
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-Hairpin mimics containing a piperidine–pyrrolidine scaffold modulate the β-amyloid aggregation process preserving the monomer species†
S.
Pellegrino
 *a,
N.
Tonali
b,
E.
Erba
a,
J.
Kaffy
b,
M.
Taverna
c,
A.
Contini
a,
M.
Taylor
d,
D.
Allsop
d,
M. L.
Gelmi
a and
S.
Ongeri
*b
*a,
N.
Tonali
b,
E.
Erba
a,
J.
Kaffy
b,
M.
Taverna
c,
A.
Contini
a,
M.
Taylor
d,
D.
Allsop
d,
M. L.
Gelmi
a and
S.
Ongeri
*b
aDISFARM-Sez. Chimica Generale e Organica “A. Marchesini”, Universitá degli Studi di Milano, via Venezian 21, 20133 Milano, Italy. E-mail: sara.pellegrino@unimi.it
bMolécules Fluorées et Chimie Médicinale, BioCIS, Univ. Paris-Sud, CNRS, Université Paris Saclay, 5 rue Jean-Baptiste Clément, 92296 Châtenay-Malabry Cedex, France. E-mail: Sandrine.ongeri@u-psud.fr
cProtéines et Nanotechnologies en Sciences Séparatives, Institut Galien Paris-Sud, Univ. Paris-Sud, CNRS, Université Paris Saclay, 5 rue Jean-Baptiste Clément, 92296 Châtenay-Malabry Cedex, France
dLancaster University, Division of Biomedical and Life Sciences, Faculty of Health and Medicine, Lancaster LA1 4YQ, UK
First published on 7th October 2016
Abstract
Alzheimer's disease is a neurodegenerative disorder linked to oligomerization and fibrillization of amyloid β peptides, with Aβ1–42 being the most aggregative and neurotoxic one. We report herein the synthesis and conformational analysis of Aβ1–42-amyloid related β-hairpin peptidomimetics, built on a piperidine–pyrrolidine semi rigid β-turn inducer and bearing two small recognition peptide sequences, designed on oligomeric and fibril structures of Aβ1–42. According to these peptide sequences, a stable β-hairpin or a dynamic equilibrium between two possible architectures was observed. These original constructs are able to greatly delay the kinetics of Aβ1–42 aggregation process as demonstrated by thioflavin-T fluorescence, and transmission electron microscopy. Capillary electrophoresis indicates their ability to preserve the monomer species, inhibiting the formation of toxic oligomers. Furthermore, compounds protect against toxic effects of Aβ on neuroblastoma cells even at substoichiometric concentrations. This study is the first example of acyclic small β-hairpin mimics possessing such a highly efficient anti-aggregation activity. The protective effect is more pronounced than that observed with molecules which have undergone clinical trials. The structural elements made in this study provide valuable insights in the understanding of the aggregation process and insights to explore the design of novel acyclic β-hairpin targeting other types of amyloid-forming proteins.
Introduction
Amyloid fibrils are self-assembled insoluble aggregates characterized by highly ordered cross-β structures. They constitute the hallmark of more than 20 serious human amyloidosis diseases, such as Alzheimer's disease (AD), Parkinson's neurodegeneration, type II diabetes and spongiform encephalopathy.1 In particular, AD is associated with the aggregation of the amyloid-β (Aβ1–42) peptide into senile plaques in the brain.2 A large number of small molecules have been proposed for their ability to inhibit or modulate Aβ1–42 aggregation and toxicity. However, the aggregation process is highly complex, and extremely difficult to control.3 Fibrils are able to generate damaging redox activity and promote the nucleation of toxic oligomers.4 Recent studies indicate that soluble transient oligomers preceding fibril formation are highly toxic species.5 Their characterization and the activity of Aβ1–42 aggregation inhibitors on these small and toxic oligomeric species is generally lacking. Thus, the development of inhibitors targeting both oligomerization and fibrillization remains challenging despite its therapeutic significance.4cPeptides are today reasonable alternatives to small molecule pharmaceuticals. They often offer greater efficacy, selectivity, specificity and a reduced risk of unforeseen side-reactions compared to small organic molecules, while some of their pharmacodynamic weaknesses can be circumvented by innovative formulations.6 A variety of small peptides that inhibit aggregation of Aβ and reduce its toxic effects have been already described.7 In particular, inhibition of Aβ-aggregation has been targeted using self-recognition elements (SREs). Indeed, molecules based on fragments of the Aβ-peptide, essentially on the nucleation sequence Aβ16–20 (KLVFF), were found promising as SREs.8 The design of macrocycles β-sheet mimics containing an unnatural tripeptide unit (Nowick's Hao) and SREs, has been a valid strategy.9 To our knowledge, the use of small acyclic β-hairpins has been very rarely explored as β-sheet binders and inhibitors of aggregation.10
Interestingly, compounds possessing several kinetically and thermodynamically accessible local minima representing conformations might be much more powerful inhibitors with respect to rigid ones in modulating protein–protein interactions.11 As Aβ-aggregation is a dynamic and complex process, we hypothesized that flexible β-hairpins could adapt themselves in the interaction with the different Aβ1–42 conformations present during the aggregation process, and in particular in the early stages of oligomerization. For that purpose, we designed two acyclic, β-hairpin mimics G1 and G2 based on the piperidine–pyrrolidine semi-rigid scaffold S1,12 developed recently as a flexible β-turn inducer (Fig. 1), and on different SREs of Aβ1–42. The nucleation sequence Aβ16–20 (KLVFF) has been introduced in the C-terminal sequence of both G1 and G2. However, the choice of the N-terminal sequence was driven by the strategy to develop both a flexible and a more structured β-hairpin. The hydrophobic sequence G33LMVG37, facing K16LVFF20 in the more flexible oligomeric structures13 has been introduced in G1. In G2, GVVIE has been chosen as a mimic of the hydrophobic sequence G38VVIA42, facing K16LVFF20 in the stable fibril structures.14 The alanine residue has been replaced by glutamic acid in order to possibly engage an ionic interaction with the facing lysine residue, thus stabilizing the β-hairpin structure (Fig. 1). The N-terminal amino acid of both G1 and G2 was either acetylated (G1a, G2a) or not (G1b, G2b), in order to evaluate the capacity of the compounds to engage electrostatic interactions with acidic residues of Aβ1–42 and with the view to increase their affinity. Several computational and experimental studies on Aβ1–42 proved in fact that, in addition to the hydrophobic interactions involving in particular the 16–21 sequence (KLVFFA), the formation of a salt-bridge between amino acids Asp23 and Lys28 of amyloid might stabilize a turn motif involving residues 24–28.13 An interaction with Glu22 might be also promoted and beneficial for the activity of the molecules.15
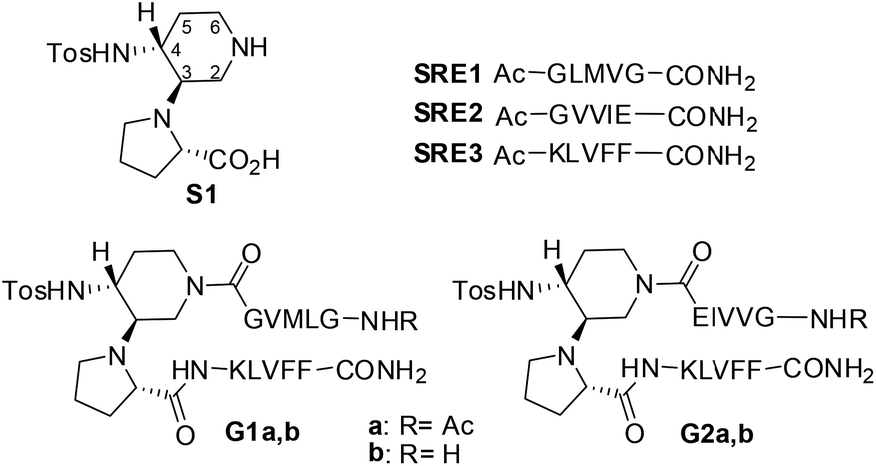 | ||
| Fig. 1 Structure of β-amyloid mimics G1 and G2 and the corresponding SREs. | ||
Results and discussion
Conformational studies and synthesis
In order to evaluate the folding propensity of the designed G1 and G2 β-hairpin mimics, as well as to get preliminary information on their conformational stability, we performed a computational study using replica exchange molecular dynamics (REMD) on G1a and G2a.16–18 Thus, we simulated peptides G1a and G2a using the ff96 force field coupled with the OBC(II) solvent model,19 (see ESI† for additional details). The secondary structure analysis by DSSP20 (Tables S1 and S2, ESI†) showed that both peptides have a relatively high tendency to form anti-parallel β-sheets. G2a seemed to form a very stable β-hairpin, with percentage values of anti-parallel β-sheet content, relatively to non-terminal amino acids, ranging from about 60 to about 90%. G1a was somehow less stable, with an anti-parallel β-sheet content averagely 20% less than G2a. In the H-bond analyses (Tables S3 and S4, ESI†) two pairs of very stable H-bonds, involving the backbone NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O atoms of residues Ile4/Leu8 and Val2/Phe10, were observed for G2a. On the other hand, the occupancies of intramolecular H-bonds detected for G1a were lower. We observed a minor populated hairpin conformation, characterized by the H-bonds involving Val4/Leu8 and Leu2/Phe10, and a major “mismatched” hairpin involving Val4/Val9 and Leu2/Phe11. The representative structures of the most populated cluster for G1a and G2a (Fig. 2) showed a mismatched β-hairpin for the former peptide, with the N-terminal strand (Gly1–Gly5) that was shifted one residue with respect to the C-terminal strand (Lys7–Phe11). Conversely, for G2a, the two strands were perfectly matched. The higher conformational flexibility of G1a, compared to G2a, was also shown by the root mean square deviation (RMSD) analysis of the corresponding REMD trajectories (Fig. S1, ESI†), confirming the possibility of an equilibrium for the former peptide between multiple β-hairpin like conformations, while a single and fairly rigid β-hairpin conformation was predicted for G2a.
O atoms of residues Ile4/Leu8 and Val2/Phe10, were observed for G2a. On the other hand, the occupancies of intramolecular H-bonds detected for G1a were lower. We observed a minor populated hairpin conformation, characterized by the H-bonds involving Val4/Leu8 and Leu2/Phe10, and a major “mismatched” hairpin involving Val4/Val9 and Leu2/Phe11. The representative structures of the most populated cluster for G1a and G2a (Fig. 2) showed a mismatched β-hairpin for the former peptide, with the N-terminal strand (Gly1–Gly5) that was shifted one residue with respect to the C-terminal strand (Lys7–Phe11). Conversely, for G2a, the two strands were perfectly matched. The higher conformational flexibility of G1a, compared to G2a, was also shown by the root mean square deviation (RMSD) analysis of the corresponding REMD trajectories (Fig. S1, ESI†), confirming the possibility of an equilibrium for the former peptide between multiple β-hairpin like conformations, while a single and fairly rigid β-hairpin conformation was predicted for G2a.
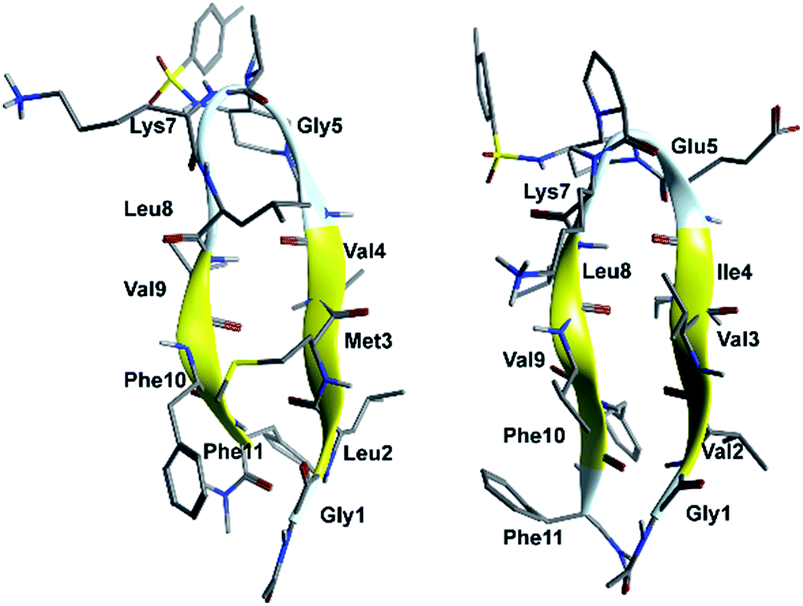 | ||
| Fig. 2 Representative structures of the most populated cluster obtained from cluster analyses of the 302.76 K trajectory of REMD simulations for peptides G1a (left) and G2a (right). | ||
Compounds G1 and G2 were thus prepared by solid phase peptide synthesis, using the Fmoc strategy (see ESI† for details).21 In order to evaluate the efficacy of G1 and G2 molecules with respect to a truncated derivative or the single arms, we also prepared derivative G3 (Fig. 3), containing the scaffold and only the Aβ (16–20) SRE, and compounds SRE1–3 corresponding to the different SREs (Fig. 1, see ESI† for details).
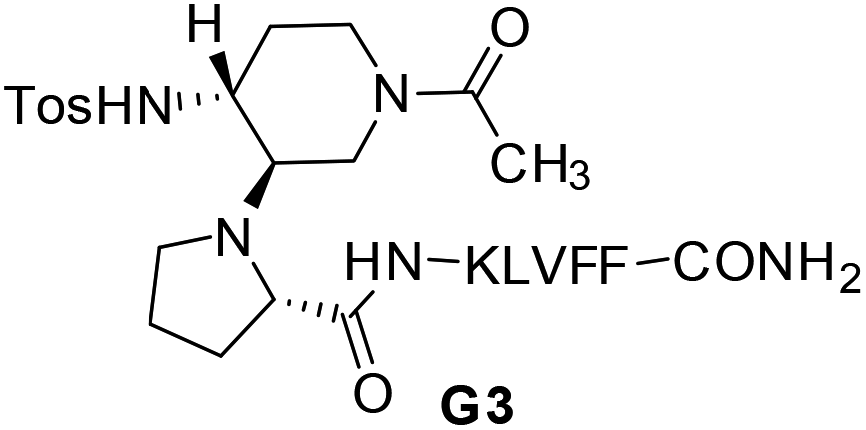 | ||
| Fig. 3 Structure of truncated mimic G3. | ||
The CD spectra of G1a and G2a were recorded in MeOH at 25 °C (Fig. 4). G1a showed a negative band at 195 nm indicating that in solution this peptidomimetic did not assume a preferred, single conformation. On the other hand, the spectrum of G2a was characterized by a strong positive Cotton effect at around 195 nm (π–π* energetic transition), and a negative band at around 215 nm (n–π* energetic transition), typical of β-sheet structures.
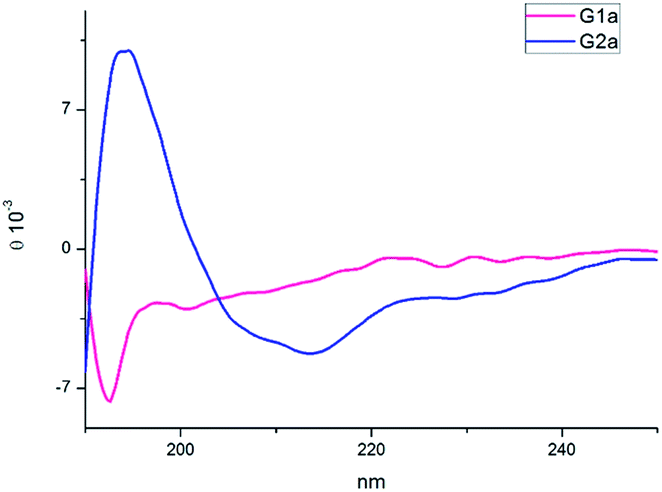 | ||
| Fig. 4 CD spectra of compounds G1a and G2a in MeOH. | ||
The different behaviour of G1a and G2a was confirmed by 1H-NMR experiments in CD3OH (Tables S6–S8 in ESI†). Compound G1a is present in solution as two different β-hairpin structures (G1a-1/G1a-2, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, Fig. 5), characterized by a different alignment of the two peptide arms. This dynamic equilibrium is proved by the presence of several negative NH/NH ROEs (Fig. S4, ESI†).22 On the other hand, 1H NMR spectrum of G2a showed a good dispersion of the NH chemical shifts indicating the presence of a stable single β-hairpin conformation characterized by a peptide arms alignment similar to G1a-2 (Fig. 5 on the bottom).23
:1 ratio, Fig. 5), characterized by a different alignment of the two peptide arms. This dynamic equilibrium is proved by the presence of several negative NH/NH ROEs (Fig. S4, ESI†).22 On the other hand, 1H NMR spectrum of G2a showed a good dispersion of the NH chemical shifts indicating the presence of a stable single β-hairpin conformation characterized by a peptide arms alignment similar to G1a-2 (Fig. 5 on the bottom).23
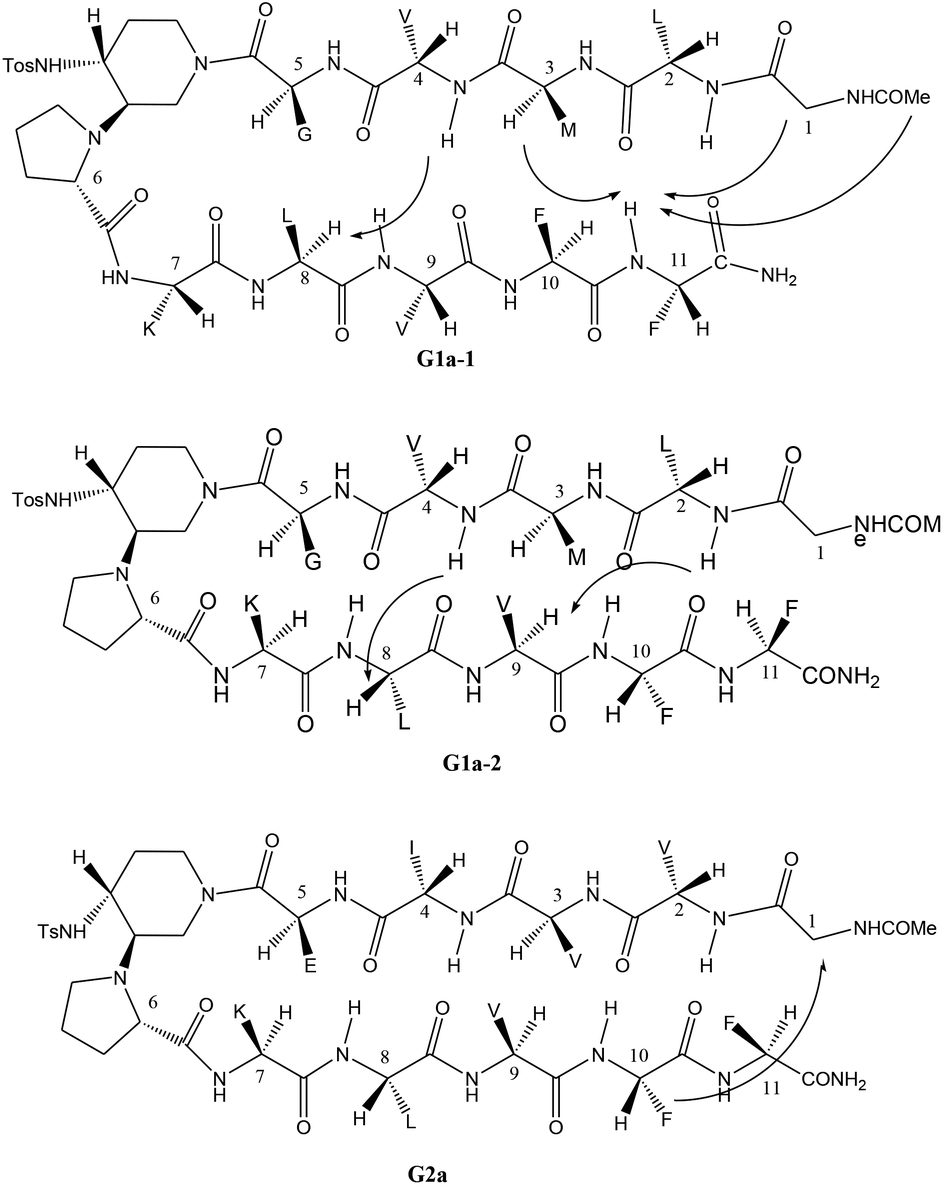 | ||
| Fig. 5 β-Hairpin structures of compounds G1a-1, G1a-2, and G2a, showing the assigned ROEs. | ||
ROESY experiments confirmed the presence of a turn structure in G1a-1, G1a-2, and G2a, as already reported for model sequences (Fig. S5 and S11, ESI†).12a
Several sequential CHα/NH ROEs, indicating β-conformations, were found for both G1a-1 and G1a-2 isomers (Fig. 5 and S6, ESI†). The different alignment of the peptide chains was proven by a ROE between NHPhe11/CHαMet3 in G1a-1, and by another one between NHLeu2/CHαVal9 in G1a-2 (for a complete discussion see ESI†).
Regarding compound G2a we could detect only one β-hairpin diagnostic ROE between CHαGly1 and the phenyl ring of Phe-10 (Fig. 5 and S12, ESI†). Several CHα signals are indeed overlapped or masked by the solvent. The presence of a β-hairpin structure was confirmed by 3JHN/CHα coupling constants that are higher than 8 Hz (Table S9, ESI†).24,25
Finally, the β-hairpin conformation was definitively confirmed for all compounds by the positive difference between experimental Hα chemical shift values and “random” ones26 (Fig. 6). Only Met-3 of G1a-1 is characterized by a negative ΔδαH value. This is probably due to the anisotropic effect27 of the aromatic ring of Phe-11 that faces Met-3, as evicted from ROESY experiments (Fig. S6A, ESI†).
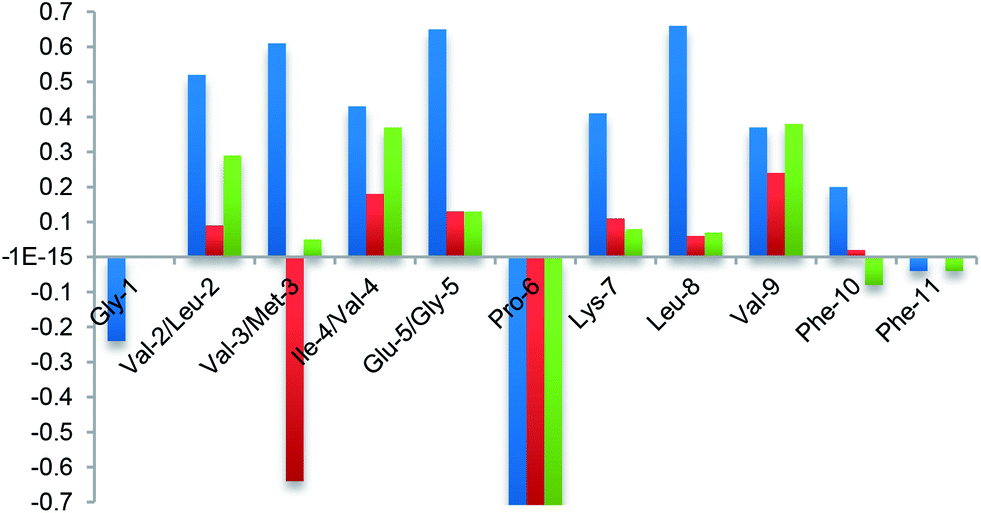 | ||
| Fig. 6 NMR analysis. Plot of difference between Hα chemical shift values in the random coil and the values determined experimentally for G2a (blue) and isomers G1a-1 (red) and G1a-2 (green) in CD3OH at 298 K. | ||
Taking together both experimental and theoretical results, we can conclude that different hairpin architectures are possible for G1a and G2a, depending on the N-terminus sequence. The GVVIE motif in G2a strongly stabilizes a single “matched” hairpin conformation. On the other hand, the GLMVG motif in G1a gave a dynamic equilibrium between two possible architectures, the “mismatched” hairpin being the more stable.
Inhibition of Aβ1–42 fibrillization
The ability of compounds G1–3 and SRE1–3 to interact with Aβ1–42 during the fibrillization process was first studied by thioflavin-T (ThT) fluorescence spectroscopy.28 The fluorescence curve for Aβ1–42 at a concentration of 10 μM followed the typical sigmoid pattern with a lag phase of 4–5 h followed by an elongation phase and a final plateau reached after 10–12 h (Fig. 7a). Two parameters were derived from the ThT curves of Aβ1–42 alone and in the presence of the evaluated compound: (1) t1/2, is defined as the time at which the half maximal ThT fluorescence is observed, which gives insight on the rate of the aggregation process; (2) the fluorescence intensity at the plateau (F) which is assumed to depend on the amount of fibrillar material formed (Table 1).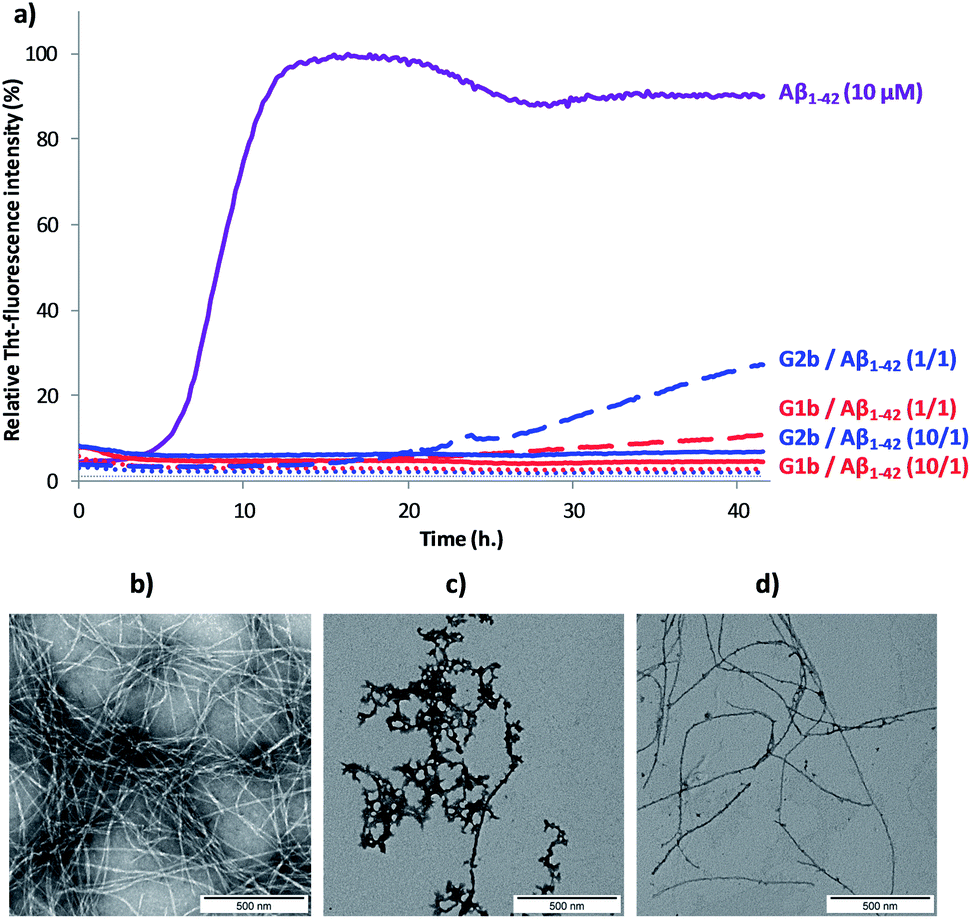 | ||
| Fig. 7 (a) Representative curves of ThT fluorescence assays over time showing Aβ1–42 (10 μM) aggregation in the absence (purple curve) and in the presence of compounds G1b (red curves) and G2b (blue curves) at compound/Aβ1–42 ratios of 10/1 and 1/1. The control curves are represented in dotted lines (G1b in red, G2b in blue and grey for buffer). Fibril formation of Aβ1–42 visualized by TEM: negatively stained images recorded after 42 h of incubation of Aβ1–42 (10 μM in 10 mM Tris·HCl, 100 mM NaCl at pH = 7.4) alone (b) or in the presence of 10 μM of G1b (c) or G2b (d). Scale bars represent 500 nm. | ||
| Compounds (Compound/Aβ ratio) | t 1/2 extensionb | Change of fluorescence intensity at the plateauc (%) |
|---|---|---|
| a NA = no aggregation, parameters are expressed as mean ± SE, n = 3–6. b See ESI for the calculation of the t1/2 extension. A compound displaying a t1/2 increase >1 is a delayer of aggregation. c See ESI for the calculation of the change of fluorescence intensity at the plateau. d Sat means that a saturation of the fluorescence signal is observed because G2a self-aggregates at 100 μM. | ||
| G1a (10/1) | NA | −97 ± 1% |
| G1a (1/1) | 2.06 ± 0.12 | −71 ± 2% |
| G2a (10/1) | Satd | Satd |
| G2a (1/1) | 1.76 ± 0.11 | −41 ± 7% |
| G1b (10/1) | NA | −97 ± 1% |
| G1b (1/1) | NA | −90 ± 2% |
| G2b (10/1) | NA | −95 ± 1% |
| G2b (1/1) | >3.56 ± 0.12 | −73 ± 3% |
Both G1 and G2 series are able to inhibit Aβ1–42 aggregation. The G1 series, containing the sequence G37VMLG33, and possessing a dynamic equilibrium between two different β-hairpin conformations, exerts a slightly superior inhibitory activity (Fig. 7 and Table 1). Furthermore, the free terminal amine is also important for Aβ1–42 aggregation suppression. Unprotected G1b and G2b were indeed able to totally suppress aggregation at compound/Aβ1–42 ratio of 10/1 and still dramatically delayed Aβ1–42 aggregation at 1/1 ratio (Fig. 7a and Table 1). Acetylated derivatives G1a and G2a retained this activity, but to a lesser extent (Table 1 and Fig. S14†). This result supports our hypothesis on the importance of establishing an ionic interaction between the N-terminal amino group and acidic residues of Aβ1–42.
No activity was observed for the isolated pentapeptides GLMVG (SRE1) and GVVIE (SRE2) (Table S11 and Fig. S14†). KLVFF (SRE3) delayed Aβ1–42 aggregation at compound/Aβ1–42 ratio of 10/1,8a,29 however in a much lesser extent than G1 and G2 series, while exerted no activity at 1/1 ratio (Table S11†). The G3 intermediate containing KLVFF linked to the piperidine–pyrrolidine scaffold S1 is more active than SRE3. These results highlight that the piperidine-pyrrolidine scaffold S1 and the pentapeptide KLVFF are both crucial for the activity, but the whole β-hairpin construct is necessary to strongly delay the Aβ1–42 aggregation kinetics.
In order to assess the selectivity on Aβ1–42 peptide, the ability of compounds G1b and G2b to interact with IAPP (islet amyloid polypeptide), an amyloid protein involved in type 2 diabetes mellitus but having another SRE,30 was also tested by the ThT-fluorescence assay under conditions similar to that described for Aβ1–42 peptide. It is noteworthy that both compounds displayed no activity on IAPP fibrillization process at compound/Aβ1–42 ratio of 1/1 and only slightly delayed it at the higher ratio (10/1) (Fig. S15†). This result suggests that the inhibition of aggregation displayed by compounds G1b and G2b on Aβ1–42 peptide is sequence specific.
Transmission electron microscopy (TEM) analyses were performed on the most promising G1a, G1b and G2b compounds. Images were recorded at 20 h and 42 h of fibrillization kinetics with samples containing 10 μM of each compound corresponding to the compound/Aβ1–42 ratio of 1/1 (Fig. 7b–d and S16†). Differences were observed in both quantity and morphology of aggregates formed. At 42 h, a very dense network of fibers displaying a typical morphology was observed for Aβ1–42 alone (Fig. 7b). In the samples containing G1a, the network of fibers was significantly less dense than in the control experiment after 20 h and 42 h. However, the fibers displayed the same morphology (Fig. S16, ESI†). In the samples containing G2b, the same trends as with G1a were observed (Fig. 7d and S16†). In samples containing G1b, we mainly observed globular aggregates after 20 h and 42 h (Fig. 7c and S16) indicating that the aggregation pathway could be different from the one observed for Aβ1–42 alone. These results validated the ThT-fluorescence data, indicating that compounds G1a, G1b and G2b dramatically slowed down the aggregation of Aβ1–42 and efficiently reduced the amount of typical amyloid fibrils.
Inhibition of Aβ1–42 oligomerization
Compounds G1b and G2b were finally studied (at compound/Aβ1–42 ratio of 1/1) by Capillary Electrophoresis (CE) using a method we recently proposed to monitor the very early steps of the oligomerization process overtime and to analyze the effect of drugs on these challenging first stages.31 We focused our attention on three kinds of species: (1) the monomer (peak ES), (2) different small metastable oligomers grouped under peak ES′ and (3) transient species formed later and which correspond to species larger than dodecamers but still soluble (peak LS). Aggregation kinetics of Aβ1–42 peptide alone (Fig. 8a and S18†) showed that overtime, the monomer ES peak decreased in favor of the oligomer peaks ES′ and LS, and that insoluble species, forming spikes in the profile, appeared after 8 hours.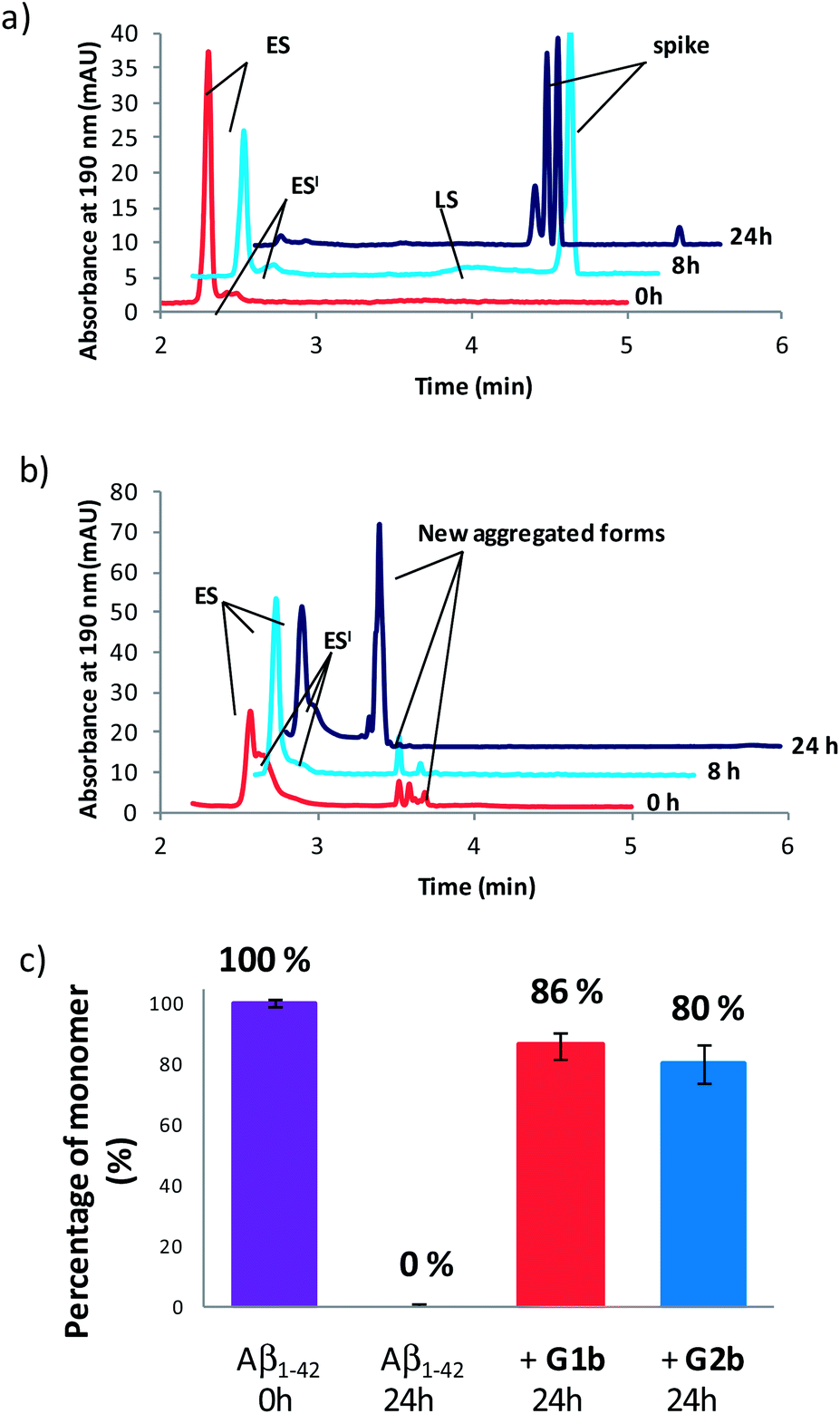 | ||
| Fig. 8 Electrophoretic profile obtained immediately (0 h, red), 8 h (blue) and 24 h (purple) after sample dissolution of Aβ1–42 peptide (100 μM) (a) alone or (b) in the presence of G1b (100 μM). (c) Peak area of the monomer (ES) related to its peak area in the sample of Aβ1–42 alone at 0 h. | ||
In the presence of G1b, the aggregation kinetics of Aβ1–42 peptide was greatly modified (Fig. 8b and S19†). Noteworthy, the monomeric species (peak ES) was dramatically stabilized. 86% of the monomer remained after 24 h in the presence of G1b, while it was no more detected in the control sample (Fig. 8c). Moreover, the larger aggregated species LS (>dodecamers) were not detected. New aggregated forms of Aβ1–42, between ES′ and LS migration times were observed on each electrophoretic profile. We checked that these new aggregated forms were not due to G1b degradation or self-assemblies (Fig. S17A†). They were probably aggregated forms with a different morphology than both LS and those giving spikes observed in Aβ1–42 control. This observation is in accordance with the TEM images where globular aggregates were observed instead of the classical dense network of fibers (Fig. 7c and S16†). In ThT-assays, no fluorescence was detected, indicating that the globular species were not characterized by highly ordered β-structures (Fig. 7a). Remarkably, the presence of the monomer was maintained even after 4 days (Fig. S19B†). We concluded that G1b is able to prevent the formation of toxic soluble oligomers of Aβ1–42 peptide and to maintain the presence of the non toxic monomer overtime.
G2b also dramatically maintained the presence of the monomer (peak ES, 80% after 24 h, Fig. 8c, S20 and S21†). However, new aggregated forms were only transiently observed but were not anymore detected after 24 h. This result was also in accordance with the TEM images where we observed a much less dense network of fibers, although the typical morphology was retained.
Protection against Aβ1–42 cell toxicity
The inhibitors were investigated to determine their ability to reduce the toxicity of aggregated Aβ1–42 to SH-SY5Y neuroblastoma cells. The addition of all compounds, to a lesser extent for G2b, showed a protective effect on cell survival (MTS assay, Fig. 9) and membrane damage (LDH membrane integrity assay, Fig. 10) in the presence of cytotoxic 5 μM Aβ1–42. Remarkably, this protective effect was seen at equimolar amounts of inhibitor to Aβ1–42 and was still significant at a very low ratio of 0.1/1 (inhibitor/Aβ1–42) in the MTS assay. Both G2a and G1b showed a slight negative effect on cell viability when incubated with cells alone, although this was negated when Aβ was present.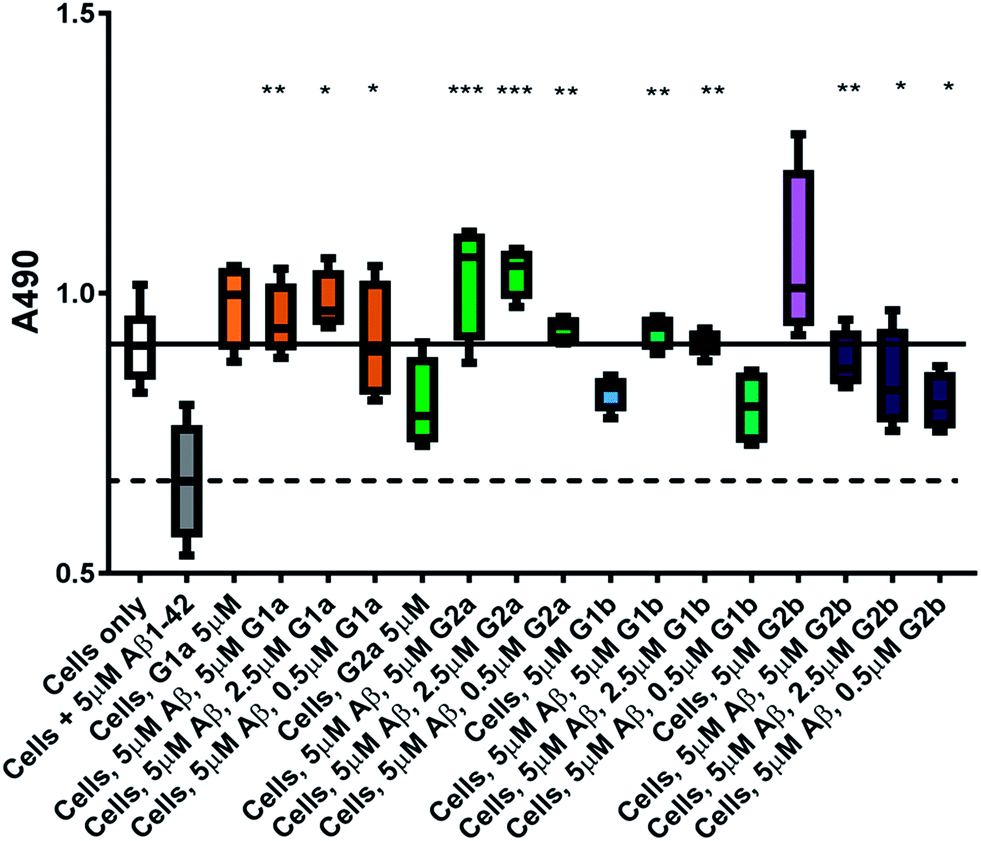 | ||
| Fig. 9 Cell viability assay results. The solid line represents the absorbance value seen for cells incubated without Aβ1–42 (white box) and the dotted line that seen for cells incubated with 5 μM Aβ1–42 (grey box). A statistically significant difference between Aβ1–42 treated cells with and without inhibitor is indicated by */**/*** corresponding to p > 0.05/0.01/0.001. n = 4 for each condition. | ||
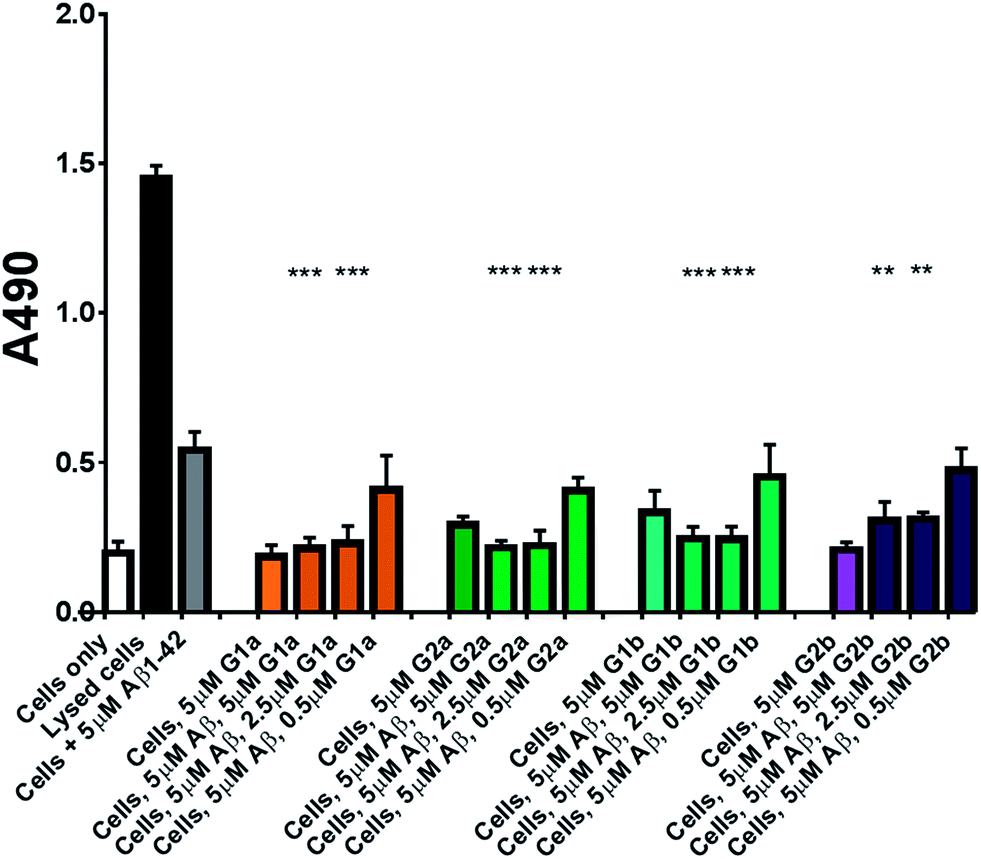 | ||
| Fig. 10 LDH based cell toxicity test. Cells were treated in the same manner as with the MTS assay, and cell proliferation was measured using the CytoTox 96® NonRadioactive Cytotoxicity Assay Protocol from Promega. Statistical analysis was performed using a Student's test comparing the results for cells exposed to 5 μM Aβ1–42 with and without inhibitor where ** = p < 0.01 and *** = p < 0.001. | ||
This protective effect is more marked than that observed with molecules which have undergone clinical trials32–34 or other molecules recently described as efficient reducers of Aβ1–42 toxicity.35 In particular, in the literature, resveratrol was reported to protect SH-SY5Y neuroblastoma cells from Aβ1–42 toxicity at 10/1 and 2/1 (resveratrol/Aβ1–42) ratios,32 scyllo-inositol was demonstrated to protect PC-12 cells at 10/1 ratio (scyllo-inositol/Aβ1–42),33 and (−)-epigallocatechin-3-gallate (EGCG) protected murine neuro-2a neuroblastoma cells at 1/1 ratio (−)-epigallocatechin-3-gallate/Aβ1–42.34 In our hands, and comparable to the published data,32 resveratrol efficiently protected SH-SY5Y neuroblastoma cells only at a ratio of 2/1 (resveratrol/Aβ1–42). A stoichiometric ratio 1/1 was less efficient than a substoichiometric ratio of G1b and G2b (0.5/1 compound/Aβ1–42) (Fig. 11). Resveratrol exhibits multi-target activity and thus is not selective for Aβ1–42 aggregation. For example, resveratrol inhibits similarly the aggregation of other amyloid proteins such as IAPP36 (EGCG also inhibits similarly Aβ1–42 and IAPP aggregation in ThT fluorescence assays37,38), which is not the case for G1b and G2b, as mentioned above. By choosing the SREs in our β-hairpin mimics, specifically according to the target amyloid proteins, we can modulate the activity and expect selective activities.
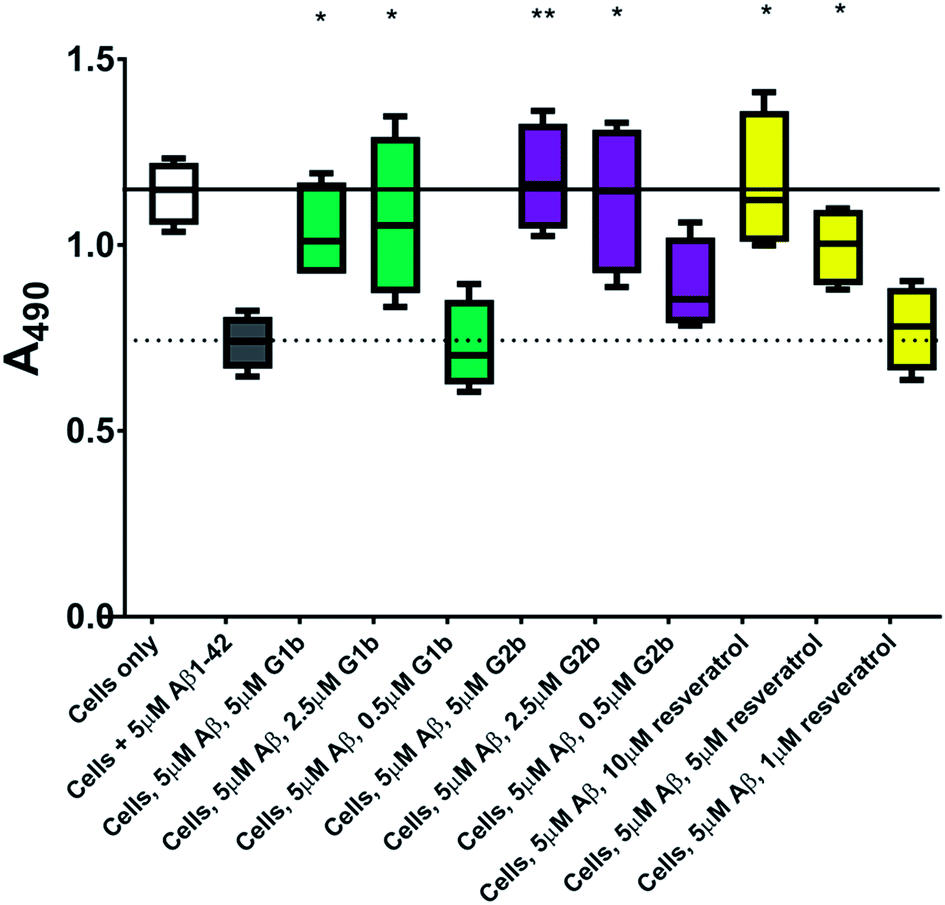 | ||
| Fig. 11 Cell viability assay results of resveratrol compared to G1b and G2b. The solid line represents the mean absorbance value seen for cells incubated without Aβ1–42 (white box) and the dotted line that seen for cells incubated with 5 μM Aβ1–42 (grey box). A statistically significant difference between Aβ1–42 treated cells with and without inhibitor is indicated by */**/*** corresponding to p > 0.05/0.01/0.001. n = 4 for each condition. | ||
Conclusion
We described new β-hairpin mimics designed on oligomeric and fibril structures of Aβ1–42 and containing a piperidine–pyrrolidine β-turn inducer. The presence of two small recognition sequences able to engage both hydrophobic and ionic interactions with Aβ1–42, dramatically increased the inhibitory effect on the fibrillization process. Furthermore, the presence of the semi-rigid piperidine–pyrrolidine scaffold S1 and of the hydrophobic sequence G33LMVG37, which allows a dynamic equilibrium between different architectures, leads to the obtainment of compound G1b able to inhibit totally the formation of amyloid fibrils. As far as we know, this study is the first example of acyclic small β-hairpin mimics possessing such a highly efficient anti-aggregation activity. This activity is much higher than isolated SREs described in the literature. Furthermore, to the best of our knowledge, this is the first example of compounds able to dramatically preserve the non toxic monomer species of Aβ1–42. This result might explain the mechanism by which β-hairpin mimics exhibit a strong protective effect on cells even at substoichiometric concentrations. The structural elements made in this study provide valuable insights to explore the design of novel acyclic β-hairpin targeting other types of amyloid-forming proteins.Acknowledgements
Géraldine Toutirais (Institut de Biologie Paris Seine (IBPS)/FR3631, Service de Microscopie Electronique, Université Pierre et Marie Curie, France) is acknowledged for her advice in TEM experiments. The Ministère de l'Enseignement Supérieur et de la Recherche (MESR) and the Alzheimer's Society UK are thanked for financial support for N. Tonali and M. Taylor/D. Allsop respectively. The Laboratory BioCIS is a member of the Laboratory of Excellence LERMIT supported by a Grant from ANR (ANR-10-LABX-33).Notes and references
- (a) M. Stefani and C. M. Dobson, J. Mol. Med., 2003, 81, 678–699 CrossRef CAS PubMed; (b) F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed; (c) A. Aguzzi and T. O'Connor, Nat. Rev. Drug Discovery, 2010, 9, 237–248 CrossRef CAS PubMed; (d) Y. S. Eisele, C. Monteiro, C. Fearns, S. E. Encalada, R. L. Wiseman, E. T. Powers and J. W. Kelly, Nat. Rev. Drug Discovery, 2015, 14, 759–780 CrossRef CAS PubMed.
- (a) M. Goedert and M. G. A. Spillantini, Science, 2006, 314, 777–780 CrossRef CAS PubMed; (b) C. Haas and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef PubMed.
- (a) F. Belluti, A. Rampa and S. Gobbi, Expert Opin. Ther. Pat., 2013, 23, 581–596 CrossRef CAS PubMed; (b) T. Härd and C. Lendel, J. Mol. Biol., 2012, 421, 441–465 CrossRef PubMed; (c) A. J. Doig and P. Derreumaux, Curr. Opin. Struct. Biol., 2015, 30, 50–56 CrossRef CAS PubMed.
- (a) J. Mayes, C. Tinker-Mill, O. Kolosov, H. Zhang, B. J. Tabner and D. Allsop, J. Biol. Chem., 2014, 289, 12052–12062 CrossRef CAS PubMed; (b) S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed; (c) M. J. Guerrero-Muñoza, D. L. Castillo-Carranza and R. Kayed, Biochem. Pharmacol., 2014, 88, 468–478 CrossRef PubMed.
- (a) C. Haas and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 101–112 CrossRef PubMed; (b) K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef CAS PubMed; (c) P. Prangkio, E. C. Yusko, D. Sept, J. Yang and M. Mayer, PLoS One, 2012, 7, e47261 CAS; (d) P. Cizas, R. Budvytytec, R. Morkuniene, R. Moldovan, M. Broccio, M. Lösche, G. Niaura, G. Valincius and V. Borutait, Arch. Biochem. Biophys., 2010, 496, 84–92 CrossRef CAS PubMed; (e) I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef CAS PubMed.
- (a) P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed; (b) F. Albericio and H. G. Kruger, Future Med. Chem., 2012, 4, 1527–1531 CrossRef CAS PubMed; (c) T. Uhlig, T. Kyprianou, F. G. Martinelli, C. A. Oppici, D. Heiligers, D. Hills, X. R. Calvo and P. Verhaert, EuPa Open Proteomics, 2014, 4, 58–69 CrossRef CAS.
- (a) S. Aileen Funke and D. Willbold, Curr. Pharm. Des., 2012, 18, 755–767 CrossRef; (b) M. Chemerovski-Glikman, M. Richman and S. Rahimipour in Topics in Heterocyclic Chemistry, Springer, Berlin, Heidelberg, 2016, pp. 1–32 Search PubMed.
- (a) L. O. Tjenberg, J. Näslund, F. Lindqvist, A. R. Karlström, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef; (b) C. I. Stains, K. Mondal and I. Ghosh, ChemMedChem, 2007, 2, 1674–1692 CrossRef CAS PubMed; (c) T. Takahashi and H. Mihara, Acc. Chem. Res., 2008, 41, 1309–1318 CrossRef CAS PubMed; (d) B. Neddenriep, A. Calciano, D. Conti, E. Sauve, M. Paterson, E. Bruno and D. A. Moffet, Open Biotechnol. J., 2011, 5, 39–46 CrossRef CAS PubMed; (e) J. Kumar, R. Namsechi and V. L. Sim, PLoS One, 2015, 10, e0129087 Search PubMed; (f) V. Parthsarathy, P. L. McClean, C. Hölscher, M. Taylor, C. Tinker, G. Jones, O. Kolosov, E. Salvati, M. Gregori, M. Masserini and D. Allsop, PLoS One, 2013, 8, e54769 CAS.
- P.-N. Cheng, C. Liu, M. Zhao, D. Eisenberg and J. S. Nowick, Nat. Chem., 2012, 4, 927–933 CrossRef CAS PubMed.
- (a) G. Hopping, J. Kellock, B. Caughey and V. Daggett, ACS Med. Chem. Lett., 2013, 4, 824–828 CrossRef CAS PubMed; (b) K. N. L. Huggins, M. Bisaglia, L. Bubacco, M. Tatarek-Nossol, A. Kapurniotu and N. H. Andersen, Biochemistry, 2011, 50, 8202–8212 CrossRef CAS PubMed; (c) Y. Hamada, N. Miyamoto and Y. Kiso, Bioorg. Med. Chem. Lett., 2015, 25, 1572–1576 CrossRef CAS PubMed.
- (a) X. Li, J. Taechalertpaisarn, D. Xin and K. Burgess, Org. Lett., 2015, 17, 632–635 CrossRef CAS PubMed; (b) E. Ko, A. Raghuraman, L. M. Perez, T. R. Ioerger and K. Burgess, J. Am. Chem. Soc., 2012, 135, 167–173 CrossRef PubMed.
- (a) S. Pellegrino, A. Contini, M. L. Gelmi, L. Lo Presti, R. Soave and E. Erba, J. Org. Chem., 2014, 79, 3094–3102 CrossRef CAS PubMed; (b) E. Erba and A. Contini, RSC Adv., 2012, 2, 10652–10660 RSC.
- M. Ahmed, J. David, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostran and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CAS.
- T. Lührs, C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17342–17347 CrossRef PubMed.
- K. Hochdörffer, J. März-Berberich, L. Nagel-Steger, M. Epple, W. Meyer-Zaika, A. H. C. Horn, H. Sticht, S. Sinha, G. Bitan and T. Schrader, J. Am. Chem. Soc., 2011, 133, 4348–4358 CrossRef PubMed.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- (a) F. Ding, D. Tsao, H. Nie and N. V. Dokholyan, Structure, 2008, 16, 1010–1018 CrossRef CAS PubMed; (b) E. Lin and M. S. Shell, J. Chem. Theory Comput., 2009, 5, 2062–2073 CrossRef CAS PubMed; (c) P. Tao, J. R. Parquette and C. M. Hadad, J. Chem. Theory Comput., 2012, 8, 5137–5149 CrossRef CAS PubMed.
- (a) S. Pellegrino, A. Contini, F. Clerici, A. Gori, D. Nava and M. L. Gelmi, Chem.–Eur. J., 2012, 18, 8705–8715 CrossRef PubMed; (b) A. Ruffoni, A. Contini, R. Soave, L. Lo Presti, I. Esposto, I. Maffucci, D. Nava, S. Pellegrino, M. L. Gelmi and F. Clerici, RSC Adv., 2015, 32643–32656 RSC; (c) I. Maffucci, S. Pellegrino, J. Clayden and A. Contini, J. Phys. Chem. B, 2015, 119, 1350–1361 CrossRef CAS PubMed; (d) I. Maffucci, J. Clayden and A. Contini, J. Phys. Chem. B, 2015, 119, 14003–14013 CrossRef CAS PubMed.
- (a) I. Maffucci and A. Contini, J. Chem. Theory Comput., 2016, 12, 714–727 CrossRef CAS PubMed; (b) A. Onufriev, D. Bashford and D. A. Case, Proteins: Struct., Funct., Bioinf., 2004, 55, 383–394 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- (a) S. Pellegrino, C. Annoni, A. Contini, F. Clerici and M. L. Gelmi, Amino Acids, 2012, 43, 1995–2003 CrossRef CAS PubMed; (b) S. Pellegrino, G. Facchetti, A. Contini, M. L. Gelmi, E. Erba, R. Gandolfi and I. Rimoldi, RSC Adv., 2016, 6, 71529–71533 RSC.
- P. Cuniasse, I. Raynal, A. Yiothakis and V. Dive, J. Am. Chem. Soc., 1997, 119, 5239–5248 CrossRef CAS.
- According to the ref. 12a, in both cases the piperidine scaffold is present as mixture of conformers, making the attribution of piperidine/proline resonances very complicated. As a result, some signals are tentatively assigned.
- (a) R. Zerella, Protein Sci., 1999, 8, 1320–1331 CrossRef CAS PubMed; (b) L. J. Smith, K. A. Bolin, H. Schwalbe, M. W. Macarthur, J. M. Thornton and C. M. Dobson, J. Mol. Biol., 1996, 255, 494–506 CrossRef CAS PubMed.
- L. De Rosa, D. Diana, A. Basile, A. Russomanno, C. Isernia, M. C. Turco, R. Fattorusso and L. D. D'Andrea, Eur. J. Med. Chem., 2014, 73, 210–216 CrossRef CAS PubMed.
- (a) D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647–1651 CrossRef CAS PubMed; (b) D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS PubMed.
- A. M. Fernandez-Escamilla, S. Ventura, L. Serrano and M. A. Nez, Protein Sci., 2006, 15, 2278–2289 CrossRef CAS PubMed.
- H. LeVine, Methods Enzymol., 1999, 309, 274–284 CAS.
- (a) M. A. Findeis, G. M. Musso, C. C. Arico-Muendel, H. W. Benjamin, A. M. Hundal, J.-J. Lee, J. Chin, M. Kelley, J. Wakefield, N. J. Hayward and S. M. Molineaux, Biochemistry, 1999, 38, 6791–6800 CrossRef CAS PubMed; (b) N. Kokkoni, K. Stott, H. Amijee, J. M. Mason and A. J. Doig, Biochemistry, 2006, 45, 9906–9918 CrossRef CAS PubMed; (c) S. M. Chafekar, H. Malda, M. Merkx, E. W. Meijer, D. Viertl, H. A. Lashuel, F. Baas and W. Scheper, ChemBioChem, 2007, 8, 1857–1864 CrossRef CAS PubMed.
- (a) M. S. Fernández, Cell Calcium, 2014, 56, 416–427 CrossRef PubMed; (b) R. Akter, P. Cao, H. Noor, Z. Ridgway, L.-H. Tu, H. Wang, A. G. Wong, X. Zhang, A. Abedini, A. Schmidt and D. P. Raleigh, J. Diabetes Res., 2016, 2798269, DOI:10.1155/2016/2798269.
- (a) D. Brinet, J. Kaffy, F. Oukacine, S. Glumm, S. Ongeri and M. Taverna, Electrophoresis, 2014, 35, 3302–3309 CrossRef CAS PubMed; (b) J. Kaffy, D. Brinet, J.-L. Soulier, I. Correia, N. Tonali, K. F. Fera, Y. Iacone, A. R. F. Hoffmann, L. Khemtemourian, B. Crousse, M. Taylor, D. Allsop, M. Taverna, O. Lequin and S. Ongeri, J. Med. Chem., 2016, 59, 2025–2040 CrossRef CAS PubMed.
- Y. Feng, X.-P. Wanga, S.-G. Yang, Y.-J. Wan, X.-T. Du, X. Zhang, X.-X. Sun, M. Zhao, L. Huang and R.-T. Liu, Neurotoxicology, 2009, 30, 986–995 CrossRef CAS PubMed.
- S. Sinha, Z. Du, P. Maiti, F.-G. Klärner, T. Schrader, C. Wang and G. Bitan, ACS Chem. Neurosci., 2012, 3, 451–458 CrossRef CAS PubMed.
- S.-J. Hyunga, A. S. DeTomaa, J. R. Brendera, S. Leec, S. Vivekanandana, A. Kochia, J.-S. Choic, A. Ramamoorthya, B. T. Ruotoloa and M. H. Lima, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3743–3748 CrossRef PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Grüning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoeg, M. Fändrich, R. F. Frank, B. Reif, S. Günther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- L.-H. Tu, L. M. Young, A. G. Wong, A. E. Ashcroft, S. E. Radford and D. P. Raleigh, Biochemistry, 2015, 54, 666–676 CrossRef CAS PubMed.
- F. Meng, A. Abedini, A. Plesner, C. B. Verchere and D. P. Raleigh, Biochemistry, 2010, 49, 8127–8133 CrossRef CAS PubMed.
- S. Chan, S. Kantham, V. M. Rao, M. K. Palanivelu, H. L. Pham, P. N. Shaw, R. P. McGeary and B. P. Ross, Food Chem., 2016, 199, 185–194 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods and additional figures and tables. NMR additional data. Description of synthetic procedures and characterization of compounds. Experimental procedure for fluorescence-detected ThT binding assay; representative curves of ThT fluorescence assays. Experimental procedure for TEM studies, CE, and cellular evaluation. See DOI: 10.1039/c6sc03176e |
| This journal is © The Royal Society of Chemistry 2017 |