
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating charge transfer excited state relaxation and spin crossover in iron coordination complexes with ligand substitution†
Wenkai
Zhang‡
a,
Kasper S.
Kjær‡
*abc,
Roberto
Alonso-Mori
d,
Uwe
Bergmann
ad,
Matthieu
Chollet
d,
Lisa A.
Fredin
 e,
Ryan G.
Hadt
f,
Robert W.
Hartsock
af,
Tobias
Harlang
b,
Thomas
Kroll
df,
Katharina
Kubiček
g,
Henrik T.
Lemke
d,
Huiyang W.
Liang
ad,
Yizhu
Liu
b,
Martin M.
Nielsen
c,
Petter
Persson
e,
Joseph S.
Robinson
d,
Edward I.
Solomon
df,
Zheng
Sun
a,
Dimosthenis
Sokaras
h,
Tim B.
van Driel
c,
Tsu-Chien
Weng
h,
Diling
Zhu
d,
Kenneth
Wärnmark
i,
Villy
Sundström
b and
Kelly J.
Gaffney
*a
e,
Ryan G.
Hadt
f,
Robert W.
Hartsock
af,
Tobias
Harlang
b,
Thomas
Kroll
df,
Katharina
Kubiček
g,
Henrik T.
Lemke
d,
Huiyang W.
Liang
ad,
Yizhu
Liu
b,
Martin M.
Nielsen
c,
Petter
Persson
e,
Joseph S.
Robinson
d,
Edward I.
Solomon
df,
Zheng
Sun
a,
Dimosthenis
Sokaras
h,
Tim B.
van Driel
c,
Tsu-Chien
Weng
h,
Diling
Zhu
d,
Kenneth
Wärnmark
i,
Villy
Sundström
b and
Kelly J.
Gaffney
*a
aPULSE Institute, SLAC National Accelerator Laboratory, Stanford University, Menlo Park, California 94025, USA. E-mail: kaspersk@gmail.com; kgaffney@slac.stanford.edu
bDepartment of Chemical Physics, Lund University, P.O. Box 12 4, 22100 Lund, Sweden
cCentre for Molecular Movies, Department of Physics, Technical University of Denmark, DK-2800, Lyngby, Denmark
dLCLS, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA
eTheoretical Chemistry Division, Lund University, P.O. Box 124, 22100 Lund, Sweden
fDepartment of Chemistry, Stanford University, Stanford, California 94305, USA
gMax Planck Institute for Biophysical Chemistry, 37077, Göttingen, Germany
hSSRL, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA
iCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, 22100 Lund, Sweden
First published on 25th August 2016
Abstract
Developing light-harvesting and photocatalytic molecules made with iron could provide a cost effective, scalable, and environmentally benign path for solar energy conversion. To date these developments have been limited by the sub-picosecond metal-to-ligand charge transfer (MLCT) electronic excited state lifetime of iron based complexes due to spin crossover – the extremely fast intersystem crossing and internal conversion to high spin metal-centered excited states. We revitalize a 30 year old synthetic strategy for extending the MLCT excited state lifetimes of iron complexes by making mixed ligand iron complexes with four cyanide (CN−) ligands and one 2,2′-bipyridine (bpy) ligand. This enables MLCT excited state and metal-centered excited state energies to be manipulated with partial independence and provides a path to suppressing spin crossover. We have combined X-ray Free-Electron Laser (XFEL) Kβ hard X-ray fluorescence spectroscopy with femtosecond time-resolved UV-visible absorption spectroscopy to characterize the electronic excited state dynamics initiated by MLCT excitation of [Fe(CN)4(bpy)]2−. The two experimental techniques are highly complementary; the time-resolved UV-visible measurement probes allowed electronic transitions between valence states making it sensitive to ligand-centered electronic states such as MLCT states, whereas the Kβ fluorescence spectroscopy provides a sensitive measure of changes in the Fe spin state characteristic of metal-centered excited states. We conclude that the MLCT excited state of [Fe(CN)4(bpy)]2− decays with roughly a 20 ps lifetime without undergoing spin crossover, exceeding the MLCT excited state lifetime of [Fe(2,2′-bipyridine)3]2+ by more than two orders of magnitude.
Introduction
Understanding how ground state molecular structure dictates the electronic excited state behavior of inorganic complexes has the potential to enhance the performance of molecular-based materials for solar energy applications. Inorganic complexes have multiple properties – potential for strong visible light absorption and photocatalytic activity – that have motivated investigations of their electronic excited state properties,1–3 but the current understanding of the interplay between ground state structure and electronic excited state dynamics remains rudimentary. The important role of metal-centered excited states, often termed ligand field excited states,4 distinguishes the electronic excited state properties of transition metal-based coordination complexes and organic dyes. While selection rules make the cross-section for direct optical excitation of ligand field excited states very small, they often control the non-adiabatic relaxation dynamics of optically generated charge transfer excited states.4,5 While these general properties indicate that controlling the relative energy of charge transfer and ligand field excited states becomes an important means to influence the electronic excited state dynamics of coordination complexes, which can be achieved by changing the coordinating ligand field strength, additional issues must be considered. Thermodynamically favorable electronic states can be avoided if the internal conversion or intersystem crossing does not have an energetically accessible path from the Franck–Condon region of the optically generated excited state. Only through a clear understanding of all the relevant electronic excited state potential energy surfaces can we acquire a thorough understanding of the molecular properties that control excited state relaxation. This would set the stage for controlling these excited state dynamics, either through the suppression of internal conversion and intersystem crossing in solar applications6–11 or accelerating spin crossover in data storage applications.12–14Characterizing the relaxation dynamics of charge transfer excited states with mechanistic detail represents an important initial step to determine how ground state molecular structure influences the dynamics of excited state internal conversion and intersystem crossing. A wide array of pump-probe measurements in pseudo-octahedral polypyridal iron coordination complexes shows that metal-to-ligand charge transfer (MLCT) excited states undergo internal conversion and intersystem crossing from the MLCT state to the metal-centered high spin quintet (5MC) excited state on a sub-picosecond time scale.7,15–29 This photo-excited spin crossover involves two electronic transitions and two electronic spin flips, a surprisingly large number of charge and spin density changes to occur so promptly. Nonetheless, the weight of experimental support for ultrafast spin crossover has eliminated all doubt about the ultrafast rate of spin crossover.7,15,18,19,21,22 More recent measurements and theoretical calculations have focused on the mechanism of spin crossover. Though still debated,30 ultrafast X-ray fluorescence and theoretical calculations provide support for a stepwise spin crossover mechanism,22,31–33 where the MLCT excited state transitions to the 5MC excited state through a metal-centered triplet state (3MC). Determining whether MLCT excited state properties and spin crossover can be controlled with ligand substitution provides the focus of this manuscript.
The need to extend the excited state lifetime of iron-based dye sensitizers has long been appreciated, but progress has been slow. Winkler and Sutin investigated mixed ligand Fe complexes based on 2,2′-bipyridine and cyanide ligands.34,35 Substituting 2,2′-bipyridine ligands with larger ligand field strength cyanide anions enables the destabilization of the ligand field excited states while stabilizing the MLCT excited state. Initial studies indicated that MLCT excited state lifetimes could be extended, but the time resolution of the measurements made the result inconclusive. Ferrere and Gregg,10,11 and later Yang et al.,8,9 used these mixed ligand complexes as photo-sensitizers in dye-sensitized solar cells. The performance of these cells failed to compete with Ru dye sensitized cells, though the cause of the poorer performance remains unclear. More recently, Sundström, Wärnmark and co-workers synthesized and characterized the excited state dynamics of a series of iron carbene complexes.36–38 These iron carbene sensitized cells showed high charge injection yields, but low power conversion due to fast electron–hole recombination.
We investigate the relaxation dynamics of MLCT excitations in [Fe(CN)4(bpy)]2− to determine the impact of ligand field strength on the relative energy of MLCT and ligand field excited states. From an electronic structure perspective, we need to robustly distinguish the relevant charge transfer and metal-centered electronic excited states and the rate with which they interconvert. We have achieved this goal by using two complementary methods to probe the ultrafast electronic relaxation dynamics initiated by a visible pump pulse. Using the X-ray Pump-Probe (XPP) endstation39 of the Linear Coherent Light Source (LCLS) we have conducted femtosecond resolution iron 3p–1s (Kβ) X-ray fluorescence measurements. The transient Kβ X-ray fluorescence allows us to track the time evolution of the Fe spin moment,40–44 as recently demonstrated for the spin crossover mechanism in [Fe(bpy)3]2+.22,27,28 This technique provides a robust means of characterizing the role of ligand field excited states. We also use femtosecond UV-visible spectroscopy to track the decay dynamics for the MLCT excited state via the 2,2′-bipyridine anion excited state absorption and electronic ground state bleach. The characteristic excited state absorption of the 2,2′-bipyridine radical anion has proven to be a useful indicator of the MLCT excited state.7,18,38 These X-ray and optical probes complement one another and lead to a robust interpretation of the observed dynamics. From these measurements we demonstrate that spin crossover is suppressed for [Fe(CN)4(bpy)]2− which leads to more than a hundred-fold increase in the MLCT excited state lifetime compared to [Fe(bpy)3]2+.
Results and discussion
Fig. 1 shows the UV-visible absorption spectrum of [Fe(bpy)3]2+ and [Fe(CN)4(bpy)]2−. Note the large red-shift of the MLCT excited state absorption features of [Fe(CN)4(bpy)]2− relative to [Fe(bpy)3]2+ indicative of MLCT excited state stabilization in [Fe(CN)4(bpy)]2− when dissolved in non-protic polar solvents.45 We have photo-excited the molecule in the lowest energy MLCT excited state absorption band.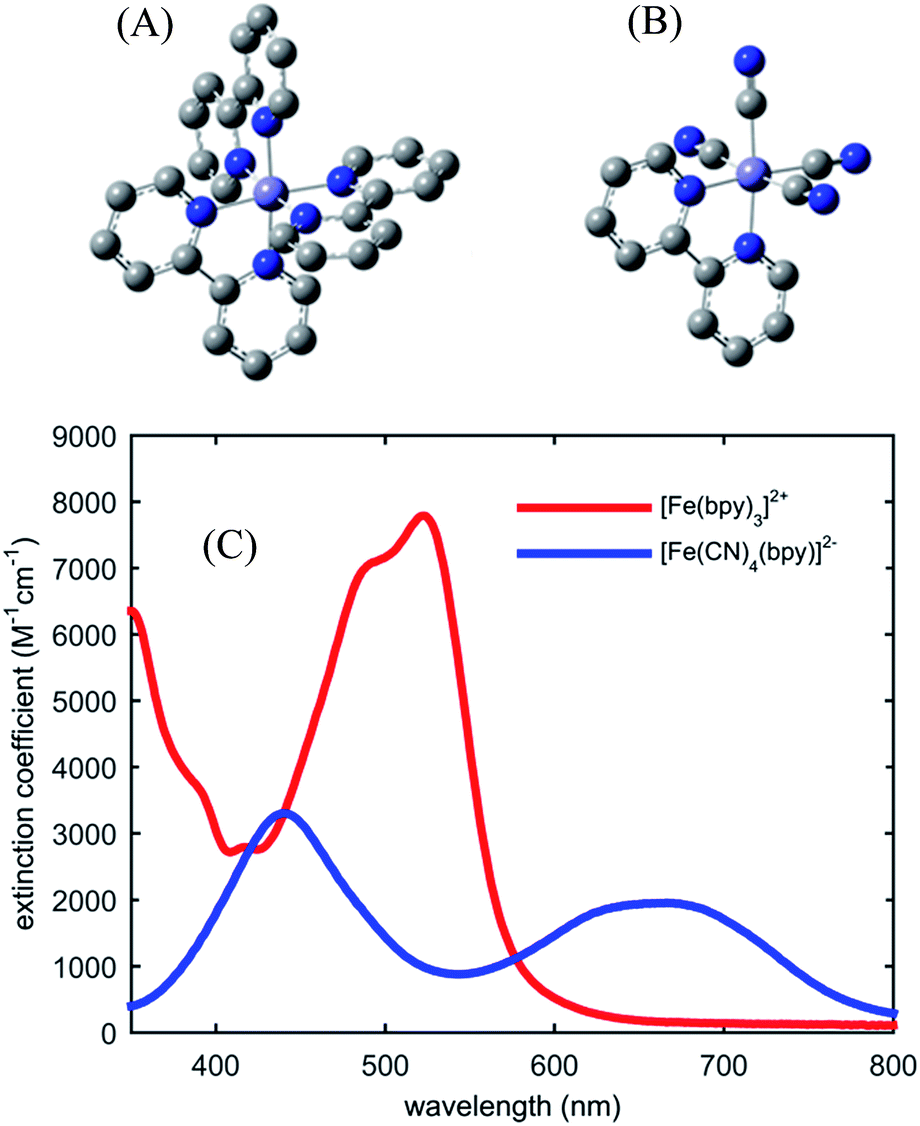 | ||
| Fig. 1 Molecular structure of investigated iron coordination complexes (A) [Fe(bpy)3]2+, (B) [Fe(CN)4(bpy)]2−. Hydrogen atoms are not shown. (C) The UV-visible absorption spectra of [Fe(bpy)3]2+ (red) and [Fe(CN)4(bpy)]2− (blue) in dimethylsulfoxide. | ||
Fig. 2(A) shows the spin-dependent Fe Kβ fluorescence spectra of Fe complexes with spin moments mirroring that of the ground state, and the possible excited states of [Fe(CN)4(bpy)]2−. The Fe Kβ fluorescence involves 3p filling of the 1s hole. The strong exchange interaction between the 3d electrons and the hole in the 3p level created by fluorescence makes the Kβ fluorescence spectrum sensitive to the 3d spin moment.40,42–44,46 The Kβ spectrum thus reflects the Fe contribution to the spin moment and shows little sensitivity to molecular symmetry for equal spin states,42,43 while the covalency of the metal–ligand bond does impact the Kβ spectrum.46 For this reason we construct reference spectra from complexes with 2,2′-bipyridine, porphyrin, phthalocyanine, and cyanide ligands to ensure similar bonding characteristics to [Fe(CN)4(bpy)]2− for use in the analysis as described in the ESI.† Subtracting the ground state reference spectrum from the spectra recorded for complexes with higher spin moment results in the reference difference spectra presented in Fig. 2(B). The amplitude and shape of the reference difference spectra provide key signatures for the 1,3MLCT (Fe S = 1/2), 3MC (Fe S = 1), 5MLCT (Fe S = 3/2), and 5MC (Fe S = 2) excited states. Since the formal spin moment of the Fe center in both 1MLCT and 3MLCT states is a doublet (Fe S = 1/2) these two states are indistinguishable in the Kβ spectrum. Furthermore, as the difference between 1,3MLCT and 3MC reference spectra is mainly one of signal amplitude, additional information on the excited state cascade has to be established to robustly separate these two states. Thus for [Fe(CN)4(bpy)]2−, where 1MLCT excitation could lead to 3MLCT, 3MC or 5MC formation with a yet to be determined quantum yield, the joint application of both UV-visible and Fe Kβ fluorescence spectroscopy proves crucial for a robust interpretation.
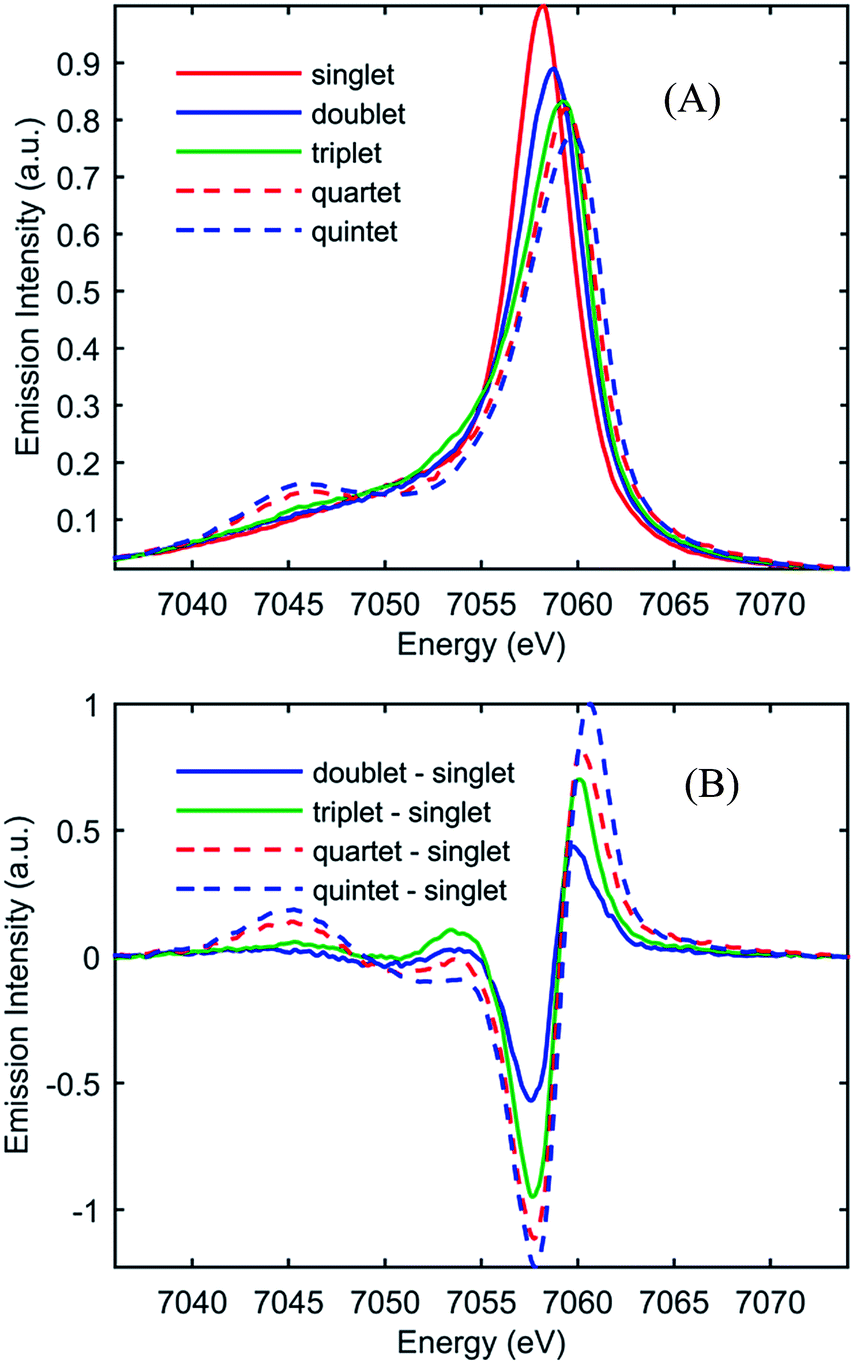 | ||
| Fig. 2 (A) Model Kβ fluorescence spectra for the ground-state and MLCT, 3MC, 5MLCT, and 5MC excited states of [Fe(CN)4(bpy)]2−. The model spectra are constructed from ground-state iron complexes with different spin moments; singlet: linear combination of [Fe(bpy)3]2+ and [Fe(CN)6]4− (red), doublet: linear combination of [Fe(bpy)3]3+ and [Fe(CN)6]3− (blue), triplet: iron(II)phthalocyanine (green), quartet: iron(III)phthalocyanine (dashed red), and quintet [Fe(phenanthroline)2(NCS)2] (dashed blue). (B) Reference difference Kβ spectra for the MLCT, 3MC, 5MLCT, and 5MC excited states constructed from the ground-state model spectra by subtracting the singlet spectrum. For detailed discussion of the modeling of the difference spectra, see the ESI.† | ||
UV-visible absorption spectroscopy tracks changes in excited state populations and spectral densities through changes in optically allowed electronic transitions. The characteristic absorption of the 2,2′-bipyridine radical anion in the near UV proves particularly valuable for the present study.18,38,47 The appearance of this excited state absorption spectral signature indicates the presence of the MLCT excited state and allows the relaxation dynamics of the MLCT state to be measured. By simultaneously measuring the ground state bleach recovery, UV-visible pump-probe spectroscopy can be used to differentiate MLCT excited state decay to metal-centered excited states versus decay to the electronic ground state.
We use a principle component analysis framework based on singular value decomposition for the UV-visible difference spectra.48 Details of the data analysis can be found in the ESI.† Global analysis of the principle components returns decay associated spectra (DAS). When the DAS can be assigned to specific molecular species or excited states the time dependent amplitudes of the DAS provide a powerful means of characterizing excited state kinetics. While distinguishing between spectral dynamics associated with changes in population from those associated with intramolecular vibrational redistribution and solvation can prove challenging, we mitigate this weakness with the time-resolved Kβ fluorescence measurements.
Fig. 3(A) shows the Kβ difference spectra for [Fe(CN)4(bpy)]2− dissolved in dimethyl sulfoxide and photo-excited at 650 nm, measured at 50 fs and 1.0 ps, while Fig. 3(B) shows a contour plot of the time dependent Kβ difference spectra out to 1.5 ps. Within the signal to noise of our measurements, only the amplitude, not the shape, of the difference spectra changes with time delay. The absence of changes in the Kβ difference spectral shape makes the extraction of kinetics straight forward. Fig. 3(C) plots the time dependent absolute value of the difference spectra along with fits to a single- and bi-exponential decay (dashed green and black lines respectively). The single exponential decay fit has a lifetime of 19 ± 2 ps. The bi-exponential does not provide a statistically significant improvement to this fit, and the analysis shows that any secondary exponential component account for <10% of the observed dynamics. The observations that the shape of the Kβ difference signal does not change, and that the difference signal amplitude is well-described by a single-exponential decay, strongly suggest that the excited state cascade is dominated by a single excited state species formed within the instrument response of our measurement. Fitting the transient spectra in Fig. 3(A) with the model difference spectra in Fig. 2 returns reduced χ2 values of 1.4, 2.7, 4.6, and 9.8 for the 1,3MLCT (Fe S = 1/2, black curve), 3MC (Fe S = 1), 5MLCT (Fe S = 3/2) and 5MC (Fe S = 2, dashed green curve) reference difference spectra. The large χ2 for the fits to the 5MLCT and 5MC difference spectra enable these excited states to be ruled out. While assigning the excited state population to a 1,3MLCT excited state provides the best fit to the experimental findings, the 3MC excited state also provides a viable fit to the experiment given the limitations associated with an analysis based on model difference spectra. Given the potential for extremely fast intersystem crossing between 1,3MLCT and 3MC excited states, we may not be temporally resolving the 1,3MLCT to 3MC transition. Consequently, the ability of a single exponential decay to fit the data cannot be used to rule out the presence of a 3MC excited state.
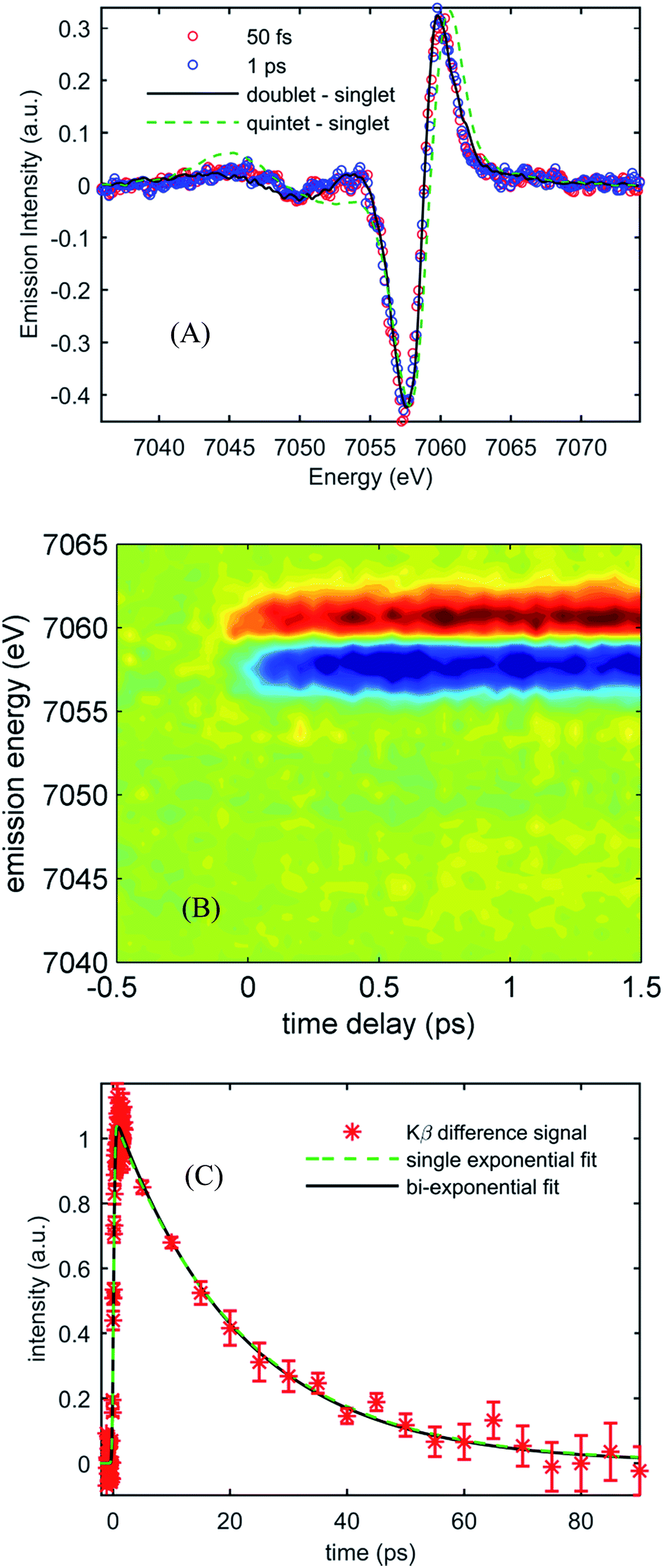 | ||
| Fig. 3 (A) Kβ transient difference spectra for 50 mM [Fe(CN)4(bpy)]2− in dimethyl sulfoxide obtained at 50 fs time delay (red circles) and 1 ps time delay (blue circles), fitted by the 1,3MLCT reference spectrum (black curve) and the 5MC reference spectrum (dashed green curve). (B) Contour plot of time-dependent optically-induced changes in Kβ fluorescence difference spectra for 50 mM [Fe(CN)4(bpy)]2− in dimethyl sulfoxide for time delays up to 1.5 ps. (C) The integrated absolute value of the Kβ fluorescence difference spectra as a function of time delay for [Fe(CN)4(bpy)]2− in dimethyl sulfoxide, as well as single and bi-exponential fits to the data. The single exponential fit returns a 19 ± 2 ps lifetime. | ||
Fig. 4(A) shows the UV-visible difference spectra measured at time delays of 50 fs and 1.0 ps for [Fe(CN)4(bpy)]2− dissolved in dimethyl sulfoxide and photo-excited at 650 nm. These spectra show an excited state absorption at 370 nm associated with the 2,2′-bipyridine radical anion, which we assign to a MLCT excited state. Unlike [Fe(bpy)3]2+, the signature absorption of the 2,2′-bipyridine radical anion does not disappear on the sub-picosecond time scale, but persists throughout the excited state lifetime. The persistence of the 370 nm excited state absorption feature is reflected in the two decay associated spectra resulting from the global analysis, shown in Fig. 4(B). The lifetimes associated with the decay associated spectra are 2.4 ± 0.4 ps and 19.0 ± 1.1 ps for DAS1 and DAS2 respectively. Fig. 4(C) presents the kinetics of the excited state absorption at 370 nm and the ground state bleach at 440 nm fit with single- and bi-exponential decays (dashed green and black lines respectively). The bi-exponential yields significantly better fits with 2.5 ± 0.5 ps and 18.5 ± 0.9 ps time constants, similar to the time constants extracted from the global analysis.
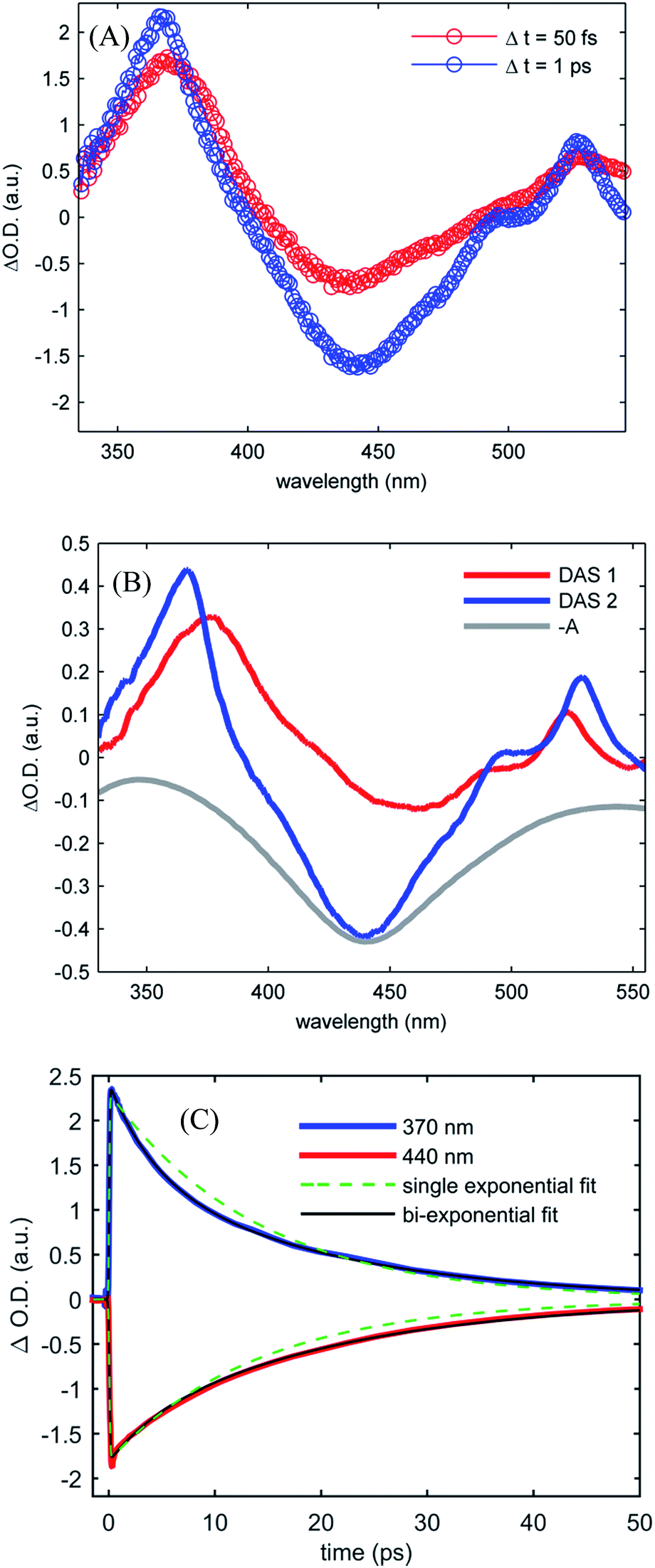 | ||
| Fig. 4 (A) Transient UV-visible absorption spectra obtained at 50 fs time delay (red curve) and 1 ps time delay (blue curve) for [Fe(CN)4(bpy)]2− in dimethyl sulfoxide. (B) The two decay associated spectra returned by global analysis of the data (red and blue curves) with lifetimes of 2.4 ± 0.4 ps and 19.0 ± 1.1 ps, are shown with the inverted ground state UV visible absorption spectrum (gray curve). (C) Kinetics of the UV visible pump-probe data at 370 nm and 440 nm (red and blue curves) with single- and bi-exponential fits (green dashed and black curves respectively) retuning 14 ps lifetime for the single exponential decay and 2.5 ± 0.5 ps and 18.5 ± 0.9 ps lifetimes for the double exponential decay. | ||
Robust assignment of the observed dynamics requires consideration of both measurements. First, the Kβ fluorescence measurements show no dynamics on the few ps time scale. The insensitivity of Kβ fluorescence spectra to molecular geometry strongly supports the conclusion that the 2.5 ± 0.5 ps dynamics result from spectral changes due to intra- and inter-molecular vibrational redistribution and solvation dynamics, not from excited electronic state population dynamics. Both measurements show a complete recovery of the ground state signal with a 19 ps lifetime, meaning that the excited state cascade is dominated by the decay of a single species. This slower decay has the characteristic absorption at 370 nm associated with the MLCT excited state and leads to a full recovery of the ground state bleach. These observations support the conclusion that the MLCT excited state decay does not lead to the formation of metal-centered excited states, a conclusion consistent with the spectral shape of the Kβ fluorescence difference spectrum. Taken together, we conclude that the MLCT excited state of [Fe(CN)4(bpy)]2− has a 19 ps lifetime.
Density functional theory (DFT) calculations were carried out to map the excited state potentials for [Fe(CN)4(bpy)]2− using Gaussian 09 (ref. 49) with PBE0/6-311G(d,p) and a complete DMSO polarizable continuum model (PCM) using the procedure described elsewhere.38 The energy levels of cyano containing transition metal complexes are influenced by the Lewis acidity of the solvent (quantified by its Gutmann acceptor number).50 While such specific solvent interactions are not accounted for within PCM, the relatively low Gutmann acceptor number of the DMSO solvent minimizes the impact of this approximation. The projected potential energy surfaces (PPES) of [Fe(CN)4(bpy)]2− appear in Fig. 5. Comparison to the potential energy surfaces calculated51 for [Fe(bpy)3]2+ with methods yielding similar results to the ones employed here shows that the substitution of 2,2′-bipyridine with the stronger ligand field CN− groups effectively increases the energy of the both the ground state and Fe-based ligand field excited states relative to the 3MLCT excited state. The 1 eV energy difference between the ground state and 3MLCT state calculated for [Fe(CN)4(bpy)]2− is considerably smaller than the 2.5 eV difference of [Fe(bpy)3]2+, consistent with the red-shift observed in the optical absorption spectrum. The calculations show 3MC and 5MC states with similar minimum energies which are significantly geometrically different to the MLCT state, making the excited state PPES qualitatively similar to those calculated for the iron carbene systems reported to have picosecond MLCT lifetimes37,38 and distinct from those calculated for [Fe(bpy)3]2+ for which the minimum energy of the 3MC and 5MC states are significantly lower than the MLCT states.31,32 The PPES of [Fe(CN)4(bpy)]2− thus provide a potential explanation for the observed relaxation dynamics; the destabilization of the 3MC and 5MC states relative to the 3MLCT state significantly extends the 3MLCT lifetime, meanwhile the very similar energies of the GS and 3MC at the 3MC minimum results in fast deactivation from the 3MC state to the ground state, inhibiting build-up of any significant fraction of excited 3MC and 5MC states in [Fe(CN)4(bpy)]2−.
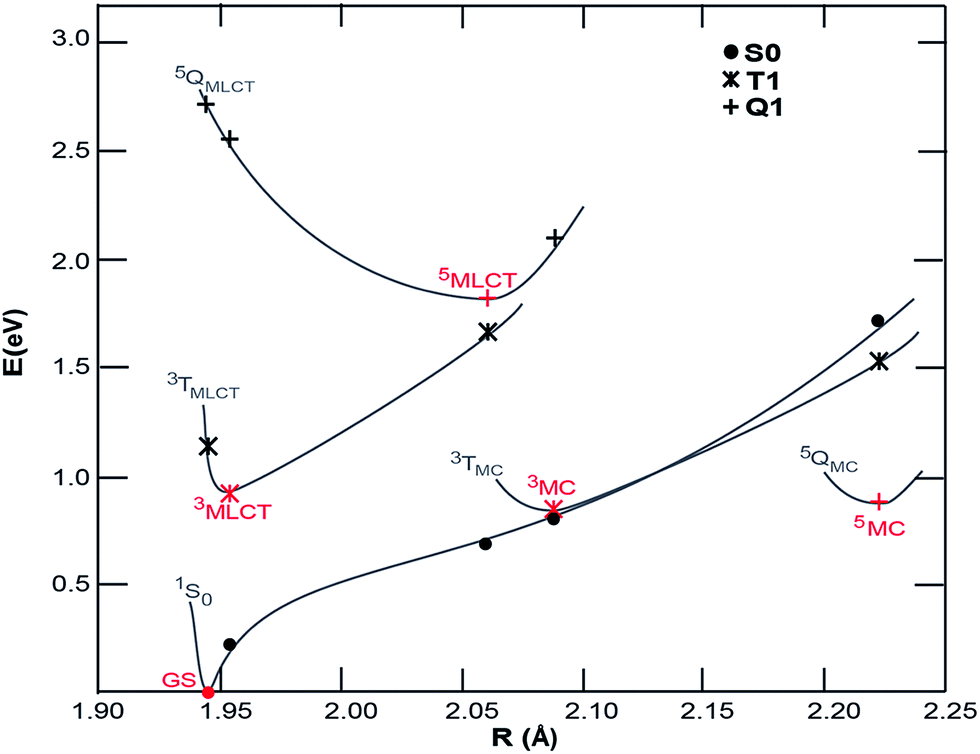 | ||
| Fig. 5 Projected potential energy surfaces (PPESs) versus the average Fe–ligand bond distances, R, for [Fe(CN)4(bpy)]2+. Red points are optimized minima of the ground state, and potential excited state configurations. Black points are single-point energies calculated at the minimum geometries (S0, singlet state; T1, triplet states; Q1, quintet states). The gray lines schematically show the PPESs. | ||
Conclusions
A combination of femtosecond resolution UV-visible and Kβ fluorescence spectroscopy has allowed the robust characterization of the electronic excited state dynamics of [Fe(CN)4(bpy)]2−. Based on the experimental data and analysis, we conclude ligand substitution increases the MLCT excited state lifetime of [Fe(CN)4(bpy)]2− by more than two orders of magnitude compared to [Fe(bpy)3]2+. In addition to the extension of the MLCT excited state lifetime, we observe no experimental evidence for 3MC or 5MC excited state formation in the time-resolved UV-visible or Kβ fluorescence spectra. Density functional theory (DFT) calculations provide a rationale for both the lengthening of the MLCT excited state lifetime and the absence of long lived metal-centered excited states, as arising from a significant destabilization of the metal-centered excited states relative to the 3MLCT state.With a lifetime of roughly 20 ps, intramolecular electronic excited state relaxation will not compete with typical rates of interfacial charge injection in dye sensitized solar cells.52–56 While the origin of the relatively poor performance of [Fe(CN)4(bpy)]2− sensitized solar cells remains uncertain, our results rule out ultrafast spin crossover as an explanation. Yang et al.8,9 demonstrated charge injection from [Fe(CN)4(bpy)]2− into TiO2, ruling out thermodynamic driving force as a potential explanation. Perhaps fast charge recombination limits the performance, as seen for iron-carbene sensitized solar cells.57
In summary, our study presents [Fe(CN)4(bpy)]2− as a bench mark molecule for interpreting the excited state dynamics of iron-centered systems designed for solar energy applications, as well as an intuitive illustration of how the manipulation of excited state levels can extend the MLCT lifetime of such systems by orders of magnitude. More importantly, we show how the joint application of femtosecond resolution optical and X-ray spectroscopies can provide a general, detailed and robust characterization of electronic excited state dynamics in complex molecular systems. Such robust characterization is of key importance for the evaluation of novel molecular systems for photosensitization and photocatalysis where UV-visible pump-probe measurements struggle to robustly characterize the nature of the involved excited states.38 Furthermore such detailed characterization can act as verification of quantum chemical simulations of excited electronic state phenomena.
Acknowledgements
Experiments were carried out at LCLS and SSRL, National User Facilities operated for DOE, OBES by Stanford University. WZ, RWH, HWL, ZS, and KJG acknowledge support from the AMOS program within the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences, Office of Science, U. S. Department of Energy. EIS acknowledges support from the NSF CHE-0948211. RGH acknowledges a Gerhard Casper Stanford Graduate Fellowship and the Achievements Rewards for College Scientists (ARCS) Foundation. TK acknowledges the German Research Foundation (DFG), grant KR3611/2-1. KSK, MMN, and TBvD acknowledge support from the Danish National Research Foundation and from DANSCATT. KSK gratefully acknowledge the support of the Carlsberg Foundation and the Danish Council for Independent Research. YL, TH, KW, LF, PP, and VS acknowledge support from the Crafoord Foundation, the Swedish Research Council (VR), the Knut and Alice Wallenberg (KAW) Foundation, the European Research Council (ERC, 226136-VISCHEM) and the Swedish Energy Agency. KK thanks the Volkswagen Foundation for support under the Peter Paul Ewald fellowship program (Az.: I/85832). PP acknowledges support from the Swedish National Supercomputing Centre and the Lund University Intensive Computation Application Research Center supercomputing facilities.Notes and references
- G. M. Brown, B. S. Brunschwig, C. Creutz, J. F. Endicott and N. Sutin, Homogeneous catalysis of the photo-reduction of water by visible-light – Mediation by a tris(2,2′-bipyridine)ruthenium(II)–cobalt(II) macrocycle system, J. Am. Chem. Soc., 1979, 101(5), 1298–1300 CrossRef CAS.
- H. B. Gray and A. W. Maverick, Solar Chemistry of Metal Complexes, Science, 1981, 214(4526), 1201–1205 CAS.
- A. F. Heyduk and D. G. Nocera, Hydrogen produced from hydrohalic acid solutions by a two-electron mixed-valence photocatalyst, Science, 2001, 293(5535), 1639–1641 CrossRef CAS PubMed.
- C. Creutz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, Lifetimes, Spectra, and Quenching of the Excited-States of Polypyridine Complexes of Iron(II), Ruthenium(II), and Osmium(II), J. Am. Chem. Soc., 1980, 102(4), 1309–1319 CrossRef CAS.
- M. Chergui, On the interplay between charge, spin and structural dynamics in transition metal complexes, Dalton Trans., 2012, 41(42), 13022–13029 RSC.
- B. Oregan and M. Gratzel, A low-cost, high-efficiency solar-cell based on dye-sensitized colloidal TiO2 films, Nature, 1991, 353(6346), 737–740 CrossRef CAS.
- J. E. Monat and J. K. McCusker, Femtosecond excited-state dynamics of an iron(II) polypyridyl solar cell sensitizer model, J. Am. Chem. Soc., 2000, 122(17), 4092–4097 CrossRef CAS.
- M. Yang, D. W. Thompson and G. J. Meyer, Dual pathways for TiO2 sensitization by Na2 Fe(bpy)(CN)4, Inorg. Chem., 2000, 39(17), 3738–3739 CrossRef CAS PubMed.
- M. Yang, D. W. Thompson and G. J. Meyer, Charge-transfer studies of iron cyano compounds bound to nanocrystalline TiO2 surfaces, Inorg. Chem., 2002, 41(5), 1254–1262 CrossRef CAS PubMed.
- S. Ferrere, New photosensitizers based upon Fe(L)2(CN)2 and Fe(L)3 (L = substituted 2,2′-bipyridine): Yields for then photosensitization of TiO2 and effects on the band selectivity, Chem. Mater., 2000, 12(4), 1083–1089 CrossRef CAS.
- S. Ferrere and B. A. Gregg, Photosensitization of TiO2 by Fe-II(2,2′-bipyridine-4,4′-dicarboxylic acid)2(CN)2: band selective electron injection from ultra-short-lived excited states, J. Am. Chem. Soc., 1998, 120(4), 843–844 CrossRef CAS.
- P. Gutlich and H. A. Goodwin, Spin crossover – an overall perspective, in Spin Crossover in Transition Metal Compounds I, ed. P. Gutlich and H. A. Goodwin, Springer-Verlag, Berlin, 2004, vol. 233, pp. 1–47 Search PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Photoinduced magnetization of a cobalt–iron cyanide, Science, 1996, 272(5262), 704–705 CAS.
- A. Hauser, Intersystem Crossing in the Fe(PTZ)6 (BF4)2 Spin Crossover System (PTZ = 1-propyltetrazole), J. Chem. Phys., 1991, 94(4), 2741–2748 CrossRef CAS.
- C. Bressler, C. Milne, V. T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Femtosecond XANES Study of the Light-Induced Spin Crossover Dynamics in an Iron(II) Complex, Science, 2009, 323(5913), 489–492 CrossRef CAS PubMed.
- A. Cannizzo, C. J. Milne, C. Consani, W. Gawelda, C. Bressler, F. van Mourik and M. Chergui, Light-induced spin crossover in Fe(II)-based complexes: the full photocycle unraveled by ultrafast optical and X-ray spectroscopies, Coord. Chem. Rev., 2010, 254(21–22), 2677–2686 CrossRef CAS.
- C. Consani, M. Premont-Schwarz, A. ElNahhas, C. Bressler, F. van Mourik, A. Cannizzo and M. Chergui, Vibrational Coherences and Relaxation in the High-Spin State of Aqueous [Fe-II(bpy)3]2+, Angew. Chem., Int. Ed., 2009, 48(39), 7184–7187 CrossRef CAS PubMed.
- W. Gawelda, A. Cannizzo, V. T. Pham, F. van Mourik, C. Bressler and M. Chergui, Ultrafast nonadiabatic dynamics of [Fe(II)(bpy)3]2+ in solution, J. Am. Chem. Soc., 2007, 129(26), 8199–8206 CrossRef CAS PubMed.
- N. Huse, H. Cho, K. Hong, L. Jamula, F. M. F. de Groot, T. K. Kim, J. K. McCusker and R. W. Schoenlein, Femtosecond Soft X-ray Spectroscopy of Solvated Transition-Metal Complexes: Deciphering the Interplay of Electronic and Structural Dynamics, J. Phys. Chem. Lett., 2011, 2(8), 880–884 CrossRef CAS PubMed.
- J. K. McCusker, K. N. Walda, R. C. Dunn, J. D. Simon, D. Magde and D. N. Hendrickson, Subpicosecond 1MLCT-5T2 Intersystem Crossing of Low-Spin Polypyridyl Ferrous Complexes, J. Am. Chem. Soc., 1993, 115(1), 298–307 CrossRef CAS.
- H. T. Lemke, C. Bressler, L. X. Chen, D. M. Fritz, K. J. Gaffney, A. Galler, W. Gawelda, K. Haldrup, R. W. Hartsock, H. Ihee, J. Kim, K. H. Kim, J. H. Lee, M. M. Nielsen, A. B. Stickrath, W. K. Zhang, D. L. Zhu and M. Cammarata, Femtosecond X-ray Absorption Spectroscopy at a Hard X-ray Free Electron Laser: Application to Spin Crossover Dynamics, J. Phys. Chem. A, 2013, 117(4), 735–740 CrossRef CAS PubMed.
- W. K. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock, T. Kroll, K. S. Kjaer, K. Kubicek, H. T. Lemke, H. W. Liang, D. A. Meyer, M. M. Nielsen, C. Purser, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. Van Driel, G. Vanko, T. C. Weng, D. Zhu and K. J. Gaffney, Tracking Excited State Charge and Spin Dynamics in Iron Coordination Complexes, Nature, 2014, 509, 345 CrossRef CAS PubMed.
- S. Nozawa, T. Sato, M. Chollet, K. Ichiyanagi, A. Tomita, H. Fujii, S. Adachi and S. Koshihara, Direct Probing of Spin State Dynamics Coupled with Electronic and Structural Modifications by Picosecond Time-Resolved XAFS, J. Am. Chem. Soc., 2010, 132(1), 61–63 CrossRef CAS PubMed.
- M. Khalil, M. A. Marcus, A. L. Smeigh, J. K. McCusker, H. H. W. Chong and R. W. Schoenlein, Picosecond X-ray absorption spectroscopy of a photoinduced iron(II) spin crossover reaction in solution, J. Phys. Chem. A, 2006, 110(1), 38–44 CrossRef CAS PubMed.
- N. Huse, T. K. Kim, L. Jamula, J. K. McCusker, F. M. F. de Groot and R. W. Schoenlein, Photo-Induced Spin-State Conversion in Solvated Transition Metal Complexes Probed via Time-Resolved Soft X-ray Spectroscopy, J. Am. Chem. Soc., 2010, 132(19), 6809–6816 CrossRef CAS PubMed.
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, Femtosecond Time-Resolved Optical and Raman Spectroscopy of Photoinduced Spin Crossover: Temporal Resolution of Low-to-High Spin Optical Switching, J. Am. Chem. Soc., 2008, 130(43), 14105–14107 CrossRef CAS PubMed.
- K. Haldrup, G. Vanko, W. Gawelda, A. Galler, G. Doumy, A. M. March, E. P. Kanter, A. Bordage, A. Dohn, T. B. van Driel, K. S. Kjaer, H. T. Lemke, S. E. Canton, J. Uhlig, V. Sundström, L. Young, S. H. Southworth, M. M. Nielsen and C. Bressler, Guest–Host Interactions Investigated by Time-Resolved X-ray Spectroscopies and Scattering at MHz Rates: Solvation Dynamics and Photoinduced Spin Transition in Aqueous [Fe(bipy)3]2+, J. Phys. Chem. A, 2012, 116(40), 9878–9887 CrossRef CAS PubMed.
- G. Vanko, P. Glatzel, V. T. Pham, R. Abela, D. Grolimund, C. N. Borca, S. L. Johnson, C. J. Milne and C. Bressler, Picosecond Time-Resolved X-Ray Emission Spectroscopy: Ultrafast Spin-State Determination in an Iron Complex, Angew. Chem., Int. Ed., 2010, 49(34), 5910–5912 CrossRef CAS PubMed.
- W. Gawelda, V. T. Pham, M. Benfatto, Y. Zaushitsyn, M. Kaiser, D. Grolimund, S. L. Johnson, R. Abela, A. Hauser, C. Bressler and M. Chergui, Structural determination of a short-lived excited iron(II) complex by picosecond x-ray absorption spectroscopy, Phys. Rev. Lett., 2007, 98(5), 057401 CrossRef PubMed.
- G. Auboeck and M. Chergui, Sub-50-fs photoinduced spin crossover in Fe(bpy)(3)(2+), Nat. Chem., 2015, 7(8), 629–633 CrossRef PubMed.
- C. de Graaf and C. Sousa, Study of the Light-Induced Spin Crossover Process of the [Fe(II)(bpy)3]2+ Complex, Chem.–Eur. J., 2010, 16(15), 4550–4556 CrossRef CAS PubMed.
- C. de Graaf and C. Sousa, On the Role of the Metal-to-Ligand Charge Transfer States in the Light-Induced Spin Crossover in Fe-II(bpy)3, Int. J. Quantum Chem., 2011, 111(13), 3385–3393 CrossRef CAS.
- C. Sousa, C. de Graaf, A. Rudavskyi, R. Broer, J. Tatchen, M. Etinski and C. M. Marian, Ultrafast Deactivation Mechanism of the Excited Singlet in the Light-Induced Spin Crossover of [Fe(2,2′-bipyridine)3]2+, Chem.–Eur. J., 2013, 19(51), 17541–17551 CrossRef CAS PubMed.
- J. R. Winkler, C. Creutz and N. Sutin, Solvent tuning of the excited-state properties of (2,2′-bipyridine)tetracyanoferrate(II) – Direct observation of a metal-to-ligand charge transfer excited-state of iron(II), J. Am. Chem. Soc., 1987, 109(11), 3470–3471 CrossRef CAS.
- J. R. Winkler and N. Sutin, Lifetimes and spectra of the excited-states of cis-dicyanobis(2,2′-bipyridine)iron(II) and cis-dicyanobis(2,2′-bipyridine)ruthenium(II) in solution, Inorg. Chem., 1987, 26(2), 220–221 CrossRef CAS.
- Y. Z. Liu, T. Harlang, S. E. Canton, P. Chabera, K. Suarez-Alcantara, A. Fleckhaus, D. A. Vithanage, E. Goransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Towards longer-lived metal-to-ligand charge transfer states of iron(II) complexes: an N-heterocyclic carbene approach, Chem. Commun., 2013, 49(57), 6412–6414 RSC.
- L. A. Fredin, M. Papai, E. Rozsalyi, G. Vanko, K. Wärnmark, V. Sundström and P. Persson, Exceptional Excited-State Lifetime of an Iron(II)-N-Heterocyclic Carbene Complex Explained, J. Phys. Chem. Lett., 2014, 5(12), 2066–2071 CrossRef CAS PubMed.
- Y. Z. Liu, K. S. Kjær, L. A. Fredin, P. Chábera, T. Harlang, S. E. Canton, S. Lidin, J. X. Zhang, R. Lomoth, K.-E. Bergquist, P. Persson, K. Wärnmark and V. Sundström, A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-triazol-5-ylidene) Ligands: Taming the MLCT Excited State of Iron(II), Chem.–Eur. J., 2014, 21(9), 3628–3639 CrossRef PubMed.
- M. Chollet, R. Alonso-Mori, M. Cammarata, D. Damiani, J. Defever, J. T. Delor, Y. P. Feng, J. M. Glownia, J. B. Langton, S. Nelson, K. Ramsey, A. Robert, M. Sikorski, S. Song, D. Stefanescu, V. Srinivasan, D. L. Zhu, H. T. Lemke and D. M. Fritz, The X-ray Pump-Probe instrument at the Linac Coherent Light Source, J. Synchrotron Radiat., 2015, 22, 503–507 CrossRef CAS PubMed.
- P. Glatzel and U. Bergmann, High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes – electronic and structural information, Coord. Chem. Rev., 2005, 249(1–2), 65–95 CrossRef CAS.
- G. Vanko, A. Bordage, P. Glatzel, E. Gallo, M. Rovezzi, W. Gawelda, A. Galler, C. Bressler, G. Doumy, A. M. March, E. P. Kanter, L. Young, S. H. Southworth, S. E. Canton, J. Uhlig, G. Smolentsev, V. Sundström, K. Haldrup, T. B. van Driel, M. M. Nielsen, K. S. Kjaer and H. T. Lemke, Spin-state studies with XES and RIXS: From static to ultrafast, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 166–171 CrossRef CAS.
- G. Vanko, T. Neisius, G. Molnar, F. Renz, S. Karpati, A. Shukla and F. M. F. de Groot, Probing the 3d spin momentum with X-ray emission spectroscopy: The case of molecular-spin transitions, J. Phys. Chem. B, 2006, 110(24), 11647–11653 CrossRef CAS PubMed.
- N. Lee, T. Petrenko, U. Bergmann, F. Neese and S. DeBeer, Probing Valence Orbital Composition with Iron K beta X-ray Emission Spectroscopy, J. Am. Chem. Soc., 2010, 132(28), 9715–9727 CrossRef CAS PubMed.
- F. de Groot, High resolution X-ray emission and X-ray absorption spectroscopy, Chem. Rev., 2001, 101(6), 1779–1808 CrossRef CAS PubMed.
- H. E. Toma and M. S. Takasugi, Spectroscopic Studies of Preferential and Asymmetric Solvation in Substituted Cyanoiron(II) Complexes, J. Solution Chem., 1983, 12(8), 547–561 CrossRef CAS.
- C. J. Pollock, M. U. Delgado-Jaime, M. Atanasov, F. Neese and S. DeBeer, K beta Mainline X-ray Emission Spectroscopy as an Experimental Probe of Metal–Ligand Covalency, J. Am. Chem. Soc., 2014, 136(26), 9453–9463 CrossRef CAS PubMed.
- A. M. Brown, C. E. McCusker and J. K. McCusker, Spectroelectrochemical identification of charge-transfer excited states in transition metal-based polypyridyl complexes, Dalton Trans., 2014, 43(47), 17635–17646 RSC.
- E. R. Henry and J. Hofrichter, Singular Value Decomposition – Application to Analysis of Experimental-Data, Methods Enzymol., 1992, 210, 129–192 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. I. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Version D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. J. Timpson, C. A. Bignozzi, B. P. Sullivan, E. M. Kober and T. J. Meyer, Influence of solvent on the spectroscopic properties of cyano complexes of ruthenium(II), J. Phys. Chem., 1996, 100(8), 2915–2925 CrossRef CAS.
- M. Papai, G. Vanko, C. de Graaf and T. Rozgonyi, Theoretical Investigation of the Electronic Structure of Fe(II) Complexes at Spin-State Transitions, J. Chem. Theory Comput., 2013, 9(1), 509–519 CrossRef CAS PubMed.
- J. B. Asbury, E. Hao, Y. Q. Wang, H. N. Ghosh and T. Q. Lian, Ultrafast electron transfer dynamics from molecular adsorbates to semiconductor nanocrystalline thin films, J. Phys. Chem. B, 2001, 105(20), 4545–4557 CrossRef CAS.
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca Jr, P. Huang, P. Chabera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Iron sensitizer converts light to electrons with 92% yield, Nat. Chem., 2015, 7(11), 883–889 CrossRef CAS PubMed.
- K. R. Siefermann, C. D. Pemmaraju, S. Neppl, A. Shavorskiy, A. A. Cordones, J. Vura-Weis, D. S. Slaughter, F. P. Sturm, F. Weise, H. Bluhm, M. L. Strader, H. Cho, M.-F. Lin, C. Bacellar, C. Khurmi, J. Guo, G. Coslovich, J. S. Robinson, R. A. Kaindl, R. W. Schoenlein, A. Belkacem, D. M. Neumark, S. R. Leone, D. Nordlund, H. Ogasawara, O. Krupin, J. J. Turner, W. F. Schlotter, M. R. Holmes, M. Messerschmidt, M. P. Minitti, S. Gul, J. Z. Zhang, N. Huse, D. Prendergast and O. Gessner, Atomic-Scale Perspective of Ultrafast Charge Transfer at a Dye-Semiconductor Interface, J. Phys. Chem. Lett., 2014, 5(15), 2753–2759 CrossRef CAS PubMed.
- E. Jakubikova and D. N. Bowman, Fe(II)–Polypyridines as Chromophores in Dye-Sensitized Solar Cells: A Computational Perspective, Acc. Chem. Res., 2015, 48(5), 1441–1449 CrossRef CAS PubMed.
- L. A. Fredin, K. Wärnmark, V. Sundström and P. Persson, Molecular and Interfacial Calculations of Iron(II) Light Harvesters, ChemSusChem, 2016, 9(7), 667–675 CrossRef CAS PubMed.
- T. Duchanois, T. Etienne, C. Cebrian, L. Liu, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, An Iron-Based Photosensitizer with Extended Excited-State Lifetime: Photophysical and Photovoltaic Properties, Eur. J. Inorg. Chem., 2015, 14, 2469–2477 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03070j |
| ‡ These authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |