stem-loop structure of cTAR DNA (a sequence complementary to the transactivation response element) produced five selected hits from a total of 4800 compounds.54 The inhibitory activity of 4 of the 5 correlated with their ability to compete with the nucleic acid for binding to NCp7.54 It is relevant that in both these surveys the inhibitory doses (to inhibit peptide/nucleic acid) were in the micromolar range. In the study using cTAR DNA the apparent dissociation constants (Kd′ for inhibitor/NCp7 binding) of two leading compounds were 7.7 × 10−6 M (designated CO7) and 1.7 × 10−5 M (designated HO2).54 The value obtained for [Pt(dien)(Xan)]2+ is in fact close to this latter value, further validating the study of this class of coordination compounds as potential inhibitors of the NCp7–RNA interaction.
Modulation of the NCp7–RNA interaction through RNA affinity has also been examined. In this respect the affinity of the compounds for DNA in presence of NCp7 is of high interest. While biophysical studies would not predict a high binding affinity for DNA in standard measurements such as Tm or EtBr competition assays the high excess of RNA in the biophysical studies may account for a possible “transfer” of the complex from peptide association to polynucleotide. The details of these reactions may depend very much on the exact experimental conditions – while the overall hypothesis is based on one site for the peptide (Trp) the polynucleotide will have multiple binding sites. This transfer would be a unique approach to inhibition of function because a priori, it is also likely to result in inhibition of the overall interaction. The results suggest that the study of the ternary system would be a novel approach to design of more effective inhibitors.
This paper has emphasized the optimization of the protein–Pt complex interactions. Nevertheless, it is of interest to briefly compare in vitro inhibition of infectivity of the prototype [Pt(dien)(9-EtGua)]2+ with the compound SP-4-2-[PtCl(NH3)(9-EtGua)(quinoline)]+ which displayed modest anti-HIV activity.55 For comparison, exploratory activity of [Pt(dien)(9-EtGua)]2+ against HIV-1 strains BaL, NL4-3 and 91-US001 strains in peripheral mononuclear blood (PBMC) cells showed only modest HIV inhibitory activity for the latter with an IC50 = 28.61 μM (Roger Ptak, personal communication). A number of extra factors come into play when considering cellular properties – amongst them the overall stability of the complex in blood and other non-selective interactions. The concept of “substitution-inert” is relative and while in principle, [PtN3(nucleobase)] compounds could stack with any available tryptophan moieties, relatively promiscuous biomolecule substitution reactions of the much more substitution-reactive Pt–Cl bond will be avoided. Optimization of the 2nd step in our concept4,22 – slow substitution by a nucleophilic cysteinate – is a valid approach for enhancement of specificity and differentiation amongst Trp moieties where only those in proximity to strong nucleophiles would enhance eventual nucleobase displacement. In summary the results presented show from first principles the systematic modulation of a fundamental biophysical property through the advantage of considering hitherto relatively understudied non-covalent interactions – more characteristic of the biodisciplines – compared to covalent biomolecule interactions, generally considered as belonging to the field of chemistry.
Acknowledgements
This work was supported by NSF CHE-1413189 and Ciencia Sem Fronteiras CAPES PVES 154/2012. We are grateful for support through Massey Cancer Center P30 CA016059. We thank R. J. Gorelick for a gift of the “full” HIV NCp7 peptide. We thank Roger Ptak (Southern Research Institute) for exploratory examination of anti-viral infectivity of [Pt(dien)(9-EtGua)]2+. Calculations were performed using high performance clusters managed by ODU Information Technology Services.References
- N. P. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- N. Farrell, Chem. Soc. Rev., 2015, 44, 8773–8785 RSC.
- J. Malina, M. J. Hannon and V. Brabec, FEBS J., 2014, 281, 987–997 CrossRef CAS PubMed.
- A. I. Anzellotti, Q. Liu, M. J. Bloemink, J. N. Scarsdale and N. Farrell, Chem. Biol., 2006, 13, 539–548 CrossRef CAS PubMed.
- S. M. Quintal, Q. Antonia dePaula and N. P. Farrell, Metallomics, 2011, 3, 121–139 RSC.
- J. G. Levin, J. Guo, I. Rouzina and K. Musier-Forsyth, Prog. Nucleic Acid Res. Mol. Biol., 2005, 80, 217–286 CAS.
- J. Darlix, J. Godet, R. Ivanyi-Nagy, P. Fossé, O. Mauffret and Y. Mély, J. Mol. Biol., 2011, 410, 565–581 CrossRef CAS PubMed.
- H. Wu, M. Mitra, M. J. McCauley, J. A. Thomas, I. Rouzina, K. Musier-Forsyth, M. C. Williams and R. J. Gorelick, Virus Res., 2013, 171, 263–277 CrossRef CAS PubMed.
- R. N. De Guzman, Z. R. Wu, C. C. Stalling, L. Pappalardo, P. N. Borer and M. F. Summers, Science, 1998, 279, 384–388 CrossRef CAS PubMed.
- N. Morellet, H. Déméné, V. Teilleux, T. Huynh-Dinh, H. de Rocquigny, M. Fournié-Zaluski and B. P. Roques, J. Mol. Biol., 1998, 283, 419–434 CrossRef CAS PubMed.
- T. L. South and M. F. Summers, Protein Sci., 2008, 2, 3–19 CrossRef PubMed.
- A. I. Anzellotti, E. S. Ma and N. Farrell, Inorg. Chem., 2005, 44, 483–485 CrossRef CAS PubMed.
- A. I. Anzellotti, C. A. Bayse and N. P. Farrell, Inorg. Chem., 2008, 47, 10425–10431 CrossRef CAS PubMed.
- A. I. Anzellotti, M. Sabat and N. Farrell, Inorg. Chem., 2006, 45, 1638–1645 CrossRef CAS PubMed.
- S. R. Spell and N. P. Farrell, Inorg. Chem., 2015, 54, 79–86 CrossRef CAS PubMed.
- S. D. Tsotsoros, A. B. Bate, M. G. Dows, S. R. Spell, C. A. Bayse and N. P. Farrell, J. Inorg. Biochem., 2014, 132, 2–5 CrossRef CAS PubMed.
- S. Quintal, A. Viegas, S. Erhardt, E. J. Cabrita and N. P. Farrell, Biochemistry, 2012, 51, 1752–1761 CrossRef CAS PubMed.
- A. Klug, Annu. Rev. Biochem., 2010, 79, 213–231 CrossRef CAS PubMed.
- A. Maynard and D. Covell, J. Am. Chem. Soc., 2001, 123, 1047–1058 CrossRef CAS PubMed.
- M. Huang, A. Maynard, J. A. Turpin, L. Graham, G. M. Janini, D. G. Covell and W. G. Rice, J. Med. Chem., 1998, 41, 1371–1381 CrossRef CAS PubMed.
- L. H. Wang, X. Y. Yang, X. Zhang, K. Mihalic, Y. Fan, W. Xiao, O. Z. Howard, E. Appella, A. T. Maynard and W. L. Farrar, Nat. Med., 2004, 10, 40–47 CrossRef CAS PubMed.
- Q. A. de Paula, S. D. Tsotsoros, Y. Qu, C. A. Bayse and N. P. Farrell, Inorg. Chim. Acta, 2012, 393, 222–229 CrossRef CAS.
- K. D. Zimmer, R. Shoemaker and R. R. Ruminski, Inorg. Chim. Acta, 2006, 359, 1478–1484 Search PubMed.
- J. A. Loo, T. P. Holler, J. Sanchez, R. Gogliotti, L. Maloney and M. D. Reily, J. Med. Chem., 1996, 39, 4313–4320 CrossRef CAS PubMed.
- J. G. Omichinski, G. M. Clore, K. Sakaguchi, E. Appella and A. M. Gronenborn, FEBS Lett., 1991, 292, 25–30 CrossRef CAS PubMed.
- M. d. C. Fernández-Alonso, F. J. Cañada, J. Jiménez-Barbero and G. Cuevas, J. Am. Chem. Soc., 2005, 127, 7379–7386 CrossRef PubMed.
- A. Bazzi, L. Zargarian, F. Chaminade, C. Boudier, H. De Rocquigny, B. Rene, Y. Mely, P. Fosse and O. Mauffret, Nucleic Acids Res., 2011, 39, 3903–3916 CrossRef CAS PubMed.
- M. Mori, U. Dietrich, F. Manetti and M. Botta, J. Chem. Inf. Model., 2010, 50, 638–650 Search PubMed.
- M. Mori, L. Kovalenko, S. Lyonnais, D. Antaki, B. E. Torbett, M. Botta, G. Mirambeau and Y. Mély, in The Future of HIV-1 Therapeutics, Springer, 2015, pp. 53–92 Search PubMed.
- K. E. Riley, M. Pitonak, P. Jurecka and P. Hobza, Chem. Rev., 2010, 110, 5023–5063 CrossRef CAS PubMed.
- J. Sponer, K. E. Riley and P. Hobza, Phys. Chem. Chem. Phys., 2008, 10, 2595 RSC.
- C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 CrossRef CAS PubMed.
- P. B. Lutz and C. A. Bayse, Phys. Chem. Chem. Phys., 2013, 15, 9397–9406 RSC.
- S. Bourbigot, N. Ramalanjaona, C. Boudier, G. F. Salgado, B. P. Roques, Y. Mély, S. Bouaziz and N. Morellet, J. Mol. Biol., 2008, 383, 1112–1128 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- T. Ishida, M. Katsuta, M. Inoue, Y. Yamagata and K. Tomita, Biochem. Biophys. Res. Commun., 1983, 115, 849–854 CrossRef CAS PubMed.
- L. R. Rutledge, L. S. Campbell-Verduyn, K. C. Hunter and S. D. Wetmore, J. Phys. Chem. B, 2006, 110, 19652–19663 CrossRef CAS PubMed.
- A. Tsipis and A. Stalikas, Inorg. Chem., 2012, 51, 2541–2559 CrossRef CAS PubMed.
- G. K. Amarasinghe, R. N. De Guzman, R. B. Turner, K. J. Chancellor, Z. R. Wu and M. F. Summers, J. Mol. Biol., 2000, 301, 491–511 CrossRef CAS PubMed.
- S. C. Keane, X. Heng, K. Lu, S. Kharytonchyk, V. Ramakrishnan, G. Carter, S. Barton, A. Hosic, A. Florwick, J. Santos, N. C. Bolden, S. McCowin, D. A. Case, B. A. Johnson, M. Salemi, A. Telesnitsky and M. F. Summers, Science, 2015, 348, 917–921 CrossRef CAS PubMed.
- W. A. Lea and A. Simeonov, Expert Opin. Drug Discovery, 2011, 6, 17–32 CrossRef CAS PubMed.
- S. Breuer, M. W. Chang, J. Yuan and B. E. Torbett, J. Med. Chem., 2012, 55, 4968–4977 CrossRef CAS PubMed.
- Y. P. Yurenko, J. Novotný, V. Sklenár and R. Marek, Phys. Chem. Chem. Phys., 2014, 16, 2072–2084 RSC.
- N. Goudreau, O. Hucke, A. Faucher, C. Grand-Maître, O. Lepage, P. R. Bonneau, S. W. Mason and S. Titolo, J. Mol. Biol., 2013, 425, 1982–1998 CrossRef CAS PubMed.
- K. B. Turner, N. A. Hagan and D. Fabris, Nucleic Acids Res., 2006, 34, 1305–1316 CrossRef CAS PubMed.
- D. M. Warui and A. M. Baranger, J. Med. Chem., 2012, 55, 4132–4141 CrossRef CAS PubMed.
- D. Garg and B. E. Torbett, Virus Res., 2014, 193, 135–143 CrossRef CAS PubMed.
- Y. Yamagata, M. Kato, K. Odawara, Y. Tokuno, Y. Nakashima, N. Matsushima, K. Yasumura, K. Tomita, K. Ihara and Y. Fujii, Cell, 1996, 86, 311–319 CrossRef CAS PubMed.
- J. Labahn, O. D. Schärer, A. Long, K. Ezaz-Nikpay, G. L. Verdine and T. E. Ellenberger, Cell, 1996, 86, 321–329 CrossRef CAS PubMed.
- A. E. Hodel, P. D. Gershon, X. Shi, S. M. Wang and F. A. Quiocho, Nat. Struct. Biol., 1997, 4, 350–354 CrossRef CAS PubMed.
- J. Marcotrigiano, A. Gingras, N. Sonenberg and S. K. Burley, Cell, 1997, 89, 951–961 CrossRef CAS PubMed.
- K. Kamiichi, M. Doi, M. Nabae, T. Ishida and M. J. Inoue, J. Chem. Soc., Perkin Trans. 2, 1987, 1739–1745 RSC.
- A. G. Stephen, K. M. Worthy, E. Towler, J. A. Mikovits, S. Sei, P. Roberts, Q. Yang, R. K. Akee, P. Klausmeyer and T. G. McCloud, Biochem. Biophys. Res. Commun., 2002, 296, 1228–1237 CrossRef CAS PubMed.
- V. Shvadchak, S. Sanglier, S. Rocle, P. Villa, J. Haiech, M. Hibert, A. Van Dorsselaer, Y. Mély and H. de Rocquigny, Biochimie, 2009, 91, 916–923 CrossRef CAS PubMed.
- D. A. Sartori, B. Miller, U. Bierbach and N. Farrell, JBIC, J. Biol. Inorg. Chem., 2000, 5, 575–583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods including experimental and characterization, molecular modelling and computational studies and control gel shifts. See DOI: 10.1039/c6sc03445d |
| This journal is © The Royal Society of Chemistry 2017 |