
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic solid fluorophores regulated by subtle structure modification: color-tunable and aggregation-induced emission†
Jing Nan
Zhang‡
a,
Hui
Kang‡
a,
Nan
Li
 a,
Shi Ming
Zhou
c,
Hua Ming
Sun
a,
Shi Wei
Yin
a,
Na
Zhao
*a and
Ben Zhong
Tang
*b
a,
Shi Ming
Zhou
c,
Hua Ming
Sun
a,
Shi Wei
Yin
a,
Na
Zhao
*a and
Ben Zhong
Tang
*b
aKey Laboratory of Macromolecular Science of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi'an, 710119, China. E-mail: nzhao@snnu.edu.cn
bDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
cHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, 230026, China
First published on 30th August 2016
Abstract
Organic solid fluorophores with a tunable emission color have been widely applied in multiple areas. However, rational design and efficient crafting of these fluorophores from a simple core skeleton is still a formidable challenge because of the undesirable concentration quenching emission effect. Herein, we present the development of two series of organic solid fluorophores based on a backbone of p-bis(2,2-dicyanovinyl)benzene. Notably, the introduction of either non-aromatic or aromatic substituents provides fluorophores with a tunable emission color. Moreover, the fluorophores with aromatic substituents exhibit additional unique optical phenomena, such as aggregation-induced emission, polymorphism-dependent emission, and reversible mechanochromic luminescence.
Introduction
Organic solid fluorophores that exhibit an intense and tunable emission, particularly those with an emission across the whole visible spectrum, have received much attention owing to their extensive application in the fabrication of organic light-emitting diodes (OLED),1 optical devices,2 organic luminescent displays,3 and fluorescent sensors.4 However, traditional organic fluorophores are usually weakly emissive or even non-emissive in the solid state, which is attributed to inherent strong intermolecular π–π stacking interactions that consume the excitation energy.5 Averting this thorny aggregation-caused quenching (ACQ) process is the key to obtaining organic solid fluorophores with superior optical properties. During the past decade, the discovery of a novel phenomenon, termed aggregation-induced emission (AIE), with a mechanism of restriction of the intramolecular rotation (RIR), has provided new insight and powerful tools for the construction of organic solid fluorophores with immunity to the ACQ process.6,7 To date, numerous elegant organic solid emitters based on diverse core skeletons have emerged that exhibit the AIE effect, and extensive technological applications have been developed.8 However, AIE systems with a tunable emission color that are based on a simple core backbone are still extremely rare.9Recently, extended π-conjugated molecules with a D–π–A structure have attracted significant interest because of their flexibility for creating organic emitters with tunable emission.10,11 The nitrile group, due to its strong electron-withdrawing ability and structural simplicity, is an ideal candidate for an excellent acceptor for such D–π–A fluorescent materials featuring advanced photoluminescence properties. More importantly, the nitrile group can induce steric hindrance and result in a twisted conformation, which enables the fluorophores to be protected from the ACQ effect.12 However, studies on regulating the emission behavior of a nitrile-containing system through altering the substituents are rare.9 Herein, we present the development of two series of symmetric p-bis(2,2-dicyanovinyl)benzene-based fluorophores by decorating the single benzene ring with a dicyanovinyl unit as the acceptor and different types of donors with diverse electronic and steric effects (Fig. 1). Interestingly, with the introduction of either non-aromatic or aromatic substituents, both classes of fluorophores yield a full-color emission. Moreover, the aromatic groups can successfully endow the fluorophores with a highly efficient solid state emission and AIE effect. Meanwhile, a specific polymorphism-dependent emission as well as reversible mechanochromic luminescence are observed for some of the AIE fluorophores.
 | ||
| Fig. 1 Chemical structures of 1a–2g. | ||
Results and discussion
The initial step of our research was to merge the components of both the donor and acceptor to form the desired compounds 1a–2g. This was readily achieved using a one-step condensation reaction between the appropriate aldehyde and malononitrile. All the compounds were easily purified using column chromatography or recrystallization with a reasonable yield.The absorption and photoluminescence properties of all the compounds in acetone and the solid state are shown in Table 1, as well as Fig. S1 and S2.† For the parent compound 1a, the absorption peak with the longest wavelength was located at around 367 nm, which was ascribed to the π–π* transition. When the substituent group at the 2,5-positions was altered from t-butyldiphenylsiloxyl to methoxyl (or from aromatic phenyl to 5-methylthienyl for series 2), the corresponding compounds 1b–1d (or 2a–2g) exhibited the longest wavelength absorption peak ranging from 428 to 457 nm (or 392 to 441 nm), which was assigned to the intramolecular charge transfer (ICT) transition from the different type of electron-donating group to the electron-accepting dicyanovinyl group.10
| Compound | Solution state | Solid (crystalline) state | ||||||
|---|---|---|---|---|---|---|---|---|
| λ abs [nm] | ε [×103 M−1 cm−1] | λ em [nm] | Φ F [%] | τ avg [ns] | λ em [nm] | Φ F [%] | τ avg [ns] | |
| a Longest wavelength absorption maximum in acetone. b Absolute fluorescence quantum yield obtained using the calibrated integrating sphere system. c Mean fluorescence lifetime (τavg) calculated using the equation τavg = A1τ1 + A2τ2. d Below the detection limit. e Two types (2d-g/2d-o) of crystal 2d. | ||||||||
| 1a | 367 | 31.12 | 426 | 0.8 | 1.10 | 467 (464) | 13.2 (3.4) | 2.05 (2.27) |
| 1b | 430 | 3.38 | 538 | 15.7 | 5.21 | 502 | 11.3 | 1.57 |
| 1c | 428 | 10.66 | 550 | 55.9 | 7.31 | 570 (574) | 2.4 (2.2) | 2.46 (3.98) |
| 1d | 457 | 9.20 | 562 | 76.3 | 10.62 | 620 (640) | 3.7 (4.8) | 7.42 (7.36) |
| 2a | 393 | 4.74 | 495 | 11.9 | 2.95 | 464 (467) | 13.3 (9.3) | 0.92 (2.87) |
| 2b | 398 | 7.30 | 545 | 1.5 | 3.18 | 514 (527) | 18.3 (6.5) | 2.72 (1.64) |
| 2c | 409 | 2.90 | 566 | 0.4 | 0.81 | 533 (538) | 45.6 (53.8) | 8.21 (8.06) |
| 2d | 418 | 4.60 | 565 | 0.9 | 1.81 | 545 (545/560)e | 13.1 (49.0/7.9)e | 6.27 (6.29/3.89)e |
| 2e | 415 | 6.40 | n.d.d | 0.5 | n.d.d | 566 (570) | 35.6 (43.1) | 7.33 (10.75) |
| 2f | 421 | 5.30 | n.d.d | 0.2 | n.d.d | 584 | 2.3 | 0.55 |
| 2g | 441 | 3.00 | 600 | 3.9 | 1.75 | 622 (593) | 11.7 (21.2) | 4.16 (4.90) |
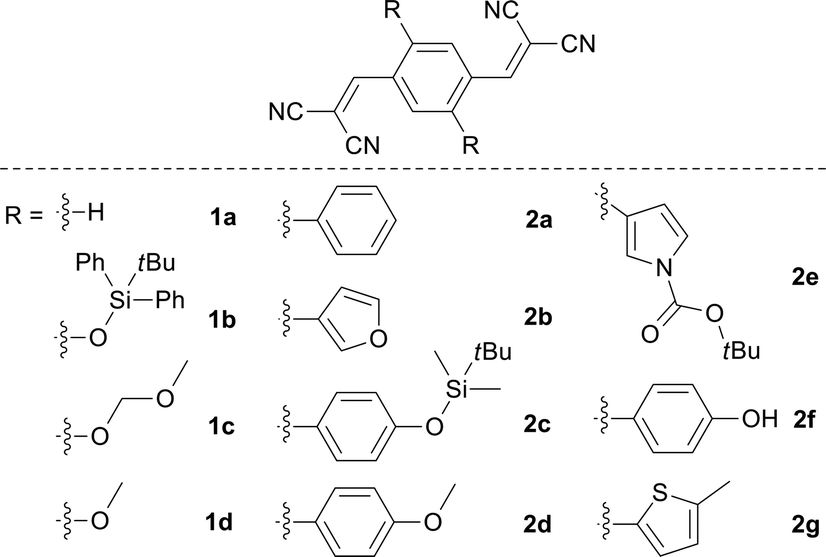
As shown in Table 1, with increase of the electron-donating ability, the emission wavelengths of 1a–1d in acetone presented a red-shift from 426 to 562 nm, while the emission peak wavelength for the solid state ranged from 467 to 620 nm. By changing the aromatic group substituent, the emission peaks of 2a–2g in acetone varied from 495 to 600 nm. Additionally, a similar red shifted emission tendency (464 to 622 nm) for the solid state was observed. It is clear that two series of solid emitters with emission over the entire color range were obtained by simply varying the type of donor (Fig. 2). The CIE chromaticity diagrams (Fig. S3 and S4, Table S3†) demonstrated the same tunable emission from CIE blue (0.1374, 0.0374) to CIE red (0.6954, 0.3045). Furthermore, single crystals for all the compounds, except 1b and 2f, were obtained by slow evaporation from an appropriate solvent (Tables S6–S8, Fig. S5†),13 and they also emitted fluorescence covering the whole visible region (464 to 640 nm for series 1 and 467 to 593 nm for series 2).
 | ||
| Fig. 2 Normalized PL spectra of 1a–1d and 2a–2g in the solid state, and photographs taken under irradiation with 365 nm UV light. | ||
To better understand the electronic transitions of all the compounds, density functional theory (DFT) calculations, using the single crystal structure determined from X-ray analysis, were carried out (Table S1†). The results indicated that both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of 1a were located over the whole molecular framework, suggesting a π–π* transition. For the other compounds, however, the HOMO was mainly located on the core benzene ring and the electron-donating moieties, while the LUMO was spread primarily over the central benzene ring and the electron-withdrawing dicyanovinyl group, which demonstrated the existence of ICT character for those compounds. Meanwhile, the HOMO and LUMO became more separated in series 2, compared with series 1, which was ascribed to the twisted conformation of series 2 originating from steric hindrance due to the introduction of the bulky aromatic group.
The dipole moment change (Δμ) between the excited state (μe) and ground state (μg) was also determined (Table S2†). The results showed that the Δμ values for series 2 were larger than for series 1, which is in agreement with the degree of separation between the HOMO and LUMO. Furthermore, 1b–2g exhibited obvious solvent effects (Fig. S6 and S7†).14 With increase of the solvent polarity (from cyclohexane to acetone or dichloromethane), the emission peak exhibited a red-shift, which indicated the existence of ICT in these compounds. However, a slight solvent effect was observed for 1a, confirming its π–π* character.
The fluorescence quantum yields (ΦF) of all the compounds in solution and the solid state (including the crystalline state) are also summarized in Table 1. The quantum yields of 1b–1d dissolved in acetone were moderate to high (15.7–76.3%), but significantly decreased for the solid state (<11.3% for the solid powder, <4.81% for the crystal). In sharp contrast, a dramatic enhancement was achieved for 1a and 2a–2g in the solid state (2.3–45.6% for the solid powder, 3.4–58.8% for the crystal) compared with in solution (0.2–11.9%). It is noteworthy that 1d and 2g exhibited red emission. More importantly, 2g retained a high quantum yield in the solid state (622 nm, 11.7%) and crystalline state (593 nm, 21.2%). Considering that red fluorophores containing π-conjugated frameworks typically have a tendency to aggregate in the solid form, which results in emission quenching, this strategy provides an extremely powerful method to fabricate organic solid emitters with a long emission wavelength. We also calculated the ratio between the solid and solution state quantum yields to verify the influence of the substituent group on the emission behavior of the compounds (Fig. 3D). It is clear that the ratio was greater than one for 1a and 2a–2g but less than one for 1b–1d. Therefore, the introduction of an aromatic group successfully reversed the trend of the quantum yields between the solution and solid phases.
 | ||
| Fig. 3 (A) PL spectra of 2e (10 μM) in acetone and acetone/water mixtures with different water fractions (fw). (B) Plot of the emission intensity versus the composition of the water mixture for 2e. (C) Particle size distribution of 2e (10 μM) in an acetone/water mixture with a fw value of 99%. (D) The ratio of the quantum yields for the solid and solution states of all the compounds. Inset in (B): photographs of 2e in acetone/water mixtures with fw values of 0 and 99% under irradiation with 365 nm UV light. | ||
A higher quantum yield for the solid state implies existence of the AIE effect. Therefore, we measured the AIE properties using acetone and water, which served as a good and poor solvent, respectively. Typically, 2e was almost non-emissive when dissolved in acetone. Increase of the water fraction (fw) resulted in a significant boost of the emission intensity at 576 nm, and yielded approximately a 150-fold enhancement when the fw was increased up to 99% (Fig. 3A and B). The observed phenomenon could be attributed to aggregation of the molecules in the mixed solution with a high fw, and thus activation of the RIR process. The particle size for 2e in the mixture with a fw of 99% was determined to be 84 nm using dynamic light scattering (DLS), confirming the formation of nano-aggregates (Fig. 3C). Furthermore, the other compounds in series 2 and 1a also displayed a similar AIE or aggregation-induced emission enhancement (AIEE) performance (Fig. S8, S12–S17†). Nevertheless, compounds 1b–1d with non-aromatic moieties exhibited an opposite phenomenon, where increase of the water fraction resulted in a gradual decrease of the emission intensity, suggesting ACQ properties (Fig. S9–S11†).
The fluorescence lifetime (τ) values of all the compounds are listed in Tables 1 and S4.† To verify the photoemission dynamics, the radiative transition rate constant (kr) as well as the nonradiative transition rate constant (knr) were calculated using the fluorescence lifetimes and the quantum yields (Table S5†). Taking 1d as an example, the knr (2.232 × 107 s−1) was close to the kr (7.185 × 107 s−1) in solution. However, the knr was enhanced to 1.297 × 108 s−1 while the kr was reduced to 4.986 × 106 s−1 in the solid state, implying that nonradiative decay was dominant in the solid state. Whereas for 2c, the knr (1.229 × 109 s−1) was remarkably greater than the kr (4.938 × 106 s−1) in solution. However, the knr was decreased to 6.626 × 107 s−1 while the kr was enhanced to 5.554 × 107 s−1 in the solid state. These results suggest that suppression of the nonradiative decay is the major reason for the higher quantum yield in the solid state.
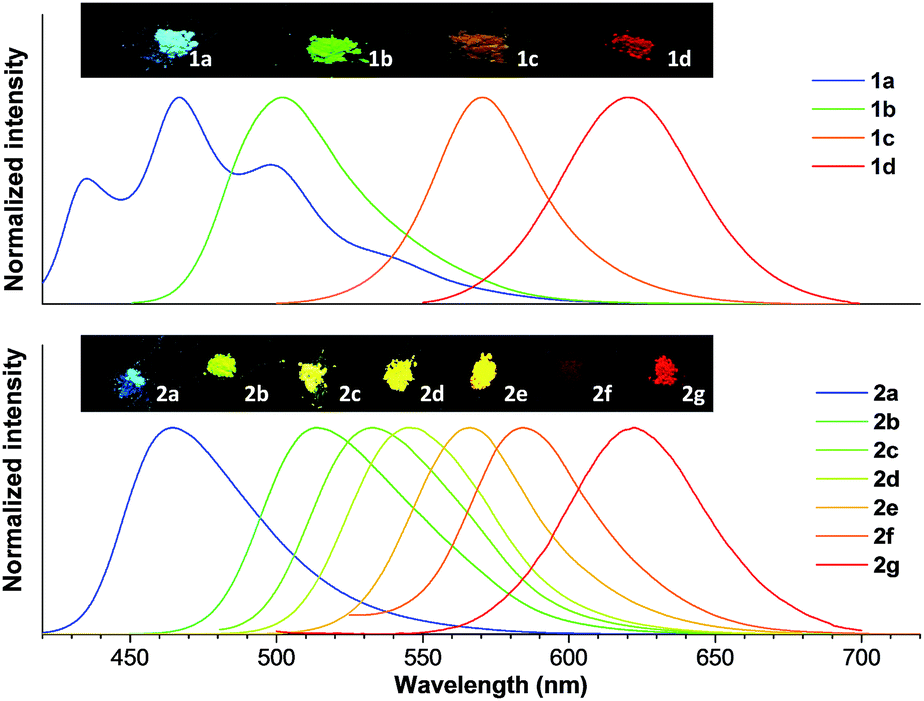
To obtain the underlying reasons for this significant substituent dependent emission behavior, inspection of the single crystals was performed. Fig. 4 presents the single crystals of 1d and 2c as examples. Crystal 1d adopted a near-planar conformation and the torsion angle between the vinyl (or methyl) group and the central benzene ring was merely 3.42° (or 3.63°). Careful evaluation of the details of the crystal packing indicated that the distance between two adjacent planes was 3.416 Å, which suggested that 1d encounters strong and consecutive intermolecular π–π stacking interactions and thus this resulted in weakening of the fluorescence in the solid state (Fig. 4A).15 Similar strong π–π stacking interactions (3.214 Å) were also observed for the crystal of 1c (Fig. S21B†), and were responsible for the reduced fluorescence in the solid state. In contrast, the crystal of 2c clearly showed a contorted conformation, where the torsion angle between the vinyl (or substituted phenyl) group and the central benzene ring was 42.14° (or 40.15°), which apparently resulted from steric congestion between the neighboring phenyl rings and dicyanovinyl moieties. Such a twisted conformation effectively enlarged the distance between the adjacent phenyl groups (>4.076 Å), and avoided the typical π–π stacking interactions (Fig. 4B). Meanwhile, crystallographic analyses of the other compounds in series 2 showed similar twisted conformations and an absence of π–π stacking interactions, which accounts for their bright luminescence in the solid state (Fig. S22–S24†). Compared with the other molecular packing arrangements in series 2, it is clear that introduction of a bulky substituent did not enlarge the distance between adjacent molecules, implying a relatively tight packing in the crystals of 2c and 2e. Additionally, multiple intermolecular C–H⋯N (or C–H⋯O) and C–H⋯π interactions were found throughout the crystal structures of 2c and 2e (Fig. S25 and S23A†), which could help to rigidify the molecular conformation, and hence result in their relatively high emission efficiency in the solid state. It is interesting to note that there were also no π–π stacking interactions in the crystal of 1a, though it exhibited an almost planar conformation (Fig. S21A†), which led to no emission quenching in the solid state.
 | ||
| Fig. 4 Single crystal structures of 1d (A) and 2c (B), and the molecular packing viewed from the side and top of adjacent molecules. | ||
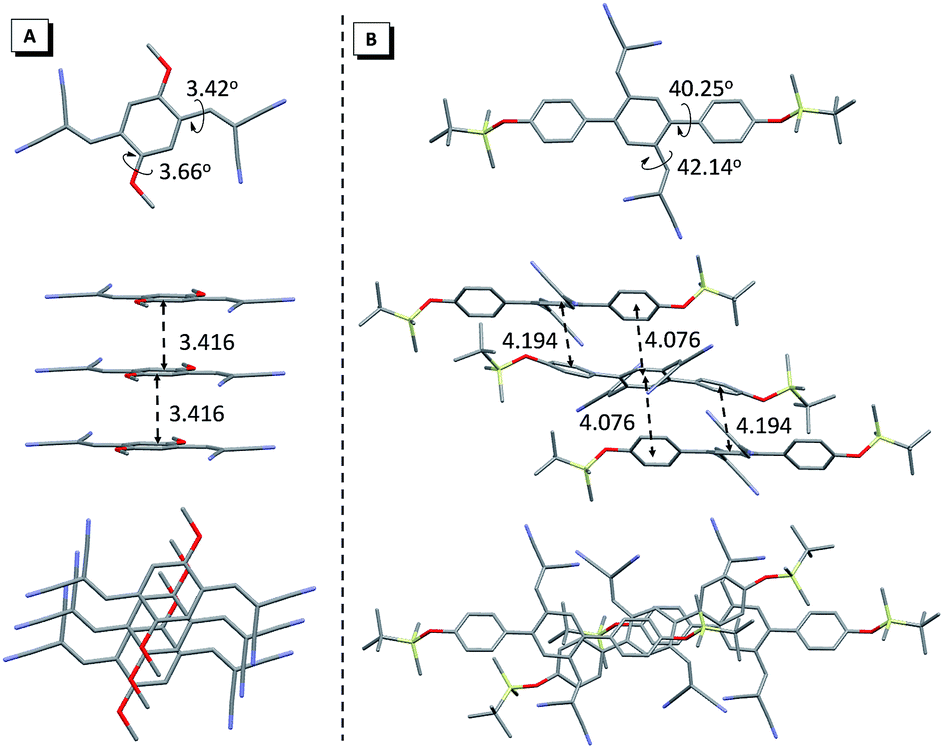
Unexpectedly, two types of 2d crystals, with crystallographically independent conformations (2d-g and 2d-o), suitable for X-ray analysis were obtained from ethyl acetate/dichloromethane and ethyl acetate/acetone mixtures. The crystal 2d-g exhibited a green emission at around 545 nm, while 2d-o showed a bathochromic shift with an orange emission at 560 nm (Fig. 5A). Both 2d-g and 2d-o adopted highly twisted conformations without π–π stacking interactions (Fig. S24†). The most pronounced difference between the two forms was in the dihedral angle between the dicyanovinyl unit and 4-methoxyl phenyl moiety, which was 46.27° for 2d-g and 43.06° for 2d-o, respectively (Fig. 5B). This difference indicated that the conformation of 2d-o was more planar than that of 2d-g, thus this endowed 2d-o with a longer emission wavelength. Moreover, the two crystals presented entirely different packing modes (Fig. 5C and D) and the amount of intermolecular C–H⋯N interactions between the cyano and phenyl or methyl groups in 2d-g was more than in 2d-o (Fig. S24†), enabling 2d-g to possess a higher quantum yield.
 | ||
| Fig. 5 (A) PL spectra of 2d-g and 2d-o. (B) A view of the dihedral angles for 2d-g and 2d-o. Molecular packing in 2d-g (C) and 2d-o (D). Inset in (A): photographs of 2d-g and 2d-o under a fluorescence microscope. | ||
It is notable that the as-prepared sample 2d exhibited an emission peak at 545 nm, which is close to that of 2d-g. However, gentle grinding induced a red-shift of the emission to 567 nm, which could be ascribed to an amorphous form of 2d (Fig. 6A). Such an amorphous state was further confirmed by the flat signal obtained from powder XRD measurement (Fig. S20B†). Meanwhile, the red-shifted emission could be attributed to collapse of the crystalline lattice during grinding, which would result in the molecules adopting a more planar conformation. An apparent phenomenon is that an amorphous solid can always be crystallized by either heating or fuming with an organic solvent vapor.16 Indeed, this was also applicable for 2d (Fig. 6B). On fuming with a dichloromethane vapor, the amorphous form of 2d converted into a crystalline form with the emission peak restored to 545 nm. Moreover, this process could be repeated several times, indicating the reversible mechanochromic luminescence properties of 2d in response to external stimuli. Similar reversible emission behavior for as-prepared powders of 2b and 2c could also be achieved through a repeated grinding/solvent fuming process (Fig. S18 and S19†).
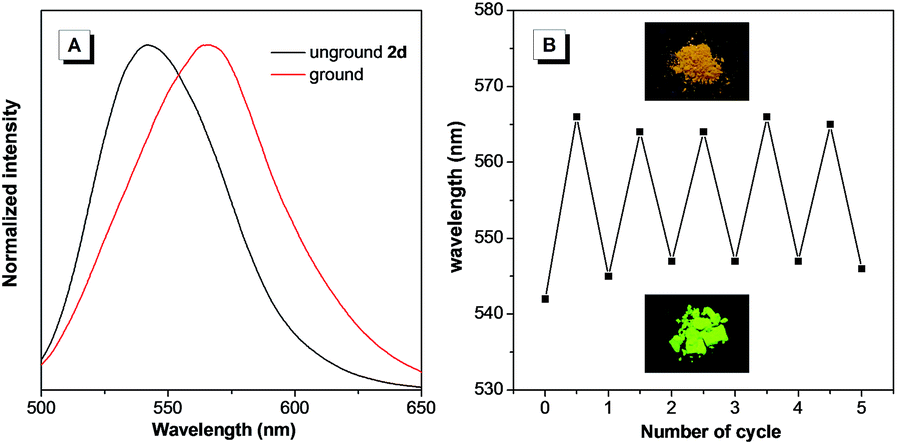 | ||
| Fig. 6 (A) PL spectra of unground and ground 2d. (B) Reversible switching of the emission of 2d through repeated grinding/fuming cycles. Inset in (B): photographs of the as-prepared 2d and after grinding, under irradiation with 365 nm UV light. | ||
Conclusions
In conclusion, two classes of p-bis(2,2-dicyanovinyl)benzene-based fluorophores were designed and synthesized. Their optical properties, as well as their single crystal packing structures, were systematically investigated. The results showed that decorating the core backbone with either non-aromatic or aromatic groups generated two kinds of organic solid emitters with tunable emission from the blue to red region. More importantly, the employment of bulky aromatic groups successfully resulted in a twisted molecular conformation. Such a contorted conformation is beneficial for the inhibition of detrimental π–π stacking interactions and assisted the fluorophores to exhibit remarkable AIE features with a high emission efficiency in the solid state. In addition, some AIE fluorophores presented polymorphism-dependent emission and reversible mechanochromic luminescence properties. Since these fluorophores exhibiting diverse emission characteristics are all derivatives of a single benzene ring, we believe such a strategy provides a rational and efficient approach for the fabrication of novel organic solid fluorophores from both fundamental and practical viewpoints.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21402115, 51403122 and 21672135), the Fundamental Research Funds for the Central Universities (GK201503028) and the Natural Science Foundation of Shaanxi Province (2015JQ5150 and 2016JQ2020). We also appreciate Dr F. Y. Liu and Dr J. Liu for helpful discussions on theoretical calculations and DLS analysis.Notes and references
- (a) L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed; (b) R. H. Friend, R. W. Gymer, A. B. Holms, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef CAS; (c) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed; (d) T. Qin, W. Wiedemair, S. Nau, R. Trattnig, S. Sax, S. Winkler, A. Vollmer, N. Koch, M. Baumgarten, E. J. W. List and K. Müllen, J. Am. Chem. Soc., 2011, 133, 1301–1303 CrossRef CAS PubMed.
- J. Zhang, W. Chen, J. A. Rojas, V. E. Jucov, V. T. Timofeeva, C. T. Parker, S. Barlow and R. S. Marder, J. Am. Chem. Soc., 2013, 135, 16376–16379 CrossRef CAS PubMed.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed.
- (a) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef PubMed; (b) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (c) L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC.
- (a) G. S. Beddard and G. Porter, Nature, 1976, 260, 366–367 CrossRef CAS; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (c) C.-K. Lim, S. Kim, I. C. Kwon, C.-H. Ahn and S. Y. Park, Chem. Mater., 2009, 21, 5819–5825 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC; (b) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- (a) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS; (b) G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. G. Shuai, B. Z. Tang, D. B. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed; (c) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Law and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed; (c) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (d) Y. Okazawa, K. Kondo, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2015, 137, 98–101 CrossRef CAS PubMed; (e) T. Shiragami, Y. Nakamura, J. Matsumoto, M. Otsuki and M. Yasuda, Phys. Chem. Chem. Phys., 2014, 16, 22046–22051 RSC; (f) X. Wang, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2014, 136, 9890–9893 CrossRef CAS PubMed; (g) J. Zhang, B. Xu, J. Chen, S. Ma, Y. Dong, L. Wang, B. Li, L. Ye and W. Tian, Adv. Mater., 2014, 26, 739–745 CrossRef PubMed; (h) R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed; (i) S. Xu, Y. Yuan, X. Cai, C. J. Zhang, F. Hu, J. Liang, G. Zhang, D. Q. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824–5830 RSC; (j) L. Peng, Y. Zheng, X. Wang, A. J. Tong and Y. Xiang, Chem.–Eur. J., 2015, 21, 4326–4332 CrossRef CAS PubMed; (k) J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef CAS; (l) G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS PubMed; (m) M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485–4491 RSC.
- (a) Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654–11657 CrossRef CAS PubMed; (b) S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS.
- (a) Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed; (b) C. T. Chen, Chem. Mater., 2004, 16, 4389–4400 CrossRef CAS.
- (a) A. Wakamiya, K. Mori and S. Yamaguchi, Angew. Chem., Int. Ed., 2007, 46, 4273–4276 CrossRef CAS PubMed; (b) M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 48, 3653–3656 CrossRef CAS PubMed; (c) E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed; (d) A. C. Shaikh, D. S. Ranade, S. Thorat, A. Maity, P. P. Kulkarni, R. G. Gonnade, P. Munshi and N. T. Patil, Chem. Commun., 2015, 51, 16115–16118 RSC; (e) Z. Song, W. Zhang, M. Jiang, H. H. Y. Sung, R. T. K. Kwok, H. Nie, I. D. Williams, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2016, 26, 824–832 CrossRef CAS.
- (a) K. A. N. Upamali, L. A. Estrada, P. K. De, X. Cai, J. A. Krause and D. C. Neckers, Langmuir, 2011, 27, 1573–1580 CrossRef CAS PubMed; (b) B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed; (c) E. Ishow, A. Brosseau, G. Clavier, K. Nakatani, P. Tauc, C. Fiorini-Debuisschert, S. Neveu, O. Sandre and A. Léaustic, Chem. Mater., 2008, 20, 6597–6599 CrossRef CAS.
- CCDC 1056239 (1a), 1056280 (1c), 1403124 (1d), 1439334 (2a), 1439332 (2b), 1443616 (2c), 1472574 (2d-g), 1499316 (2d-o), 1442948 (2e) and 1442404 (2g) contain the ESI crystallographic data for this paper.†.
- The solvent effects with 2f could not be measured due to its poor solubility.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- (a) Z. He, L. Zhang, J. Mei, T. Zhang, J. W. Y. Lam, Z. Shuai, Y. Q. Dong and B. Z. Tang, Chem. Mater., 2015, 27, 6601–6607 CrossRef CAS; (b) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed; (c) K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed; (d) C. Dou, L. Han, S. Zhao, H. Zhang and Y. Wang, J. Phys. Chem. Lett., 2011, 2, 666–670 CrossRef CAS; (e) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed; (f) M.-S. Yuan, D.-E. Wang, P. Xue, W. Wang, J.-C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467–2477 CrossRef CAS; (g) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, optical spectra, crystal data and DFT calculations. CCDC 1056239, 1056280, 1403124, 1439334, 1439332, 1443616, 1472574, 1499316, 1442948 and 1442404. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02875f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |