
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectronic tuning of self-healing fluorophores for live-cell and single-molecule imaging†
Qinsi
Zheng
a,
Steffen
Jockusch
c,
Zhou
Zhou
b,
Roger B.
Altman
b,
Hong
Zhao
b,
Wesley
Asher
d,
Michael
Holsey
d,
Signe
Mathiasen
d,
Peter
Geggier
d,
Jonathan A.
Javitch
de and
Scott C.
Blanchard
*ab
aTri-Institutional Training Program in Chemical Biology, USA
bDepartment of Physiology and Biophysics, Weill Cornell Medicine, New York, NY, USA. E-mail: scb2005@med.cornell.edu
cDepartment of Chemistry, Columbia University, New York, NY, USA
dDepartments of Psychiatry and Pharmacology, Columbia University College of Physicians & Surgeons, USA
eDivision of Molecular Therapeutics, New York State Psychiatric Institute, USA
First published on 7th September 2016
Abstract
Bright, long-lasting organic fluorophores enable a broad range of imaging applications. “Self-healing” fluorophores, in which intra-molecularly linked protective agents quench photo-induced reactive species, exhibit both enhanced photostability and biological compatibility. However, the self-healing strategy has yet to achieve its predicted potential, particularly in the presence of ambient oxygen where live-cell imaging studies must often be performed. To identify key bottlenecks in this technology that can be used to guide further engineering developments, we synthesized a series of Cy5 derivatives linked to the protective agent cyclooctatetraene (COT) and examined the photophysical mechanisms curtailing their performance. The data obtained reveal that the photostability of self-healing fluorophores is limited by reactivity of the COT protective agent. The addition of electron withdrawing substituents to COT reduced its susceptibility to reactions with molecular oxygen and the fluorophore to which it is attached and increased its capacity to participate in triplet energy transfer. Exploiting these insights, we designed and synthesized a suite of modified COT-fluorophores spanning the visible spectrum that exhibited markedly increased intra-molecular photostabilization. Under ambient oxygen conditions, the photostability of Cy3 and Cy5 fluorophore derivatives increased by 3- and 9-fold in vitro and by 2- and 6-fold in living cells, respectively. We further show that this approach can improve a silicon rhodamine fluorophore. These findings offer a clear strategy for achieving the full potential of the self-healing approach and its application to the gamut of fluorophore species commonly used for biomedical imaging.
Introduction
Fluorescence microscopy continues to undergo revolutionary advancements in both sensitivity and resolution, highlighted by the development of single-molecule and super-resolution fluorescence imaging methods.1,2 Photo-induced dark states (blinking) and fluorophore degradation (photobleaching) often limit progress in these areas, as the quality and duration of experimental signals are proportional to the total number of photons emitted and detected.3–7 Strategies that mitigate such processes are therefore in high demand.3–5The most common photo-protection strategies to increase photon emission involves the removal of molecular oxygen from imaging buffers8,9 and the addition of small-molecule protective agents, such as cyclooctatetraene (COT),10 nitrobenzyl alcohol (NBA),11 trolox,12,13 methyl viologen, ascorbic acid, and mixtures thereof,14 to solution.3 While such methods have the capacity to increase fluorophore performance in a range of biological settings, their utility can be restricted by perturbations to the systems under investigation. This includes potential toxicities arising from oxygen depletion and the high-concentrations of protective agents needed to mediate photoprotection (ca. 1 mM).15 Such issues are particularly challenging in live-cell imaging applications.7,16
To circumvent these limitations, modified organic fluorophores have been engineered that are covalently linked to specific protective agents (PAs).5,17,18 Such fluorophores exhibit substantially enhanced brightness and photostability both in vitro and in living cells as a result of intra-molecular quenching (self-healing) of photo-induced triplet and radical states.5,19–24
The self-healing strategy is based on the principles of intra-molecular reactions, which posit that the covalent attachment of protective agents to the fluorophore can increase their effective concentration up to ten orders of magnitude (ca. 107 M) beyond their aqueous solubility limit.25,26 Consequently, intra-molecularly linked protective agents can more effectively compete with reactants (e.g. molecular oxygen and reactive oxygen species (ROS)) that are potentially destructive to fluorophore excited states, including triplet states (Scheme 1), to improve fluorophore photostability in both deoxygenated and oxygenated conditions. However, the effective concentrations of protective agents in current self-healing fluorophores (ca. 1–100 mM, see ESI†) are nearly 108-times lower than this predicted limit. Hence, while up to ∼50-fold improvements in fluorophore performance can be achieved in deoxygenated solutions, the impact is relatively modest (up to ∼5 fold)5,17 under ambient molecular oxygen concentrations (∼0.3 mM)27 that are required for many biological imaging applications.7,16,28 Correspondingly, a key goal towards the optimization of self-healing fluorophores is to increase the effective concentration of the covalently attached PAs to the predicted value (∼107 M). In so doing, fluorophore triplet states could in principle be quenched to extents that make fluorophores nearly inert to photo-induced reactions with molecular oxygen.5,29
 | ||
| Scheme 1 Probable photophysical and photochemical processes governing the performance of Cy5–COT fluorophores. S0: ground state; S1: singlet excited state; T1: triplet excited state; hν: excitation energy; 3COT: triplet state of the COT molecule; P: photobleached products, which are likely achieved through multi-step processes (red arrows). | ||
Here we describe photophysical investigations into the self-healing mechanism that have enabled us to identify critical bottlenecks to current technologies, including unwanted reactions of the PA species with both the fluorophore and solution components. Exploiting the insights obtained through these investigations, we generated self-healing cyanine-class fluorophores spanning the visible spectrum that exhibit marked photostability enhancements over previous technologies.5,17In vitro, the fluorophore generated that is spectrally equivalent to Cy5 exhibits a net 10- and 100-fold increase in total number of photons emitted under ambient oxygen and deoxygenated conditions, respectively. In the context of living cells, fluorophores spectrally analogous to Dy549 and Alexa Fluor 647 (AF647), commonly used for live cell imaging due to their low non-specific binding,30 exhibit 2- and 6-fold increases in the number of photons detected under ambient oxygen conditions, respectively. The design principles afforded by these investigations correspondingly mark an important step towards delivering the full potential of the self-healing strategy.
Results and discussion
The lifetime of the Cy5–COT(n) triplet state correlates with linker length
Self-healing fluorophores can be stabilized through intra-molecular triplet energy transfer to protective agents (e.g. COT).21 In line with early investigations on the distance dependence of triplet energy transfer in other organic compounds,25,31 we have shown for self-healing fluorophores that the rates of triplet energy transfer are more rapid at shorter distances.21 However, such investigations only examined two distinct linker lengths. To systematically examine the linker dependence of triplet energy transfer, we synthesized a series of Cy5–COT fluorophores in which COT was covalently attached in proximity to the Cy5 via polymethylene linkers of defined-lengths.31,32 In total, six Cy5–COT(n) fluorophores were generated, in which the number (n) of carbon atoms separating the COT and the Cy5 fluorophore is 1, 2, 3, 4, 5, or 10 (Chart 1; Materials and methods). Using these compounds, we examined the relationship between each fluorophore's triplet state lifetime and photostability both in ambient oxygen and in deoxygenated solutions. | ||
| Chart 1 Structures of Cy5–COT(n) and Cy5–bisCOT(3) fluorophores used in this study. | ||
In previous work,21 we employed laser flash photolysis to determine the lifetime (τT) of the Cy5 triplet state (3Cy5*) using solution benzophenone (BP) as a sensitizer. This approach generated sensitized 3Cy5* at a timescale of approximately 0.5 μs, limited by the diffusion of triplet benzophenone (3BP*) and Cy5 (Fig. S1†). While providing an estimate of the τT of 3Cy5* and enabling qualitative comparisons of the triplet lifetimes of intra-molecularly photostabilized fluorophores,21 this method proved insufficient to accurately determine the τT below 1 μs.
To address this shortcoming, we have used an intra-molecular sensitizing approach to more efficiently populate the Cy5 triplet state. To do so, we covalently linked the triplet state sensitizer, thioxanthone (9-oxothioxanthene, OTX), to Cy5 and Cy5–COT(n) fluorophores (Fig. 1A, Chart S1†). OTX was selected as an intra-molecular sensitizer because it has high triplet quantum yield33 and a higher triplet state energy than Cy5.33 OTX could also be selectively excited at non-interfering wavelengths (e.g. 355 nm).34 Using this approach, the rate of triplet–triplet energy transfer from OTX to Cy5 was increased by nearly an order of magnitude (ca. 0.05 μs), independent of concentration. Correspondingly, triplet lifetime measurements could be performed at substantially lower dye concentrations (ca. 1–10 μM), where the τT of Cy5 was revealed to be approximately twice as long (∼110 μs) as previously estimated (Fig. 1B).21 Hence, all τT measurements reported here were made using OTX-conjugated versions of the Cy5–COT(n) fluorophores (Chart S1; Fig. S2†), from which intra-molecular triplet energy transfer rates were calculated (1/τT). Triplet state lifetime measurements performed using OTX–Cy5–COT(10) (3.1 ± 0.1 μs) and Cy5–COT(10) (3.2 ± 0.3 μs) populated via solution benzophenone indicated that the observed rates were independent of the sensitizing method (Fig. S3†).
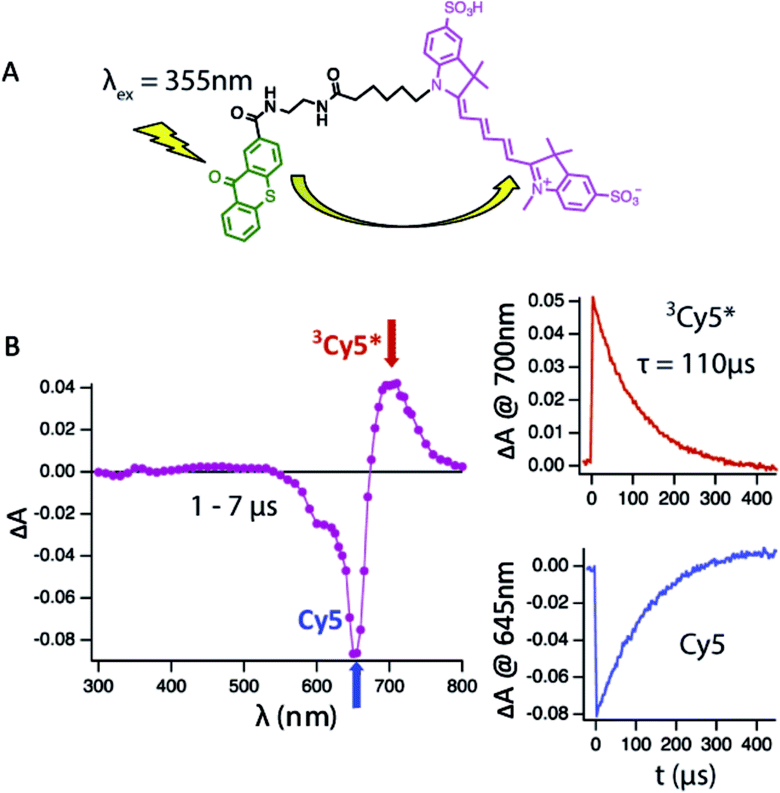 | ||
| Fig. 1 Transient absorbance measurements of triplet state lifetimes. (A) Reaction scheme for intramolecular sensitization of the Cy5 fluorophore with 9-oxothioxanthene (OTX); (B) transient absorption spectra recorded at 1–7 μs after the laser pulse (355 nm, 5 ns pulse width) of deoxygenated acetonitrile solutions of OTX–Cy5 (10 μM). The insets show kinetic traces at appropriate observation wavelengths. | ||
Cy5–COT(n) fluorophores with shorter linker lengths generally exhibited shorter τT, with a sharp drop below a length of four atoms (Table 1, Fig. 2A). These data are consistent with previous intra-molecular triplet energy transfer studies, where it was suggested that energy transfer preferentially occurs via a through-bond mechanism at short spacing (length < 4 atoms), whereas intra-molecular collisional interactions dominate when longer linkers (length > 4 atoms) are employed.31,35,36 The most rapid triplet quenching rates were observed when COT was separated from Cy5 by just a single carbon atom (1/τT = 6.5 × 106 s−1). Hence, Cy5–COT(1) exhibited a triplet state lifetime of ∼150 ns. This 1/τT is, however, nearly 2–3 orders of magnitude lower than expected for the through-bond energy transfer mechanism.31,35,36 This finding suggests that the rates of triplet energy transfer observed for the intra-molecularly photostabilized Cy5–COT(n) fluorophores may be influenced by additional constraints. Such factors likely include the rate-limiting formation of planar conformations of the COT molecule that are required for its “non-vertical” transition into the aromatic triplet state.37–40
| τ T (μs) | Counts in deoxygenated buffer (×106) | Counts in ambient oxygen (×104) | |
|---|---|---|---|
| Cy5 | 110 ± 5 | 0.04 ± 0.02 | 0.5 ± 0.1 |
| Cy5–COT(1) | 0.15 ± 0.01 | 0.19 ± 0.02 | 2.4 ± 0.2 |
| Cy5–COT(2) | 0.19 ± 0.01 | 1.7 ± 0.1 | 2.1 ± 0.2 |
| Cy5–COT(3) | 0.40 ± 0.02 | 2.1 ± 0.1 | 2.1 ± 0.2 |
| Cy5–COT(4) | 1.1 ± 0.1 | 1.7 ± 0.1 | 1.9 ± 0.1 |
| Cy5–COT(5) | 0.75 ± 0.04 | 1.7 ± 0.1 | 1.9 ± 0.2 |
| Cy5–COT(10) | 3.1 ± 0.1 | 1.3 ± 0.1 | 1.9 ± 0.2 |
| Cy5–COT(13) | 13 ± 2 | 0.8 ± 0.1 | 1.6 ± 0.3 |
| Cy5–bisCOT(3) | 0.22 ± 0.01 | 3.1 ± 0.1 | 4.4 ± 0.3 |
| Cy5–AC(4) | 0.077 ± 0.005 | 4.4 ± 0.1 | 4.4 ± 0.3 |
| Cy5–AC(5) | 0.28 ± 0.01 | 3.6 ± 0.2 | 4.1 ± 0.1 |
| Cy5–AC(11) | 0.33 ± 0.01 | 2.8 ± 0.1 | 3.5 ± 0.3 |
 | ||
| Fig. 2 Relationship between triplet state lifetime and the observed photon counts of intra-molecularly photostabilized Cy5 fluorophores. (A) The inverse of triplet state lifetime (1/τT) and (B) the average number of photons detected before photobleaching in single-molecule measurements as a function of the linker length between Cy5 and COT. The measurement for Cy5–COT(13) is from our previous study.21 | ||
Linker-length dependence of Cy5–COT(n) photostability
To examine the relationship between triplet state lifetime and photostability, each fluorophore was imaged at single-molecule resolution through total internal reflection fluorescence (TIRF) microscopy, where total photon yields prior to photobleaching (photon count) could be examined for thousands of individual fluorophores simultaneously (methods).17Using this approach, we first quantified the total number of photons detected for each Cy5–COT(n) fluorophore prior to photobleaching under deoxygenated conditions.9 As previously reported,17 Cy5–COT(n) fluorophores exhibited marked increases in total photon count relative to the parent Cy5 molecule. Consistent with the notion that photobleaching occurs through the triplet state (Scheme 1), the photon counts observed correlated with the rate of triplet state quenching (1/τT) for Cy5–COT(n) fluorophores with n > 2 (Table 1, Fig. 2). However, for Cy5–COT(2) and Cy5–COT(1) the total photon counts were notably uncorrelated with 1/τT (Table 1, Fig. 2B). Although Cy5–COT(1) possesses the shortest triplet state lifetime (τT = 150 ns), this fluorophore exhibited the lowest photon count (1.9 × 105). We therefore conclude that the benefits of COT-mediated, intra-molecular triplet state quenching are compromised at short linker lengths.
To understand these observations, we considered the most probable photophysical and photochemical pathways for Cy5 stabilization and photo-induced degradation.21,41–43 Fluorophores may photobleach through the first triplet state (T1), or they may absorb multiple photons and degrade through higher excited states (Sn or Tn, where n > 1).43,44 For the latter case, the rate of photobleaching is expected to increase with excitation power.43,44 We therefore determined the photon counts for each Cy5–COT(n) fluorophore over a range of illumination intensities. In these experiments, the inverse of photon counts, which is proportional to the photobleaching quantum yield (ΦB), was observed to increase linearly with illumination intensity (Fig. S4†). Such findings indicate that higher-order excited states (Sn or Tn, n > 1) contribute to the effective rate of photobleaching.
Accordingly, we reasoned that fluorophores principally photobleach through their first triplet state at illumination intensities <100 W cm−2 (T1) (Scheme 1). Under these conditions, the photobleaching rate from T1, defined as the inverse of the quantum yield of photobleaching through the triplet state (1/ΦB,T), is predicted to increase linearly with 1/τT, the rate of triplet energy transfer from Cy5 to COT (Scheme S1†). Correspondingly, the photon count should be linearly dependent on 1/τT if photobleaching solely occurs from T1. Such correlations were indeed observed for Cy5–COT(n) fluorophores, where n was >2 (Fig. 3A, red dots). However, Cy5–COT(1) and Cy5–COT(2) fluorophores deviated substantially from this trend (Fig. 3A). This finding substantiated the notion that these two fluorophores exhibit a pronounced increase in photobleaching probability through one or more additional pathways. Such degradation pathways may specifically arise from the proximity of COT to the Cy5 fluorophore, where at short distances, ground-state or excited-state COT molecules may react with Cy5 to promote photobleaching. In this context, we note that the lifetime of the COT triplet state (3COT*) is similar to that of 3Cy5* (ca. ∼ 100 μs),45 making it sufficiently long-lived for chemical reactions that lead to degradation.
 | ||
| Fig. 3 Observed correlations between fluorophore photostability, triplet state lifetime and number of linked COT molecules. (A) The photon count of Cy5–COT(n) fluorophores in deoxygenated solutions (80 W cm−2) vs. 1/τT, Cy5–COT(n > 2), and Cy5–bisCOT(3) (red circles); Cy5–COT(1) and Cy5–COT(2) (black rectangles). (B) The photon count of Cy5(0), Cy5–COT(n) (1) and Cy5–bisCOT(2) fluorophores in ambient oxygen, vs. the number of COT molecules covalently attached. | ||
Addition of a second COT molecule to Cy5 further increases photostability
To confirm that the reduced photostability of Cy5–COT(1) and Cy5–COT(2) fluorophores arises from physical proximity of COT to the Cy5 and not from an unexpected consequence of the shortened triplet state lifetimes, we synthesized Cy5–bisCOT(3) in which Cy5 was attached to two COT molecules via three-carbon linkers (Chart 1). Here, the second COT molecule is expected to reduce the Cy5 triplet state while maintaining a constant linker length. Consistent with this expectation, Cy5–bisCOT(3) exhibited a τT of 0.22 μs (Table 1, Fig. S5†), an approximately two-fold reduction in triplet state lifetime compared to Cy5–COT(3) (τT = 0.40 μs). This impact translated into a 50% increase of the fluorophore's photostability in deoxygenated buffers, the highest photon counts for any of the Cy5–COT(n) fluorophores examined (Table 1, Fig. 3A). These findings corroborate the notion that Cy5 photostability increases linearly with 1/τT (Fig. 3A). They also confirm that the reduced photon counts exhibited by Cy5–COT(1) and Cy5–COT(2) do not arise from their shortened triplet state, but instead arise from the proximity of COT to Cy5.Cy5–COT(n) fluorophores exhibit improved photostability under ambient oxygen conditions
Having examined the photophysical processes that determine Cy5–COT(n) performance under deoxygenated conditions, we next sought to quantify their photostability under ambient oxygen conditions. Consistent with the model that COT-mediated reductions in Cy5 triplet state lifetime protect the Cy5 fluorophore from damaging reactions with molecular oxygen (Scheme 1), each of the Cy5–COT(n) fluorophores exhibited approximately 3- to 5-fold increases in total photon counts compared to the parent Cy5 fluorophore (Table 1). In line with the additive COT impacts observed in deoxygenated buffers, the total photon count for the Cy5–bisCOT(3) fluorophore was increased by an additional 2–3 fold to approximately 9 fold compared to the parent Cy5 molecule (Table 1). Such findings support the prediction that the performance of self-healing fluorophores parallels the rates of intra-molecular triplet energy transfer. They also indicate that the rate of COT-mediated triplet energy transfer substantially exceeds the effective rate of damaging reactions with molecular oxygen and/or reactive oxygen species (ROS).5,41These data support the notion that it is possible, in principle, to achieve rates of fluorophore triplet quenching that are rapid enough to improve performance even in the presence of ambient oxygen, where in its limit, fluorophore performance could potentially reach the levels observed under deoxygenated conditions.5 Considered in this context, it is notable that Cy5 photostability in ambient oxygen correlates linearly with the number of COT molecules attached (Fig. 3B), whereas linear correlations were not observed as a function of triplet state lifetime (Fig. S6,†Table 1). This finding suggests that each COT molecule extends the Cy5 fluorophore photon count by an equal amount. Such observations indicate that COT may be inactivated through reactions with molecular oxygen or ROS followed by Cy5 photobleaching. Evidence supporting such an inactivation mechanism was directly observed in deoxygenated conditions, where ∼5% of single-molecule fluorescence traces exhibit abrupt transitions to states exhibiting behaviours akin to the parent Cy5 fluorophore followed by rapid photobleaching (Fig. S7†). We therefore hypothesized that the performance of self-healing fluorophores may be limited by an inherent instability of the COT protective agent.
Tuning COT's impact on fluorophore photostability
Exploiting this understanding, we reasoned that the photostability of self-healing fluorophores could be improved by stabilizing the protective agent itself. Based on previous work indicating that the reaction of COT with photo-generated singlet oxygen (1O2) is reduced when COT is substituted with an electron withdrawing group (EWG),46 we synthesized a Cy5 derivative linked to an amide-substituted COT molecule (AC) via an 11-atom linker (Cy5–AC(11) (Chart 2)). Consistent with the prediction that the amide EWG protects COT from reaction with 1O2 and/or other ROS, Cy5–AC(11) exhibited a two-fold increase in photon count compared to Cy5–COT(13) under ambient oxygen conditions (Fig. 4A). Cy5–AC(11) also showed enhanced photostability in oxygen-depleted buffer, exhibiting a 3-fold increase in photon count compared to Cy5–COT(13) (Fig. 4B). These distinctions could not be attributed to the 2-atom difference in linker length. | ||
| Chart 2 Structures of Cy5–COT(13), Cy5–AC(n) fluorophores used in this study. | ||
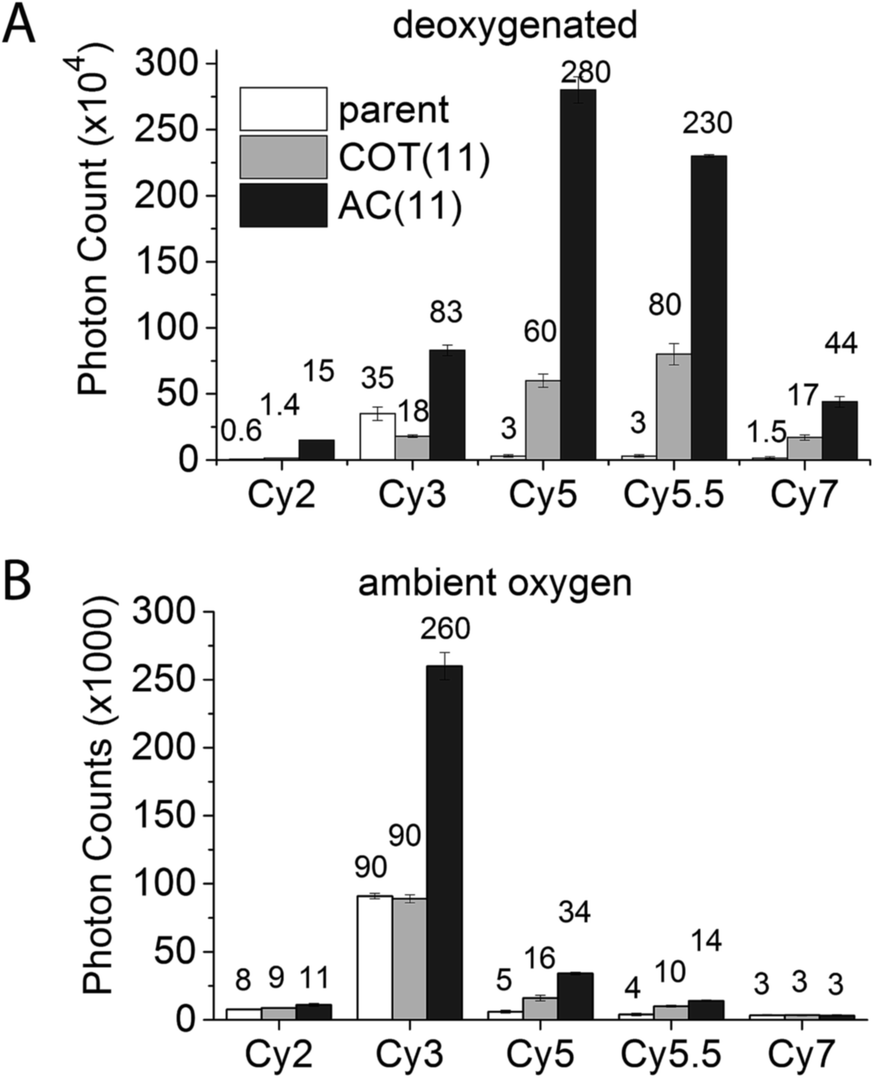 | ||
| Fig. 4 COT substituted with an amide electron-withdrawing group increases the photostability of cyanine-class organic fluorophores. Photon counts of cyanine fluorophores linked with COT or amide-COT in (A) deoxygenated buffer or (B) in buffer with ambient oxygen. | ||
Enhancements in performance exceeding those observed for unsubstituted COT were also observed for AC derivatives of Cy2, Cy3, Cy5.5, and Cy7, which span the visible spectrum (Fig. 4). For the Cy3 fluorophore, for which COT has been shown to have only marginal impact,18 AC derivatization improved its photon count nearly 3-fold in both oxygenated and deoxygenated solutions.
As for the Cy5–COT(n) fluorophores, further improvements in photostability were achieved by shortening the linker length (Chart 2). In ambient oxygen, Cy5–AC(4) and Cy5–AC(5) exhibited an ∼8–9 fold increase in photon counts compared to Cy5 and a 100% increase in photon count compared to Cy5–COT fluorophores of similar linker length (Fig. 5; Table 1). By contrast, shortening linker length from 13 atoms to 4 atoms did not significantly change Cy3 performance (data not shown). As previously demonstrated for the COT protective agent,17 the attached AC moiety had only minor (3–4 nm) bathochromic shifts on the absorption and emission spectra of the fluorophores (Table S1†), which we attribute to solvent or substitution effects.
 | ||
| Fig. 5 Amide-COT with a shortened linker further improves fluorophore photostability. Photon counts of Cy5, Cy5–COT(13), Cy5–COT(3), and Cy5–AC(4) (A) in oxygen-depleted buffer or (B) in buffer with ambient oxygen. | ||
The improved photostability of AC-linked fluorophores may arise from the reduced reactivity of AC with oxygen species. Alternatively, AC may more efficiently quench the triplet state than COT. Consistent with this latter hypothesis, we found that the triplet state lifetimes of Cy5–AC(n) (n = 4, 5, 11) derivatives were shorter than the equivalent Cy5–COT(n) fluorophores (Table 1, Fig. S8†). Nonetheless, the improved photostability for Cy5–AC(n) in ambient oxygen could not be completely attributed to the shortened triplet state, as they generated 100% more photons than the Cy5–COT(n) fluorophores exhibiting comparable τT (i.e. Cy5–COT(1), Cy5–COT(2), and Cy5–COT(3); Table 1). Hence, the improved photostability of AC-linked fluorophores likely arises from a combination of its reduced reactivity with oxygen species as well as its capacity to mediate more rapid triplet energy transfer with the fluorophore.
Although the mechanism of more efficient triplet state quenching by AC is not entirely clear, we speculate that the amide group changes the potential energy surface of COT in a manner that impacts the rate of “non-vertical” triplet energy transfer.38 As evidenced by the upfield chemical shifts observed by NMR and the decreased hydrophobicities of AC-linked fluorophores (see NMR spectra in ESI and Fig. S9†), this impact is likely to directly relate to electron withdrawing effects of the amide substituent. Investigations quantifying the impact and performance of COT derivatives bearing EWGs of varied strengths and placements will be needed to further substantiate this possibility.
Utility of AC dyes for live-cell imaging
We next examined if the improved self-healing strategy could be extended to cellular imaging. Such applications have previously utilized the cyanine fluorophore derivatives Dy549 and AF647, which bear a total of four sulfonate groups (two more than the Cy3 and Cy5 fluorophores) to reduce non-specific binding to cellular membranes.30 Hence, to compare the performances of Cy3–AC(4) and Cy5–AC(4) dyes to Dy549 and AF647, two additional sulfonate groups were introduced into the AC dyes (termed Cy3(4S)–AC(4) and Cy5(4S)–AC(4), respectively), making their hydrophobicity comparable to Dy549 and AF647 (Fig. S9†).After incubating living Chinese hamster ovary (CHO) cells stably expressing an N-terminally SNAPfast-tagged, human dopamine D2 receptor short isoform (SNAPf-D2s) with 500 nM benzylguanine activated Dy549, Cy3(4S)–AC(4), AF647, or Cy5(4S)–AC(4) fluorophores (supplementary methods in ESI†), objective-based single-molecule TIRF imaging revealed that each molecule exhibits comparable levels of specific labelling (Fig. 6A). Strikingly, both Cy5(4S)–AC(4) and Cy3(4S)–AC(4) dyes exhibited marked improvements in overall performance. In both fixed and living cells, the photobleaching time of Cy5(4S)–AC(4) was ∼6-fold longer than AF647 (Fig. 6B and C; ESI Movie 1†). Cy3(4S)–AC(4) exhibited 90% and 60% increases in photobleaching time in the fixed and living cells, respectively (Fig. S10; ESI Movie 2†). Such improvements were additionally accompanied by an approximately 20% increase in brightness (Fig. S11†).
 | ||
| Fig. 6 Cy5(4S)–AC(4) improves cellular imaging. (A) Number of fluorophores per cell for cells expressing SNAPf-D2s (SNAPf-D2s) compared to cells that do not (mock). (B) Comparison of photobleaching time for Cy5(4S)–AC(4) and AF647 in fixed cells and in living cells. (C) Single-molecule TIRF image time-sequences of living SNAPf-D2s-expressing CHO cells labeled with AF647 (top) or Cy5(4S)–AC(4) (bottom). | ||
We next sought to examine whether the intra-molecular photostabilization strategy extends to intracellular organic fluorophores. Silicon rhodamine (SiR), a fluorogenic, membrane-permeable dye that exhibits good photostability, has been widely used for advanced fluorescent microscopy in a cellular context.47–50 Both SiR–BG and SiR–AC–BG (Fig. S12A†) were first examined in the context of purified SNAP proteins immobilized on quartz surfaces. In these settings, SiR–AC exhibited a 90% increase in photon counts in ambient oxygen, and a 16-fold increase in deoxygenated conditions (Table S2†). To test whether AC also improve the performance of SiR in cellular contexts, we labelled a Gαi1-SNAPf fusion protein expressed in live CHO cells with either SiR–BG or SiR–AC–BG. Gα is an N-terminally myristoylated component of the heterotrimeric G protein complex, which localizes to cellular membranes to couple to activated G-protein coupled receptors and other targets. Low background labelling, good labelling efficiency and high-fluorescence intensity were observed for both fluorophores (Fig. S12B and C†), indicating SiR–AC is suitable for cellular imaging. SiR–AC also exhibited significantly improved (30%) photostability in fixed cells (Fig. S12D†). Following identical procedures, control experiments performed with SiR conjugated to COT showed very high non-specific cellular labelling (Fig. S13†), suggesting that SiR–COT adopts a fluorescent form when it is non-specifically bound. These data demonstrate that AC conjugation may be a general approach to improve fluorophores for intra-cellular imaging.
Conclusion
The present investigations advance our understanding of the self-healing mechanism and provide important insights on how to optimize the beneficial impacts of intra-molecular triplet state quenching to improve organic fluorophore performance. Our findings indicate that the probability of photobleaching from the triplet state can be substantially reduced by chemically engineering the protective agents employed to: [1] increase the rate at which they quench fluorophore triplet states; [2] make them resistant to oxidative damage; and [3] increase the rates at which they relax from the triplet state to the ground state. Exploiting these insights, we found that a simple structural modification – the addition of a modest EWG to the COT protective agent – further improves photostability across the visible and near-infrared spectrum. In ambient oxygen at the linker lengths tested, amide-COT improves the photostability up to 9 fold in vitro, and 6 fold in living cells. This improvement is expected to greatly expand the number and type of cellular processes that are accessible by single-molecule imaging. We anticipate additional improvements may be possible through further optimizations of both the linker length and composition.Future studies will be needed to examine the extent to which COT with EWGs of distinct strengths impact fluorophore performance. In principle, chemical engineering of COT and/or the introduction of alternative, more efficient, triplet state quenchers may potentially increase the rate of triplet energy transfer by multiple orders of magnitude. Combined with a reduced susceptibility to reactions with oxygen species, these efforts may ultimately afford “ultra-stable” organic fluorophores that exhibit bright, long-lived fluorescence over a broad range of biological imaging conditions and environments.5 The data presented also reveal that the probability of organic fluorophore photobleaching from higher-order excited states is non-trivial at elevated illumination intensities. This finding poses substantial challenges for imaging at high temporal resolution (ca. 100–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 frames per second), suggesting the need for fluorophore engineering strategies focused on this consideration. Progress towards overcoming both bottlenecks will be needed to unlock the full potential of the self-healing mechanism and to enable the intra-molecular photostabilization strategy to be generalized to other structurally distinct classes of organic fluorophores (e.g. rhodamines) commonly employed for cellular imaging.50,51
000 frames per second), suggesting the need for fluorophore engineering strategies focused on this consideration. Progress towards overcoming both bottlenecks will be needed to unlock the full potential of the self-healing mechanism and to enable the intra-molecular photostabilization strategy to be generalized to other structurally distinct classes of organic fluorophores (e.g. rhodamines) commonly employed for cellular imaging.50,51
Materials and methods
Chemical synthesis of self-healing fluorophores
The synthesis of Cy5–COT(n), Cy5–bisCOT(3), Cy5–AC(n) fluorophores and their N-hydroxysuccinimide (NHS) and oxothioxanthone (OTX) derivatives are described in the ESI.†Measurements of triplet state lifetime
Deoxygenated acetonitrile solutions containing 10 μM OTX-derivatives of Cy5 and Cy5–COT fluorophores were irradiated with light pulses from a Nd:YAG laser at 355 nm (5 ns pulse width) to generate transient absorption kinetic traces across the visible spectrum. As previously described,21 the 700 nm transient absorption was assigned to the triplet state of the Cy5 fluorophore. 10 × 10 mm quartz cells and right angle pump/probe geometry were used.Measurements of single-molecule fluorescence
As previously described, single-molecule fluorescence measurements were performed using a prism-based total-internal-reflection microscope.11,17 Fluorophores were conjugated to a biotinylated 21-base pair double-stranded DNA oligonucleotide and purified by hydrophobic interaction chromatography as previously described.18 The performance of each fluorophore was evaluated in this context over extended periods by immobilizing the labelled oligonucleotides within passivated quartz microfluidic chambers via a biotin–streptavidin interaction and imaging in T50 buffer (10 mM Tris acetate (pH 7.5) and 50 mM KCl) in the presence of ambient oxygen. 5 mM β-mercaptoethanol, 1 mM 3,4-dihydroxybenzoic acid (PCA), and 50 nM protocatechuate 3,4-deoxygenase (PCD) (Sigma-Aldrich) were added for imaging in deoxygenated conditions. All single-molecule data were analyzed using automated software built in-house using Matlab (MathWorks).11,17Acknowledgements
This work was supported by the National Institutes of Health (GM098859-01A1 to SCB; MH054137 & DA022413 to JAJ) and the National Science Foundation (CHE 11-11392 to SJ).Notes and references
- C. Joo, H. Balci, Y. Ishitsuka, C. Buranachai and T. Ha, Annu. Rev. Biochem., 2008, 77, 51–76 CrossRef CAS PubMed.
- B. Huang, H. Babcock and X. Zhuang, Cell, 2010, 143, 1047–1058 CrossRef CAS PubMed.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- E. M. S. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC.
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43, 1044–1056 RSC.
- Y. Sako, M. Hiroshima, C.-G. Pack, K. Okamoto, K. Hibino and A. Yamamoto, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2012, 4, 183–192 CrossRef CAS PubMed.
- Z. Liu, L. D. Lavis and E. Betzig, Mol. Cell, 2015, 58, 644–659 CrossRef CAS PubMed.
- R. E. Benesch and R. Benesch, Science, 1953, 118, 447–448 CAS.
- C. E. Aitken, R. A. Marshall and J. D. Puglisi, Biophys. J., 2008, 94, 1826–1835 CrossRef CAS PubMed.
- S. C. Blanchard, R. L. Gonzalez, H. D. Kim, S. Chu and J. D. Puglisi, Nat. Struct. Mol. Biol., 2004, 11, 1008–1014 CAS.
- R. Dave, D. S. Terry, J. B. Munro and S. C. Blanchard, Biophys. J., 2009, 96, 2371–2381 CrossRef CAS PubMed.
- A. N. Glazer, FASEB J., 1988, 2, 2487–2491 CAS.
- I. Rasnik, S. A. McKinney and T. Ha, Nat. Methods, 2006, 3, 891–893 CrossRef CAS PubMed.
- J. Vogelsang, R. Kasper, C. Steinhauer, B. Person, M. Heilemann, M. Sauer and P. Tinnefeld, Angew. Chem., Int. Ed., 2008, 47, 5465–5469 CrossRef CAS PubMed.
- J. L. Alejo, S. C. Blanchard and O. S. Andersen, Biophys. J., 2013, 104, 2410–2418 CrossRef CAS PubMed.
- J. J. Sakon and K. R. Weninger, Nat. Methods, 2010, 7, 203–205 CrossRef CAS PubMed.
- R. B. Altman, D. S. Terry, Z. Zhou, Q. Zheng, P. Geggier, R. A. Kolster, Y. Zhao, J. A. Javitch, J. D. Warren and S. C. Blanchard, Nat. Methods, 2011, 9, 68–71 CrossRef PubMed.
- R. B. Altman, Q. Zheng, Z. Zhou, D. S. Terry, J. D. Warren and S. C. Blanchard, Nat. Methods, 2012, 9, 428–429 CrossRef CAS PubMed.
- P. Tinnefeld and T. Cordes, Nat. Methods, 2012, 9, 426–427 CrossRef CAS PubMed.
- S. C. Blanchard, Nat. Methods, 2012, 9, 427–428 CrossRef CAS.
- Q. Zheng, S. Jockusch, Z. Zhou, R. B. Altman, J. D. Warren, N. J. Turro and S. C. Blanchard, J. Phys. Chem. Lett., 2012, 3, 2200–2203 CrossRef CAS PubMed.
- J. H. M. van der Velde, E. Ploetz, M. Hiermaier, J. Oelerich, J. W. de Vries, G. Roelfes and T. Cordes, ChemPhysChem, 2013, 14, 4084–4093 CrossRef CAS PubMed.
- Q. Zheng, Ph.D. thesis, Weill Medical College of Cornell University, 2016.
- J. H. M. van der Velde, J. Oelerich, J. Huang, J. H. Smit, A. Aminian Jazi, S. Galiani, K. Kolmakov, G. Guoridis, C. Eggeling, A. Herrmann, G. Roelfes and T. Cordes, Nat. Commun., 2016, 7, 10144 CrossRef CAS PubMed.
- T. C. Bruice and U. K. Pandit, J. Am. Chem. Soc., 1960, 82, 5858–5865 CrossRef CAS.
- F. C. Lightstone and T. C. Bruice, J. Am. Chem. Soc., 1996, 118, 2595–2605 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC/Taylor & Francis, Boca Raton, 3rd edn, 2006 Search PubMed.
- I. Konig, A. Zarrine-Afsar, M. Aznauryan, A. Soranno, B. Wunderlich, F. Dingfelder, J. C. Stuber, A. Pluckthun, D. Nettels and B. Schuler, Nat. Methods, 2015, 12, 773–779 CrossRef CAS PubMed.
- R. R. Nani, J. A. Kelley, J. Ivanic and M. J. Schnermann, Chem. Sci., 2015, 6, 6556–6563 RSC.
- P. J. Bosch, I. R. Corrêa Jr, M. H. Sonntag, J. Ibach, L. Brunsveld, J. S. Kanger and V. Subramaniam, Biophys. J., 2014, 107, 803–814 CrossRef CAS PubMed.
- P. J. Wagner and P. Klán, J. Am. Chem. Soc., 1999, 121, 9626–9635 CrossRef CAS.
- P. Klán and P. J. Wagner, J. Am. Chem. Soc., 1998, 120, 2198–2199 CrossRef.
- X. Allonas, C. Ley, C. Bibaut, P. Jacques and J. P. Fouassier, Chem. Phys. Lett., 2000, 322, 483–490 CrossRef CAS.
- S. Jockusch, O. Zeika, N. Jayaraj, V. Ramamurthy and N. J. Turro, J. Phys. Chem. Lett., 2010, 1, 2628–2632 CrossRef CAS.
- D. O. Cowan and A. A. Baum, J. Am. Chem. Soc., 1971, 93, 1153–1162 CrossRef CAS.
- P. J. Wagner and G. M. El-Taliawi, J. Am. Chem. Soc., 1992, 114, 8325–8326 CrossRef CAS.
- P. G. Wenthold, D. A. Hrovat, W. T. Borden and W. C. Lineberger, Science, 1996, 272, 1456–1459 CAS.
- L. M. Frutos, O. Castaño, J. L. Andrés, M. Merchán and A. U. Acuña, J. Chem. Phys., 2004, 120, 1208–1216 CrossRef CAS PubMed.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2010 Search PubMed.
- J. Widengren, A. Chmyrov, C. Eggeling, P. A. Lofdahl and C. A. Seidel, J. Phys. Chem. A, 2007, 111, 429–440 CrossRef CAS PubMed.
- X. Kong, E. Nir, K. Hamadani and S. Weiss, J. Am. Chem. Soc., 2007, 129, 4643–4654 CrossRef CAS PubMed.
- C. Eggeling, J. Widengren, L. Brand, J. Schaffer, S. Felekyan and C. A. M. Seidel, J. Phys. Chem. A, 2006, 110, 2979–2995 CrossRef CAS PubMed.
- T. N. Das and K. I. Priyadarshini, J. Chem. Soc., Faraday Trans., 1994, 90, 963–968 RSC.
- W. Adam, G. Klug, E.-M. Peters, K. Peters and H. G. von Schnering, Tetrahedron, 1985, 41, 2045–2056 CrossRef CAS.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z.-G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed.
- G. Lukinavicius, L. Reymond, E. D'Este, A. Masharina, F. Gottfert, H. Ta, A. Guther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H.-D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef CAS PubMed.
- G. Lukinavicius, C. Blaukopf, E. Pershagen, A. Schena, L. Reymond, E. Derivery, M. Gonzalez-Gaitan, E. D'Este, S. W. Hell, D. Wolfram Gerlich and K. Johnsson, Nat. Commun., 2015, 6, 8497 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, J. Chen, J. P. Slaughter, Z. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- Q. Zheng, S. Jockusch, G. G. Rodríguez-Calero, Z. Zhou, H. Zhao, R. B. Altman, H. D. Abruña and S. C. Blanchard, Photochem. Photobiol. Sci., 2016, 15, 196–203 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chart S1, Fig. S1 to S13, Scheme S1, Tables S1 and S2, note on calculating the effective concentration of protective agents, methods of cellular imaging and details the synthesis and characterization of compounds. Supplementary Movie 1: Single-molecule TIRF movies of living SNAPf-D2s-expressing CHO cells labeled with AF647 (left) or Cy5(4S)-AC(4) (right). Supplementary Movie 2: Single-molecule TIRF movies of living SNAPf-D2s-expressing CHO cells labeled with Dy549 (left) or Cy3(4S)-AC(4) (right). See DOI: 10.1039/c6sc02976k |
| This journal is © The Royal Society of Chemistry 2017 |