
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron supported on bioinspired green silica for water remediation†
Khalid M.
Alotaibi
a,
Lewis
Shiels
b,
Laure
Lacaze
b,
Tanya A.
Peshkur
c,
Peter
Anderson
c,
Libor
Machala
d,
Kevin
Critchley
 e,
Siddharth V.
Patwardhan
*f and
Lorraine T.
Gibson
*a
e,
Siddharth V.
Patwardhan
*f and
Lorraine T.
Gibson
*a
aDepartment of Pure and Applied Chemistry, WestCHEM, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: lorraine.gibson@strath.ac.uk; Tel: +44 (0)141 548 2224
bDepartment of Chemical and Process Engineering, University of Strathclyde, 75 Montrose Street, Glasgow, G1 1XJ, UK
cScottish Environmental Technology Network (SETN), Faculty of Engineering, University of Strathclyde, 204 George Street, Glasgow, G1 1XW, UK
dRegional Centre of Advanced Technologies and Materials, Palacký University, Šlechtitelů 27, 78371 Olomouc, Czech Republic
eMolecular & Nanoscale Physics Group, School of Physics & Astronomy, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
fDepartment of Chemical and Biological Engineering, University of Sheffield, Sheffield S1 3JD, UK. E-mail: s.patwardhan@sheffield.ac.uk; Tel: +44 (0)114 222 7593
First published on 13th September 2016
Abstract
Iron has been used previously in water decontamination, either unsupported or supported on clays, polymers, carbons or ceramics such as silica. However, the reported synthesis procedures are tedious, lengthy (involving various steps), and either utilise or produce toxic chemicals. Herein, the use of a simple, rapid, bio-inspired green synthesis method is reported to prepare, for the first time, a family of iron supported on green nanosilica materials (Fe@GN) to create new technological solutions for water remediation. In particular, Fe@GN were employed for the removal of arsenate ions as a model for potentially toxic elements in aqueous solution. Several characterization techniques were used to study the physical, structural and chemical properties of the new Fe@GN. When evaluated as an adsorption platform for the removal of arsenate ions, Fe@GN exhibited high adsorption capacity (69 mg of As per g of Fe@GN) with superior kinetics (reaching ∼35 mg As per g sorbent per hr) – threefold higher than the highest removal rates reported to date. Moreover, a method was developed to regenerate the Fe@GN allowing for a full recovery and reuse of the adsorbent in subsequent extractions; strongly highlighting the potential technological benefits of these new green materials.
Introduction
On the Earth's surface, only 2.53% of the total water is freshwater and only a third of this freshwater is accessible for drinking.1 Contamination of such a scarce commodity, by various pollutants such as toxic metal ions is a serious problem.2 This study focussed on the development of novel ‘green’ materials for the decontamination of aqueous systems using As(V) as a model toxic pollutant. Arsenic contamination, specifically inorganic arsenic species, has caused severe health problems in many countries such as Bangladesh, Canada, Cambodia, China, Hungary, India, and the USA; it is classified as a group 1 carcinogen.1,3–6 Chronic exposure to low arsenic levels has been linked to health complications, including cancer of the lung, kidney, skin, and bladder, as well as other potential effects including neurologic impairment.7,8 It is found in the environment as a naturally occurring substance, with the more toxic inorganic species being more abundant.9 In natural waters, two forms of inorganic arsenic are common: arsenite As(III) or arsenate As(V). As(III) is usually more mobile and toxic than As(V), and difficult to remove from water using techniques such as ion exchange, coagulation/precipitation and reverse osmosis.10 Therefore, the removal of As(III) is usually achieved after oxidisation to As(V), which can be adsorbed onto iron oxides, activated alumina, activated carbon or polymer resins.11–16Iron compounds have shown great promise in selective removal of arsenic from water. It is known that iron oxides, hydroxides and oxyhydroxides (e.g. ferrihydrite) selectively adsorb As(V) through the formation of mono- and bi-dentate “Fe–As” complex as shown in exemplar reaction below.12,17,18
| ≡FeOH + H2AsO4− + H+ → ≡FeH2AsO4 + H2O | (1) |
The formation of these complexes is dependent on solution pH, arsenic concentration, iron surface chemistry and the presence of other metal ions.17 Nano-iron particles with large active surface areas have been shown to provide high arsenic adsorption capacity, presumably though increased surface area.19,20 However, the use of iron nanoparticle powders is found to be prone to aggregation which not only leads to reduced efficiency,21 but also restricts their application in water treatment systems due to a rapid loss of iron particles into the drinking water.22 This necessitates the use of a support which holds the iron-nanoparticle during its application. High surface area supports that have been used include zeolites, activated carbons, and mesoporous silica.22–24 When supported, aggregation of iron particles reduced significantly, the material had improved stability and better dispersion of iron nanoparticles leading to more efficient catalysts in decontamination applications.25–29 Porous silica has been of wide interest as a support for dispersing iron particles.30,31 Several methods have been proposed for the synthesis of mesoporous silica materials,32,33 but secondary pollution problems are often created at the end of the process when unused reactants (e.g. toxic alkoxysilanes) are discarded.34,35 Most reported syntheses involve lengthy, multistep procedures (11–72 h), high temperatures (80–120 °C) and extreme pH conditions (pH ∼ 1) creating additional problems. Thus, considerable efforts have been made to develop eco-friendly routes, taking into consideration the whole life-cycle of mesoporous silica materials from extraction of the raw material to disposal at the end of their life.
Porous silica materials can be prepared using bioinspired green routes that mimic the biomineralisation process.36,37 The popularity of this method is down to the ease of synthesis, the use of mild conditions and less toxic reagents. The understanding of the mechanism of biological silica formation in organisms has led to silica being produced in vitro under environmentally friendly mild reaction conditions, while maintaining a high level of control over the product.38 Here synthetic “additives” are used to produce silica rapidly and under mild conditions.39 Furthermore, bioinspired methods allow better control of the chemical and physical properties of the products with one-pot synthesis of hybrid materials such as optical materials, catalysts and biomaterials.38,40–43
In this study the versatility of green nanoparticles (GN) for water remediation is examined using arsenic as a target pollutant. The synthesis is simple, fast, and uses non-toxic reagents to produce porous green nanoparticles (GN) encapsulating iron. Although the preparation of nanoparticles encapsulated in silica using bioinspired routes has been demonstrated for quantum dots, iron oxide particles and gold particles,41,44–46 iron supported on silica (Fe@GN) has not been reported before, let alone for applications in environmental remediation. As shown in Table 1, there are significant advantages of the Fe@GN preparation method, compared to Fe-mesosilica (MS). The novel GN products were synthesised and characterised (see ESI†) in order to determine their chemical, textural and surface properties. The new materials were assessed at different extraction pH, before being used, and regenerated, for the removal of arsenic(V) from environmental samples under batch or continuous flow.
| Conditions | Fe-MS | Fe@GN | |
|---|---|---|---|
| Step 1 | Chemicals | Alkoxysilane, surfactant, ethanol | Sodium silicate, additive, iron precursor, water |
| T, t, pH | 60–100 °C, 2–5 d, pH 2 or 10 | 20 °C, 15 min, pH 7 | |
| Step 2 | Chemicals | Iron precursor, acetone or ethanol | n/a |
| t | 5–24 h | n/a |
Experimental
Materials and reagents
Sodium metasilicate (Na2SiO3·5H2O, Fisher Scientific), pentaethylenehexamine (PEHA, C10H28N6 Sigma Aldrich), iron nitrate (Sigma Aldrich), borohydride solution (NaBH4 Sigma Aldrich), sodium arsenate dibasic heptahydrate (99.995%, Sigma Aldrich), hydrochloric acid (HCl, 36 wt%, Fisher Scientific), nitric acid (HNO3, 65 wt%, Fisher Scientific) and ethanol absolute (Sigma Aldrich) were used as supplied. Glassware was soaked in 5% HNO3 overnight and cleaned with deionized water before use. Deionized water was used throughout this work.Synthesis of GN and Fe@GN
The bioinspired green nanosilica, GN, was synthesized using a method reported by Ewlad-Ahmed et al.47 A solution of sodium metasilicate in deionized water was acidified using 1 M HCl and mixed with a solution of PEHA and, if required, the final solution pH adjusted with 1 M HCl to 7.0 ± 0.1. The final concentrations of silicate and nitrogen (from the amine) were each 30 mM. Rapid precipitation of silica was visible within 1–2 min. The resultant as-synthesized precipitate was collected after 15 min by filtration, washed three times with deionized water and dried in air at room temperature (hereafter referred to as “D-GN”) before calcination at 550 °C for 5 h, to produce GN.Fe@GN was prepared by dissolving the desired amount of iron nitrate in water prior to addition to the silicate–PEHA mixture immediately before the pH was set to 7. After mixing the solution, the pH was adjusted to 7.0 ± 0.1 as above. The solution was left for approximately 15 min producing as-synthesized iron-silica precipitate which was collected by filtration, washed three times with deionized water and dried in air at room temperature (hereafter referred to as “D-Fe@GN”) before calcination at 550 °C for 5 h, to produce Fe@GN.
Reduction of GN
0.5 M of sodium borohydride solution was added in excess to Fe@GN or D-Fe@GN and the solution was left to react for 1 h. Since the reduction reaction produced hydrogen gas, the cap of vial was removed frequently to avoid pressure build up. The solution was centrifuged for 15 min at 8000 rpm and the supernatant was disposed of. The reduced Fe@GN was then washed with ethanol and centrifuged. This method was repeated twice and the particles were dried in oven at 85 °C for 2 h to produce R-Fe@GN.Characterisation and analysis
Nitrogen adsorption–desorption isotherms were collected using a Micromeritics ASAP 2420 surface area and porosity analyser after sample degassing for 6 h at 200 °C. The Brunauer–Emmett–Teller (BET) surface areas were calculated using experimental points at a relative pressure (P/P0) of 0.05–0.25. The total pore volume was calculated from the N2 amount adsorbed at the P/P0 of 0.99, and the average pore size distribution was calculated using the Barret–Joyner–Halenda (BJH) model from a 28-point BET surface area plot. Desorption isotherms were used to calculate the pore diameters.Elemental analysis was carried out using an Exeter Analytical CE440 elemental function to provide the functional group (carbon and nitrogen) and hydrogen content of the studied materials. Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) data were obtained using an ABB MB3000 instrument and analysed as described elsewhere.47,48 The transmission 57Fe Mössbauer spectra were collected using a Mössbauer spectrometer in a constant acceleration mode with a 57Co(Rh) source. The isomer shift values were related to metallic α-Fe at room temperature (RT). The measurements were performed at RT and 5 K in a zero external magnetic field and at 5 K in an external magnetic field of 5 T, applied parallel to the direction of the gamma-rays propagation. Low temperature and in-field measurements were obtained using a cryomagnetic system by Oxford Instruments.
X-ray photoelectron spectroscopy data were obtained using a Thermo Electron Corporation ESCA Lab 250 instrument with a chamber pressure maintained below 1 × 10−9 mbar during acquisition. A monochromated Al Kα X-ray source (15 kV 150 W) irradiated the samples, with a spot diameter of approximately 0.5 mm. The spectrometer was operated in large area XL magnetic lens mode using pass energies of 150 and 20 eV for survey and detailed scans, respectively. The spectra were obtained with an electron takeoff angle of 90°. Charge compensation was applied using a low energy flood gun. High-resolution spectra were fitted using Avantage (Thermo VG software package) peak fitting algorithms.
Adsorption isotherms for the maximum adsorption capacity evaluation
A Thermo Scientific iCAP 6200 ICP-OES was used for the determination of arsenic concentration in aqueous solutions. Calibration standards (0.1–1 μg cm−3) were prepared in 5% HNO3. A quality control check solution was prepared at 0.1 μg cm−3 to ensure the accuracy of the method used and was repeatedly analysed after 10 samples. The method detection limit (MDL) for arsenic was 0.004 μg cm−3 using a wavelength of 189 nm.To study the maximum adsorption capacity of arsenic on the prepared GN, the Langmuir and Freundlich isotherm models were applied to experimental data. Extraction experiments used 25 cm3 spiked solutions (40, 60, 80 or 100 μg cm−3) of As(V) and 50 mg of each GN adsorbent. Solutions were held at 20° C, pH 3 and stirred at 250 rpm for 120 min. The amount of As(V) extracted at equilibrium; qe (mg g−1) was calculated according to the following equation:
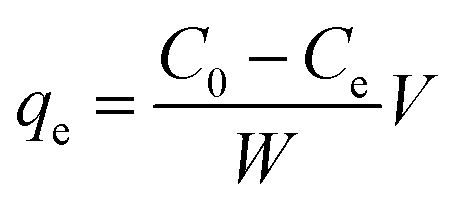 | (2) |
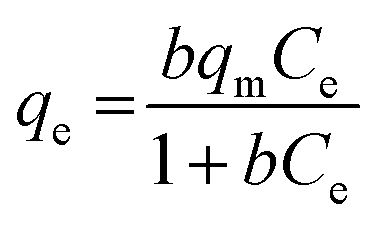 | (3) |
| qe = KfCe1/n | (4) |
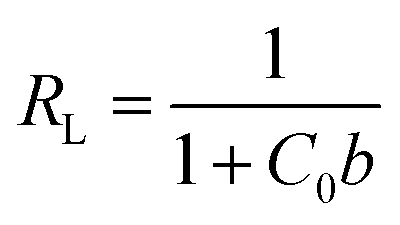 | (5) |
The efficiencies of sorbents in real water samples
To evaluate the environmental performance of sorbents with real samples, a river water sample (collected from the Bothlin river in Glasgow) and a tap water sample (taken from a building in the Glasgow harbour terraces in Glasgow) were analysed. The river and tap water samples were known to contain high concentrations of metal ions, but not arsenic (see Table S1†) and so they were spiked to produce solutions containing 1 μg cm−3 of As(V). In each experiment 25 mg of Fe@GN was added to solutions which were stirred for 2 h. Then the solutions were filtered and the aliquots were analysed by ICP-OES to examine the extraction efficiency. Then the material was washed with 0.1 M sodium hydroxide to examine the possibility to recover metals ions from the sorbent after extraction.Examination of sorbent performance in dynamic mode
An accurate mass, 50 mg, of Fe@GN was mixed with 2 g of glass beads (1.5 mm) and packed in a self-made polypropylene column (6 cm3) plugged with a modified filter paper at both ends to give a final height of approximately 2 cm. In order to equilibrate, clean and neutralize the column before use, diluted solutions of hydrochloric acid and doubly distilled water were passed over the sorbent bed. Then sample solutions containing As(V) were prepared with a pH value being adjusted to 3 using 1 M HCl and 1 M ammonia solutions. Each solution was allowed to pass through the column at flow rate of 1, 2, 3, 5, 7, or 9 cm3 min−1 controlled by a peristaltic pump. Finally, the removed arsenic ions were recovered using 0.1 M sodium hydroxide. The eluates were then collected prior to analysis by ICP-OES.Results and discussion
Materials characterisation
Our initial investigations using iron loading from 0–50% indicated a gradual increase in the surface area from ∼20 m2 g−1 to ∼240 m2 g−1 with increasing iron loading, and typically samples with higher iron content exhibited higher pore volumes.52 Therefore, here we decided to focus on 50% iron loaded samples in order to allow systematic investigations.With 50% Fe iron loading, five samples were prepared for decontamination assessment studies: 3 with iron (D-Fe@GN, Fe@GN and R-Fe@GN) and 2 without iron (D-GN and GN) where a prefix “D” indicates dried at room temperature, “R” indicates reduced and no prefix suggests calcined samples (see Table 2). The samples prepared without Fe (D-GN and GN) both demonstrated Type II isotherms, with the GN samples also illustrating a H4 hysteresis that was attributed to the presence of a small amount of mesopores (Fig. S1a and b†).53 The porosity results for all samples are summarised in Table 2. Sample D-GN had the lowest surface area at 12 m2 g−1 (and no pore volume or pore size, presumably due to template inclusion), which increased significantly, as expected, to 347 m2 g−1 after the removal of the bioinspired additive to produce a material with a pore size and pore volume of 3.5 nm and 0.23 cm3 g−1, respectively. When iron was incorporated into the synthesis mixture the silica framework of GN appeared to greatly alter and the materials produced illustrated Type IV N2 sorption isotherms, as shown in Fig. S1c† suggesting that all Fe containing GN were mesoporous. Even without the removal of the additives, D-Fe@GN had a surface area of approximately 244 m2 g−1 and an average pore size of approximately 18 nm; the latter being an interesting and remarkable feature that can provide accessibility to bulky analytes. This characteristic of D-Fe@GN could be attributed to the influence of the Fe3+ ions on silica condensation reactions, manifesting in a material with a significantly different microstructure compared to the D-GN sample.
| Sample | Fe wt% | Finishing | Surface areaa (m2 g−1) | Pore sizeb (nm) | Pore volumec (cm3 g−1) |
|---|---|---|---|---|---|
| a Calculated by the BET model from sorption data in a relative pressure range from 0.05 to 0.25. b Calculated by the BJH model from the desorption branches of isotherms. c Calculated from N2 amount adsorbed at a relative pressure P/P0 of 0.99. | |||||
| D-GN | 0 | Air dried | 12 | Nil | Nil |
| GN | 0 | Calcined | 347 | 3.5 | 0.23 |
| D-Fe@GN | 50 | Air dried | 244 | 18.6 | 0.75 |
| Fe@GN | 50 | Calcined | 203 | 18.6 | 0.6 |
| R-Fe@GN | 50 | Reduced | 129 | 18 | 0.47 |
The presence of nitrogen and carbon in the air dried samples confirmed the retention of PEHA in samples D-GN and D-Fe@GN, see elemental analysis data in Table 3. Interestingly, when the samples with and without iron were compared, the amine loading reduced from 4.49 for D-GN to 2.66 mmol g−1 for D-Fe@GN (reduction in both N and C content; Table 3). This implied that the presence of iron in the synthesis solution had a direct effect on amine loading, perhaps through ionic interactions. Calcination appeared to completely remove PEHA; carbon and nitrogen content reduced to undetectable levels (GN and Fe@GN). Furthermore, the reduction by sodium borohydride seem to have increased the hydrogen content, perhaps through hydration of the samples.
| Sample | % C | % H | % N | L 0 (mmol g−1) |
|---|---|---|---|---|
| a Degree of residual PEHA (L0 millimoles of nitrogen per gram of silica). | ||||
| D-GN | 9.36 | 3.06 | 6.29 | 4.49 |
| GN | 0 | 1.08 | 0 | |
| D-Fe-GN | 5.31 | 2.92 | 3.73 | 2.66 |
| Fe-GN | 0 | 0.2 | 0 | |
| R-Fe-GN | 0 | 1.08 | 0 | |
In order to further investigate the composition of inorganic components and the oxidation state of iron, XPS analyses were performed. XPS analysis provided the evidence of silicon and iron in the samples (Table 4 and Fig. 1a). Upon chemical reduction, oxygen (not shown) and hydrogen content increased (Table 3), consistent with the possibility of hydration upon reduction. Further analysis of the Fe 2p1 peak for the R-Fe-GN (Fig. 1b) indicated that most of the Fe species are likely to be either in the Fe2+ or Fe3+ oxidation state, while the lack of a strong satellite peak suggests it is more likely Fe3+. A weak shoulder at binding energy of 707 eV indicates the presence of very little amount of Fe0 metal, if any. This result is surprising because it was expected that upon chemical reduction, most iron will be converted to zero valent metal. These results were further validated by using Mössbauer spectroscopy.
 | ||
| Fig. 1 (a) Representative XPS survey scans for two iron containing samples. (b) High resolution Fe 2p1 spectrum for R-Fe-GN sample. | ||
Mössbauer spectroscopy provided additional information on the chemical nature of the iron in the samples (also see ESI†). Room temperature Mössbauer spectra (not presented) of the both samples (before and after the sodium borohydride treatment) indicated a presence of octahedral trivalent iron atoms in the structure of ferric oxide or (oxy)hydroxide. No zero valent and/or divalent iron were detected in the spectra, even for samples reduced using sodium borohydride for extended periods. More detail information was obtained from low temperature (T = 5 K) and in-field (B = 5 T) spectra (Fig. 2). The zero-field spectrum of the sample before the sodium borohydride treatment (Fig. 2a) reflects unusually low magnetic ordering temperature of the ferric oxide or (oxy)hydroxide phase and thus a presence of very small nanoparticles (<10 nm). The 5 K zero-field spectrum of the sodium borohydride treated sample (Fig. 2b) shows the presence of nanoparticles which were smaller in comparison with the “non-treated” sample. The superparamagnetic regime was confirmed by in-field Mössbauer spectra (Fig. 2c and d). The quadrupole shifts of the both sextets (Fig. 2c and d) were close to zero indicating amorphous Fe2O3 or Fe(OH)3. The Mössbauer spectroscopy data confirmed that nanoparticles of ferric oxide or hydroxide were incorporated, and were probably uniformly distributed, within the silica matrix. The long-term sodium borohydride treatment did not result in a reduction of the ferric oxide phase at all. It did however result in a particle size decrease.
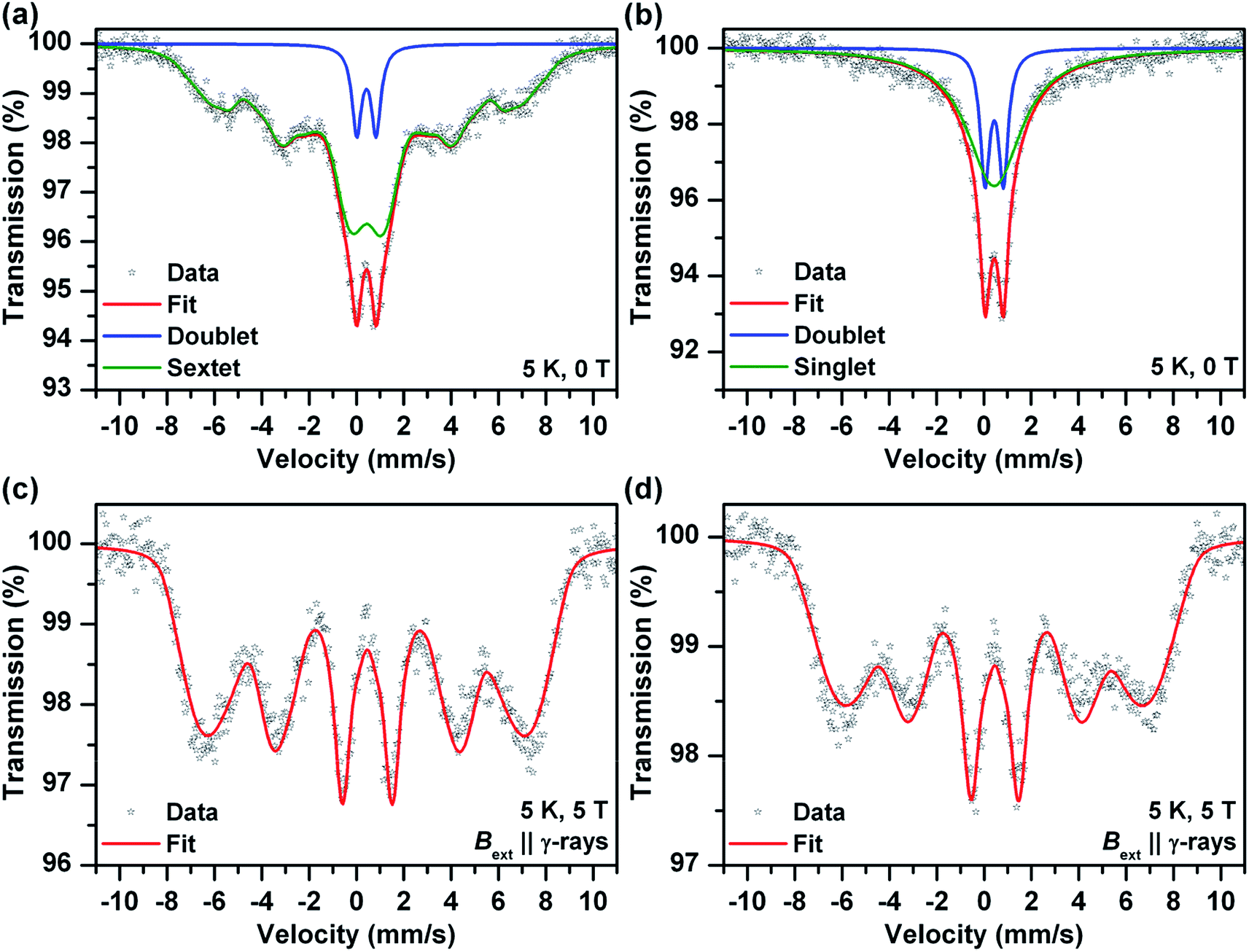 | ||
| Fig. 2 Zero-field and low temperature (5 K) Mössbauer spectrum of the sample before (a) and after (b) sodium borohydride treatment. In-field (5 T) and low temperature (5 K) Mössbauer spectrum of the sample before (c) and after (d) sodium borohydride treatment. | ||
Further characterisation of the D-GN and D-Fe@GN samples was performed using ATR-FTIR (Fig. 3a). The results supported the formation of silica (Si–O–Si peaks at ∼1100 cm−1 and 800 cm−1). The presence of PEHA was also detected from amine peaks located in the region of 1500–1700 cm−1. Although the shape of the siloxane peak at ∼1100 cm−1 was as expected for the sample without iron (D-GN), the shape of this peak was found to significantly change for the sample containing iron (D-Fe@GN). This suggested that the inclusion of iron affected the materials produced at a molecular level and further supported the observed differences in porosities discussed above between samples with and without iron. In order to further probe the effect of iron on silica, peak deconvolution protocol was applied to the ∼1100 cm−1 peak of both samples (Fig. S2a and b†). Analysis of the D-Fe@GN sample peak indeed highlighted drastic differences, in particular, the presence of additional peaks at ∼954 cm−1 and ∼870 cm−1. In the literature, these peaks have been commonly found in sol–gel materials and were attributed to Fe–O–Si/Fe–O–OH.54,55 Furthermore, as the iron content was increased from 0% to 50%, the area under the Fe–O–Si peak was found to increase (Fig. 3b), thus further strengthening the formation of iron oxide–silica composite material.
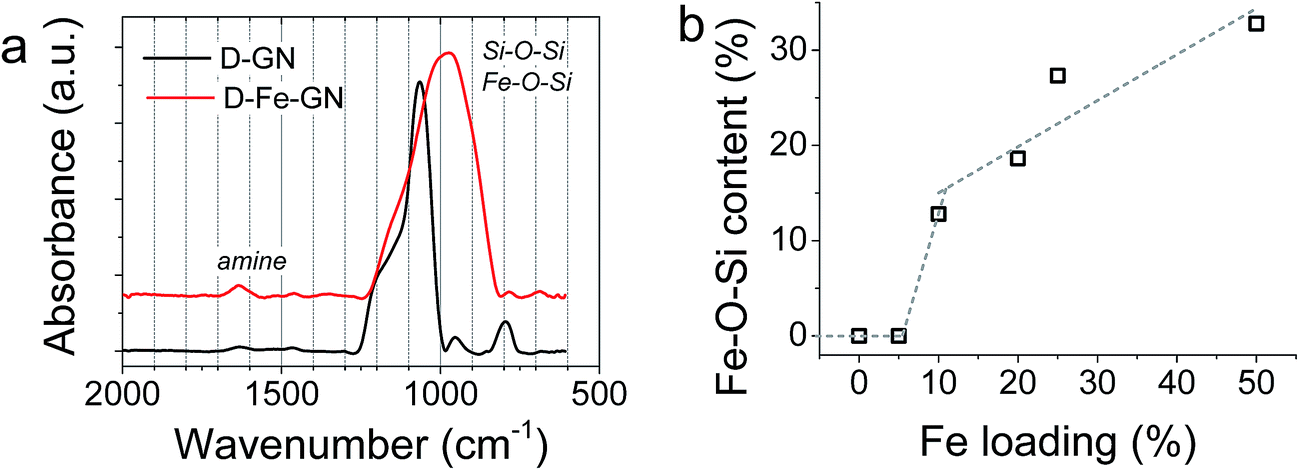 | ||
| Fig. 3 (a) ATR-FTIR spectra for samples with and without iron. (b) iron content, measured from the area under the Fe–O–Si peak, as a function of iron loading. For the 5% iron sample, the iron content was below the detection limit. | ||
In summary, the materials characterisation suggested that the presence of iron profoundly affected the chemical nature of the GN samples, in addition to the physical properties, as observed from the porosity measurements. The surface area was found to depend on iron loading, calcination and chemical reduction. Further, chemical reduction increased the hydration of samples. It was clear that iron was not in a zero valent form, but rather in an FeII or, more likely, FeIII oxidation state, presented as well-dispersed, <10 nm particles of iron oxide, hydroxide and/or oxyhydroxide, in addition to Si–O–Fe.
The selective removal of arsenic using Fe@GN
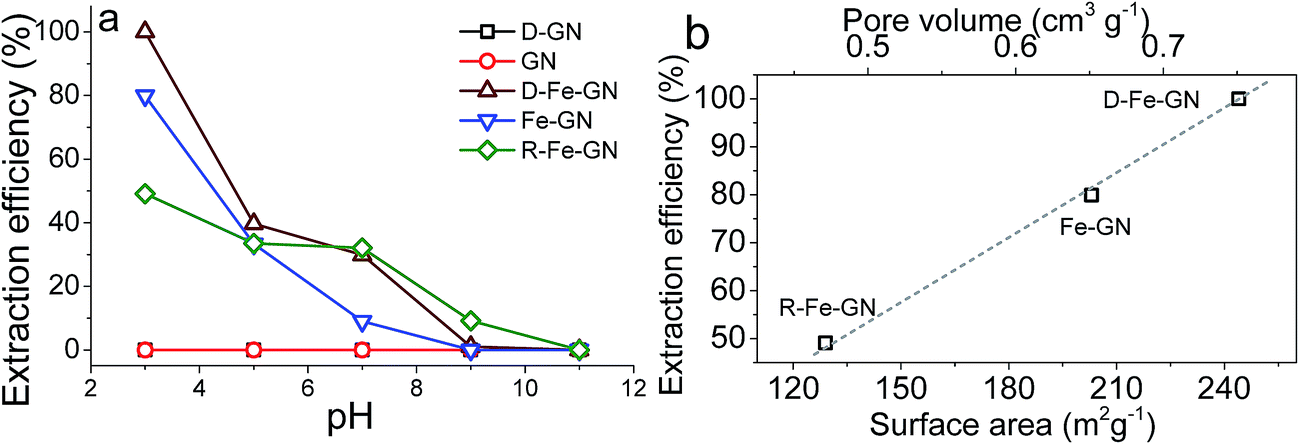 | ||
| Fig. 4 (a) Effect of pH on the extraction of arsenic ions from water using variety of GNs and iron modified GN. (b) Extraction efficiency as a function of surface area and pore volume of iron containing samples. | ||
The highest extraction efficiencies were observed at pH 3, which corroborates with the literature.17 Under acidic conditions, and specifically at pH 3, the dominant As(V) species is H2AsO4−.56 Under acidic pH condition, iron oxyhydroxides, such as those present in our samples, possess positive charge (typically >+1).17 Furthermore, the extraction efficiency of 100% at pH 3 may also indicate the possibility of co-operative effects from PEHA and iron towards the adsorption of this bulky inorganic anion. The surface amines, detected by FTIR and XPS, will be protonated at pH 3, further facilitating interactions between the negatively charged arsenic ions with the GN surface.57 It was interesting to note that R-Fe@GN exhibited the lowest arsenic extraction efficiency amongst the iron containing samples. Materials characterisation revealed that R-Fe@GN had lower surface area compared to the un-reduced Fe@GN sample (Table 2). Furthermore, XPS analyses suggested that upon reduction, some iron was lost, thus reducing the total iron content of the R-Fe@GN sample. These two observations – reduced surface area and reduced iron content – perhaps help explain why R-Fe@GN had the lowest arsenic extraction efficiency. This was further supported when the surface area and pore volume for all 3 iron containing samples were compared with extraction efficiencies (Fig. 4b). It was clear that both iron content and high surface area were crucial for As(V) removal – the former was known to actively interact with arsenic while the latter maximises the mass transport.
Adsorption isotherms for As(V) on D-Fe@GN
Focusing on the most promising adsorption platform for As(V) removal at pH 3, the adsorption capacity value was determined and compared with other iron-encapsulated materials quoted in the literature. The experimental data is shown in Fig. 5 as the amount of As(V) adsorbed at equilibrium (qe) as a function of equilibrium solution concentration (Ce). Langmuir and Freundlich isotherm models correspond to homogeneous and heterogeneous adsorbent surfaces, respectively.49 The Langmuir isotherm was originally derived for the adsorption of gas molecules onto smooth and ideal solid surface, while for aqueous systems, Freundlich isotherm is most adequate and hence it was preferred.58,59 The experimental adsorption data of As(V) on D-Fe@GN had a good fit with both the Langmuir and Freundlich models (Fig. 5 and Table 5), which indicated more than one type of interaction between the arsenic ions and the surface of Fe-GN.60 As discussed in the introduction section, it is well-known that iron has specific selectivity towards chemisorbing As(V). In addition to iron, our samples contain amine groups from the additive (PEHA). Under the acidic conditions used for arsenic removal (pH 3), these amines will be highly protonated, thus providing additional sites for arsenic binding on silica surface. The calculated RL value was in the range of 0 and 1, suggesting favourable adsorption properties for arsenic species onto D-Fe@GN.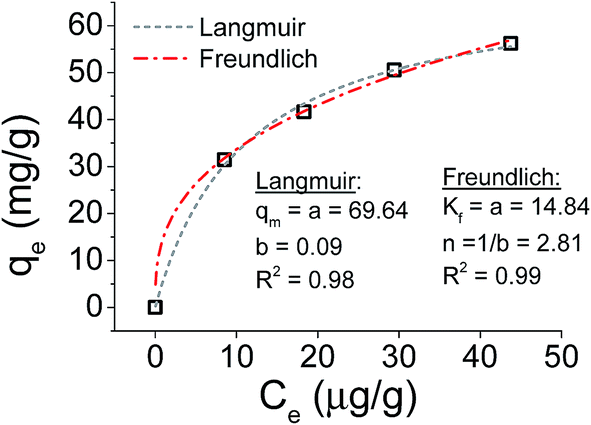 | ||
| Fig. 5 Adsorption isotherms for As(V) on D-Fe-GN fitted using Freundlich and Langmuir isotherms. | ||
| Method | Langmuir | Freundlich | |||
|---|---|---|---|---|---|
| q m (mg g−1) | b (L mg−1) | R L | K f (mg g−1) | n (L mg−1) | |
| As made Fe-GNs | 69.64 | 0.09 | 0.2 | 14.84 | 2.81 |
A comparative evaluation of Fe-GNs and other iron containing low cost adsorbents for arsenic removal is listed in Table 6. It was observed that the adsorption capacity of D-Fe-GN (69.64 mg g−1) was higher than many other sorption materials reported in the literature for the removal of As(V) (e.g., the maximum adsorption capacity of the iron oxide nanoparticles immobilized on activated carbon was 35.34 mg g−1). Despite the extensive use of activated carbon (AC) in the water and wastewater treatments, AC is perhaps not ideal as it is difficult to separate powdered AC from aquatic system when it becomes exhausted.61 Furthermore, the regeneration of AC by chemical or thermal procedures are expensive and can result in sorbent loss.62 A number of candidates, e.g. Zr(IV)-loaded chelating resin, akaganeite nanocrystals and polyethylene mercaptoacetimide exhibit superior performance compared to GN, however, these materials have been reported to require several hours for equilibration. In contrast, D-Fe-GN provide a rapid adsorption platform, reaching 34.82 mg g−1 h−1 – a threefold increase when compared to the highest capacity reported in the literature. To the best of our knowledge, the highest iron loading reported in the literature for GAC was 33.6%,63 and as higher iron oxide loading provides better adsorption capacities, the material produced here with an iron loading of 50% is higher than any other loading reported in the literature.
| Sorbent | Capacity (mg g−1) | Equilibration time (h) | Capacity (mg g−1 h−1) | Ref. |
|---|---|---|---|---|
| Activated carbon, Fe modified | 35.34 | 48 | 0.74 | 64 |
| Clinoptilolite, Fe modified | 30.21 | 48 | 0.63 | 64 |
| GAC-Fe (0.05 M) | 2.96 | 24 | 0.12 | 65 |
| Fe10SBA-15 | 12.68 | 24 | 0.53 | 66 |
| Cellulose loaded with iron oxyhydroxide | 15.6 | 24 | 0.65 | 67 |
| Synthetic siderite | 31 | 3 | 10.33 | 68 |
| Zr(IV)-loaded chelating resin (Zr-LDA) | 88.73 | 24 | 3.70 | 69 |
| Akaganeite β-FeO(OH) nanocrystals | 141.3 | 24 | 5.89 | 70 |
| Poly ethylene mercaptoacetimide | 105.75 | 20 | 5.29 | 71 |
| Magnetite–maghemite nanoparticles | 6 | 3 | 2.00 | 72 |
| D-Fe-GN | 69.64 | 2 | 34.82 | This work |
Assessment of Fe-GN performance in real samples
To examine the performance of iron containing GN samples for As(V) extraction in the presence of other cations, 25 mg of D-Fe@GN was added to 100 cm3 samples of distilled water, tap water or river water, each spiked with As(V) to a solution concentration of 1 μg cm−3. The results given for stirred batch experiments, in Table S2† indicate that extraction efficiencies of almost 100% were achieved for the removal of As(V) from all samples studied herein (<0.005 As detected after extraction). These exciting results implied that the coordination bonds between D-Fe@GN and the As(V) anions were not appreciably hindered by the presence of other coordinating ions at high concentration. Significantly, upon washing the As(V) loaded D-Fe@GN samples with 0.1 M sodium hydroxide >96% As(V) was recovered. This suggested a strong potential of these new materials in water treatment applications where regeneration and reusability of sorbents, in addition to good adsorption capacities is crucial to their economic success.Examination of sorbent performance in packed-bed under continuous flow systems
Although the batch extraction results provide the fundamental information related to sorbent behaviour and metal sorption performance, a continuous mode of operation would be preferred in large scale water treatment applications with perceived advantages including high yields, simple operation, easy regeneration of packed bed, and ease of scale up from a laboratory to industrial or environmental application.73 It has been reported that regeneration of iron compounds after As(V) adsorption is challenging but critically important for a long-term performance.12 Hence, the extraction of As(V) was also studied in continuous flow at a maximum flow rate of 9 cm3 min−1 (the flow limits of the system used). The results shown in Fig. 6 indicate that almost 100% extraction efficiencies and 100% recovery efficiencies were achieved over the flow rate range studied. More importantly this system involved reuse and clean-up of the same sample cartridge which (as shown in Fig. 6) has been used 6 times without any detrimental effects on either extraction or As(V) recovery values.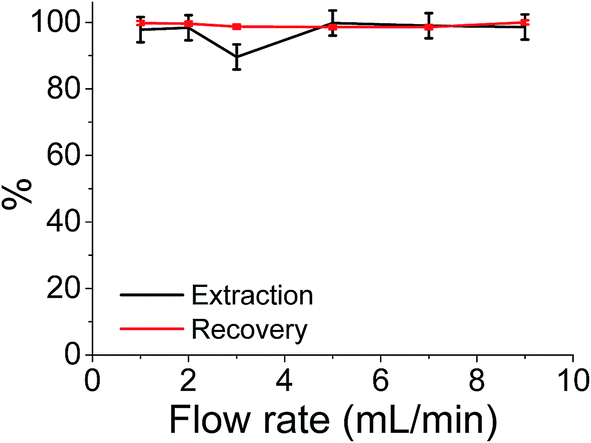 | ||
| Fig. 6 The extraction or recover of arsenic ions under different flow rate. | ||
Conclusions
We report the successful use of a new family of iron supported on green silicas for water remediation. The preparation of Fe@GN materials exhibits the advantages of eco-friendly process with a substantial reduction in secondary pollution. The addition of iron during synthesis was found to control the silica framework produced. Their use for the removal of As(V) – a most difficult PTE in aqueous systems – was evaluated which revealed that Fe@GN exhibited high adsorption capacity with superior kinetics. These new materials can be easily regenerated and retain their As(V) removal efficiencies upon reuse, thus providing a significant advantage over existing adsorption technologies for the removal of As(V). These advantages represent a significant progress into environmental remediation technologies.Acknowledgements
The Saudi Arabian Government is thanked for funding a PhD studentship for KA. Department of Chemical and Process Engineering, ERASMUS exchange scholarship (to LL) and the EPSRC vacation bursary (to LS) are also thanked for their financial support. Leeds EPSRC Nanoscience and Nanotechnology Research Equipment Facility is thanked for access to XPS analysis and PACER award (Strathclyde) is thanked for supporting collaboration with Palacký University. We thank Dr T. Yip for help with FTIR. The Mössbauer spectroscopy work has been also supported by the project LO1305 of the Ministry of Education, Youth and Sports of the Czech Republic, by the Operational Program Education for Competitiveness – European Social Fund (projects CZ.1.07/2.3.00/20.0017 and CZ.1.07/2.3.00/20.0058 of the Ministry of Education, Youth and Sports of the Czech Republic), and by the Technology Agency of the Czech Republic “Competence Centres” (project TE01020218). The authors thank Jiri Tucek (Palacký University in Olomouc, Czech Republic) for the help with Fig. 2 presenting Mössbauer spectra.References
- J. C. Ng, J. Wang and A. Shraim, Chemosphere, 2003, 52, 1353–1359 CrossRef CAS PubMed.
- Water and Sanitation Related Diseases and the Environment: Challenges, Interventions and Preventive Measures, ed. J. M. H. Selendy, Wiley-Blackwell, 2011 Search PubMed.
- S. Bhattacharjee, S. Chakravarty, S. Maity, V. Dureja and K. K. Gupta, Chemosphere, 2005, 58, 1203–1217 CrossRef CAS PubMed.
- C. F. Harvey, Science, 2002, 298, 1602–1606 CrossRef CAS PubMed.
- J. C. Ng, J. P. Wang, B. Zheng, C. Zhai, R. Maddalena, F. Liu and M. R. Moore, Toxicol. Appl. Pharmacol., 2005, 206, 176–184 CrossRef CAS PubMed.
- C. K. Jain and I. Ali, Water Res., 2000, 34, 4304–4312 CrossRef CAS.
- EPA, Technologies and Costs for Removal of Arsenic from Drinking Water, 2000 Search PubMed.
- C. K. Jain and R. D. Singh, J. Environ. Manage., 2012, 107, 1–18 CrossRef CAS PubMed.
- M. Bissen and F. H. Frimmel, Acta Hydrochim. Hydrobiol., 2003, 31, 9–18 CrossRef CAS.
- M. E. Pena, G. P. Korfiatis, M. Patel, L. Lippincott and X. Meng, Water Res., 2005, 39, 2327–2337 CrossRef CAS PubMed.
- X. Guan, J. Ma, H. Dong and L. Jiang, Water Res., 2009, 43, 5119–5128 CrossRef CAS PubMed.
- D. Mohan and C. U. Pittman, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- I. Ali and V. K. Gupta, Nat. Protoc., 2007, 1, 2661–2667 CrossRef PubMed.
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed.
- I. Ali, M. Asim and T. A. Khan, J. Environ. Manage., 2012, 113, 170–183 CrossRef CAS PubMed.
- I. Ali, Sep. Purif. Rev., 2010, 39, 95–171 CrossRef CAS.
- A. Jain, K. P. Raven and R. H. Loeppert, Environ. Sci. Technol., 1999, 33, 1179–1184 CrossRef CAS.
- S. Dixit and J. G. Hering, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- W. X. Zhang, J. Nanopart. Res., 2003, 5, 323–332 CrossRef CAS.
- L. S. Zhong, J. S. Hu, H. P. Liang, A. M. Cao, W. G. Song and L. J. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS.
- S. C. N. Tang and I. M. C. Lo, Water Res., 2013, 47, 2613–2632 CrossRef CAS PubMed.
- H. Zhu, Y. Jia, X. Wu and H. Wang, J. Hazard. Mater., 2009, 172, 1591–1596 CrossRef CAS PubMed.
- C.-S. Jeon, K. Baek, J.-K. Park, Y.-K. Oh and S.-D. Lee, J. Hazard. Mater., 2009, 163, 804–808 CrossRef CAS PubMed.
- L. Zeng, Water Res., 2003, 37, 4351–4358 CrossRef CAS PubMed.
- L.-N. Shi, Y.-M. Lin, X. Zhang and Z.-L. Chen, Chem. Eng. J., 2011, 171, 612–617 CrossRef CAS.
- D. Li, D. H. Wu, X. Wang, L. D. Lu and X. J. Yang, Mater. Res. Bull., 2001, 36, 2437–2442 CrossRef CAS.
- L. B. Hoch, E. J. Mack, B. W. Hydutsky, J. M. Hershman, I. M. Skluzacek and T. E. Mallouk, Environ. Sci. Technol., 2008, 42, 2600–2605 CrossRef CAS PubMed.
- C. Uezuem, T. Shahwan, A. E. Eroglu, K. R. Hallam, T. B. Scott and I. Lieberwirth, Appl. Clay Sci., 2009, 43, 172–181 CrossRef.
- S. M. Ponder, J. G. Darab and T. E. Mallouk, Environ. Sci. Technol., 2000, 34, 2564–2569 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- G. E. Fryxell, J. Liu, T. A. Hauser, Z. Nie, K. F. Ferris, S. Mattigod, M. Gong and R. T. Hallen, Chem. Mater., 1999, 11, 2148–2154 CrossRef CAS.
- T. J. Barton, L. M. Bull, W. G. Klemperer, D. A. Loy, B. McEnaney, M. Misono, P. A. Monson, G. Pez, G. W. Scherer, J. C. Vartuli and O. M. Yaghi, Chem. Mater., 1999, 11, 2633–2656 CrossRef CAS.
- P. Selvam, S. K. Bhatia and C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237–3261 CrossRef CAS.
- C. Pantani, N. Spreti, M. C. Maggitti and R. Germani, Bull. Environ. Contam. Toxicol., 1995, 55, 179–186 CrossRef CAS PubMed.
- B. Isomaa, J. Reuter and B. M. Djupsund, Arch. Toxicol., 1976, 35, 91–96 CrossRef CAS PubMed.
- R. L. Brutchey and D. E. Morse, Chem. Rev., 2008, 108, 4915–4934 CrossRef CAS PubMed.
- M. Hildebrand, Chem. Rev., 2008, 108, 4855–4874 CrossRef CAS PubMed.
- S. V. Patwardhan, Chem. Commun., 2011, 47, 7567–7582 RSC.
- S. V. Patwardhan, S. J. Clarson and C. C. Perry, Chem. Commun., 2005, 1113–1121 RSC.
- C. Forsyth and S. V. Patwardhan, J. Mater. Chem. B, 2013, 1, 1164 RSC.
- S. V. Patwardhan and C. C. Perry, Silicon, 2010, 2, 33–39 CrossRef CAS.
- C. R. Steven, G. A. Busby, C. Mather, B. Tariq, M. L. Briuglia, D. A. Lamprou, A. J. Urquhart, M. H. Grant and S. V. Patwardhan, J. Mater. Chem. B, 2014, 2, 5028–5042 RSC.
- S. Davidson, D. A. Lamprou, A. J. Urquhart, M. H. Grant and S. V. Patwardhan, ACS Biomater. Sci. Eng., 2016 DOI:10.1021/acsbiomaterials.1026b00224.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS PubMed.
- M. R. Knecht and D. W. Wright, Chem. Mater., 2004, 16, 4890–4895 CrossRef CAS.
- R. R. Naik, M. M. Tomczak, H. R. Luckarift, J. C. Spain and M. O. Stone, Chem. Commun., 2004, 1684–1685 RSC.
- A. M. Ewlad-Ahmed, M. A. Morris, S. V. Patwardhan and L. T. Gibson, Environ. Sci. Technol., 2012, 46, 13354–13360 CrossRef CAS PubMed.
- C. Drummond, R. McCann and S. V. Patwardhan, Chem. Eng. J., 2014, 244, 483–492 CrossRef CAS.
- G. Purna Chandra Rao, S. Satyaveni, A. Ramesh, K. Seshaiah, K. S. N. Murthy and N. V. Choudary, J. Environ. Manage., 2006, 81, 265–272 CrossRef CAS PubMed.
- T. Y. Guo, Y. Q. Xia, G. J. Hao, M. D. Song and B. H. Zhang, Biomaterials, 2004, 25, 5905–5912 CrossRef CAS PubMed.
- J. Pan, X. Zou, X. Wang, W. Guan, Y. Yan and J. Han, Chem. Eng. J., 2010, 162, 910–918 CrossRef CAS.
- S. L. Goh, ZeroValent Iron (ZVI) Supported By Bio-Inspired Silica for Environmental Remediation, MSc, Supervisor: S. V. Patwardhan, University of Strathclyde, 2011 Search PubMed.
- W. D. Einicke, H. Uhlig, D. Enke, R. Glaser, C. Reichenbach and S. G. Ebbinghaus, Colloids Surf., A, 2013, 437, 108–112 CrossRef CAS.
- Y. Fu, S. L. Yu, Y. Z. Yu, L. P. Qiu and B. Hui, J. Environ. Sci., 2007, 19, 678–688 CrossRef CAS.
- K. Siskova, J. Tucek, L. Machala, E. Otyepkova, J. Filip, K. Safarova, J. Pechousek and R. Zboril, J. Nanopart. Res., 2012, 14, 805 CrossRef.
- W. R. Cullen and K. J. Reimer, Chem. Rev., 1989, 89, 713–764 CrossRef CAS.
- L. Da Sacco and A. Masotti, Mar. Drugs, 2010, 8, 1518–1525 CrossRef CAS PubMed.
- S. Sohn and D. Kim, Chemosphere, 2005, 58, 115–123 CrossRef CAS PubMed.
- D. Do Duong, Adsorption Analysis, Imperial College Pr, 1998 Search PubMed.
- S. Zhang, X.-Y. Li and J. P. Chen, Carbon, 2010, 48, 60–67 CrossRef CAS.
- K. M. Shareef, World J. Agric. Sci., 2009, 5, 819–831 CAS.
- J. Qu, J. Environ. Sci., 2008, 20, 1–13 CrossRef CAS.
- W. Chen, R. Parette, J. Zou, F. S. Cannon and B. A. Dempsey, Water Res., 2007, 41, 1851–1858 CrossRef CAS PubMed.
- K. Payne and T. Abdel-Fattah, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2005, 40, 723–749 CrossRef CAS.
- Z. Gu, J. Fang and B. Deng, Environ. Sci. Technol., 2005, 39, 3833–3843 CrossRef CAS PubMed.
- M. Jang, E. W. Shin and J. K. Park, Proceedings of the Water Environment Federation, 2002, vol. 2002, pp. 40–54 Search PubMed.
- X. Guo and F. Chen, Environ. Sci. Technol., 2005, 39, 6808–6818 CrossRef CAS PubMed.
- H. Guo, Y. Li and K. Zhao, J. Hazard. Mater., 2010, 176, 174–180 CrossRef CAS PubMed.
- T. Balaji, T. Yokoyama and H. Matsunaga, Chemosphere, 2005, 59, 1169–1174 CrossRef CAS PubMed.
- P. M. Solozhenkin, E. A. Deliyanni, V. N. Bakoyannakis, A. I. Zouboulis and K. A. Matis, J. Min. Sci., 2003, 39, 287–296 CrossRef.
- P. M. Styles, M. Chanda and G. L. Rempel, React. Funct. Polym., 1996, 31, 89–102 CrossRef CAS.
- S. R. Chowdhury, E. K. Yanful and A. R. Pratt, Environ. Earth Sci., 2011, 64, 411–423 CrossRef CAS.
- I. Ali, Sep. Purif. Rev., 2014, 43, 175–205 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02937j |
| This journal is © The Royal Society of Chemistry 2017 |