
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Greigite Fe3S4 as a new anode material for high-performance sodium-ion batteries†
Qidong
Li‡
a,
Qiulong
Wei‡
a,
Wenbin
Zuo
b,
Lei
Huang
a,
Wen
Luo
a,
Qinyou
An
a,
Vasiliy O.
Pelenovich
b,
Liqiang
Mai
 *a and
Qingjie
Zhang
*a
*a and
Qingjie
Zhang
*a
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan, 430070, China. E-mail: mlq518@whut.edu.cn; zhangqj@whut.edu.cn
bSchool of Physics and Technology, Wuhan University, Wuhan 430070, China
First published on 1st August 2016
Abstract
Transition metal dichalcogenide materials have been considered as promising anode materials for rechargeable sodium-ion batteries because of their high specific capacity and low cost. Here, we demonstrate an iron sulfide Fe3S4 as a new anode material for a rechargeable sodium-ion battery. The involved conversion mechanism has been proved when the as-prepared Fe3S4 was used as the host material for sodium storage. Remarkably, a compound FeSx with quantum size generated by conversion reaction overcame the kinetic and thermodynamic constraints of chemical conversion to achieve superior cycling and rate capability. As a result, the as-prepared Fe3S4 electrode delivers a high reversible specific capacity of 548 mA h g−1 at 0.2 A g−1, together with an excellent cycling stability of 275 mA h g−1 after 3500 cycles at 20 A g−1.
Introduction
Owing to the increasing demand for sustainable and renewable power sources, much effort has been devoted to energy storage innovation over the past decades. Due to their high energy density and fast recharge capability, lithium-ion batteries (LIBs) have been successfully applied in many aspects of our daily life.1,2 In particular, the application of LIBs in electric vehicles (EVs) and hybrid EVs has reduced significantly our energy dependence on ‘one-off’ resources. Nevertheless, concerns about LIBs have arisen both in terms of their cost and the supply limits of lithium resources in recent years.3 Alternatively, sodium-ion batteries (SIBs) have recently attracted considerable interest owing to the low cost, wide distribution and abundant resource of sodium.4,5 However, compared to Li+ ion, the larger ionic radius and molar mass of Na+ ion often lead to inferior cyclability and lower specific capacity.6,7 There are still many challenges to exploit host materials for sodium with high capacity, fast charge–discharge, and long cycle life, especially for anode materials.6The emerging transition metal dichalcogenides (TMD) materials which have been researched in electrochemistry8–12 for many years have drawn extensive attention for SIBs in recent years.13–17 These TMD materials often involve a multi-step reaction mechanism (intercalation and conversion, such as MoS2) which contributes a high specific capacity but with poor cycling life.18 Among these TMD materials, iron sulfides (FeS,13 FeS2 (ref. 15, 19 and 20)) have been researched in LIBs and SIBs numerous times owing to their high capacity, low cost and environmental friendliness. Unfortunately, the limited cycling life of iron sulfides severely restricts their real application in energy storage.21,22 Wang et al. constructed the multi-functional yolk–shell FeS@C structure to improve the cycling stability, but which could only prolong the cycling life to 300 cycles.13 Through controlling the cut-off voltage to avoid the conversion reaction, Hu et al. have improved the cycling life of iron sulfides to a quite high level (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) but with inferior capacity.15 The key point to achieve high capacity and stability simultaneously is to sustain the high reversibility of the conversion reactions. Ultrafine nanoparticles have proved to be advantageous in this respect, which is attributed to nanoparticles having a size comparable to the diffusion length of the cation in the host-materials, leading to highly reversible and efficient conversion reaction.14 However, it still remains a challenge to make common materials to reach the quantum size.23
000 cycles) but with inferior capacity.15 The key point to achieve high capacity and stability simultaneously is to sustain the high reversibility of the conversion reactions. Ultrafine nanoparticles have proved to be advantageous in this respect, which is attributed to nanoparticles having a size comparable to the diffusion length of the cation in the host-materials, leading to highly reversible and efficient conversion reaction.14 However, it still remains a challenge to make common materials to reach the quantum size.23
Greigite Fe3S4, an important semi-metallic magnetic material, has been widely used in paleomagnetism, electrochemistry, biomedicine and environmental magnetic studies.24,25 However, to the best of our knowledge, there is no report on Fe3S4 as the anode of SIBs. Herein, we demonstrate Fe3S4 as a promising host-material for sodium storage. The involved conversion reaction pulverizes the Fe3S4 particles to quantum size during the sodiation/desodiation processes, resulting in a high capacity and superior stability. The synthesized Fe3S4 particles display a discharge capacity of 548 mA h g−1 in a wide operating voltage between 0.5 and 3 V. Meanwhile, the remarkable long-term cyclic stability (275 mA h g−1 after 3500 cycles at 20 A g−1) and excellent rate capability (233 mA h g−1 at 40 A g−1) assure its great potential for practical utilization. This high reversible conversion mechanism presents a new method to enable SIBs possessing both high capacity and long-cycle stability.
Results and discussion
As the counterpart of the oxide magnetite Fe3O4, greigite Fe3S4 contains 32 atoms of sulfur and 24 atoms of iron per unit cell. There are two sublattices of iron atoms where the Fe3+ ions occupy tetrahedral A-sites and both Fe2+ and Fe3+ ions occupy octahedral B-sites (Fig. 1a).25Fig. 1b shows the X-ray diffraction (XRD) pattern of the as-prepared Fe3S4. All diffraction peaks are fully consistent with JCPDS no. 89-1998, showing a cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The morphology of the as-prepared Fe3S4 is characterized by scanning electron microscopy (SEM) (Fig. 1c and d). The particles present octahedral features and the particle size is 100–200 nm. Fig. 1f clearly shows two lattices: (111) and (1−1−1), which are parallel to the surface of the octahedron. The intersection angle of these two lattice plane is measured to be 109.5°, which is consistent with the theoretical value. Combining the crystal structure of Fe3S4 (Fig. 1a), it is speculated that the exposed faces are the {111} family of crystal planes.
m space group. The morphology of the as-prepared Fe3S4 is characterized by scanning electron microscopy (SEM) (Fig. 1c and d). The particles present octahedral features and the particle size is 100–200 nm. Fig. 1f clearly shows two lattices: (111) and (1−1−1), which are parallel to the surface of the octahedron. The intersection angle of these two lattice plane is measured to be 109.5°, which is consistent with the theoretical value. Combining the crystal structure of Fe3S4 (Fig. 1a), it is speculated that the exposed faces are the {111} family of crystal planes.
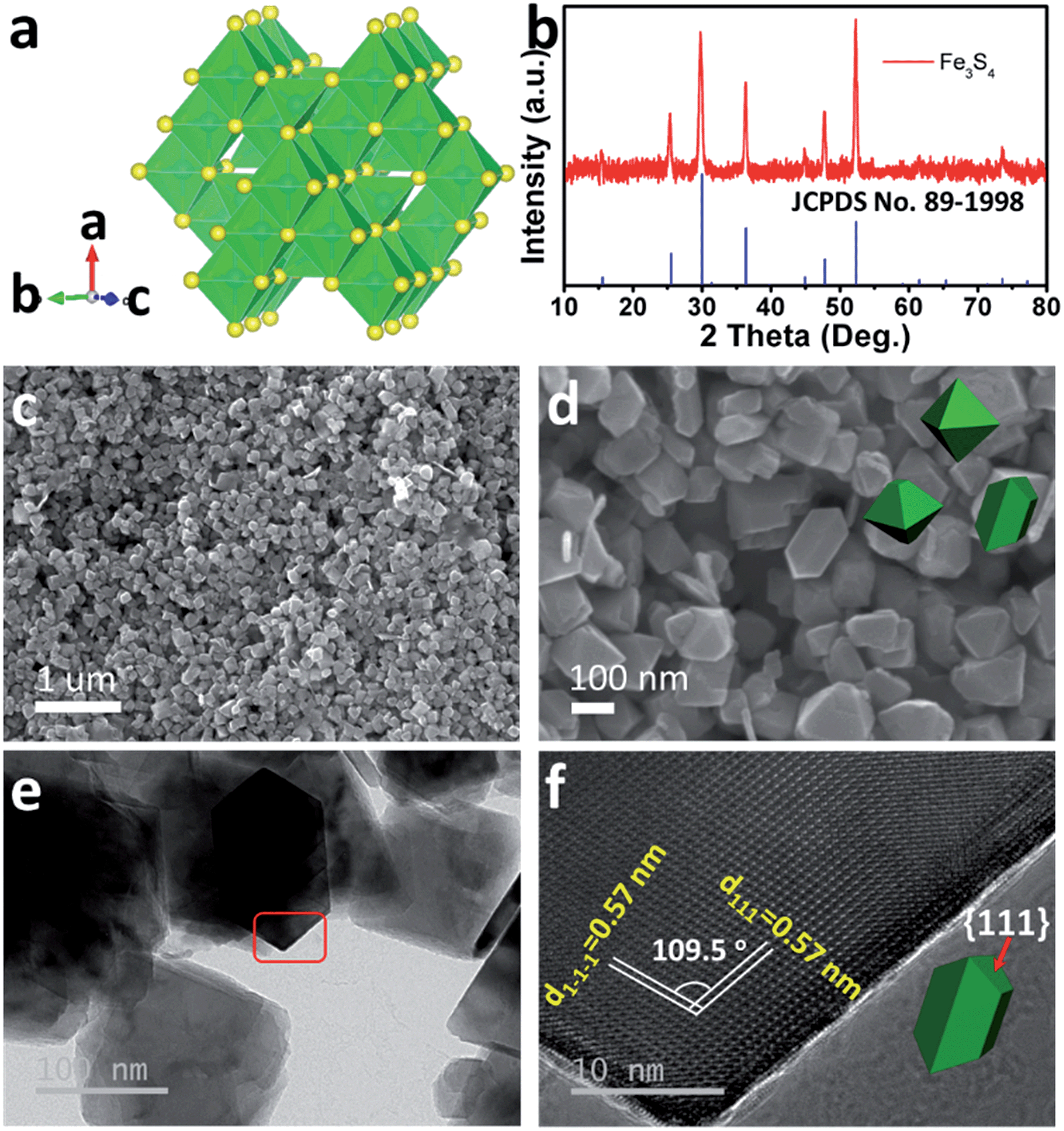 | ||
| Fig. 1 Structure characterization of the as-prepared Fe3S4. (a) Crystal structure and (b) XRD pattern of the as-prepared Fe3S4. (c and d) SEM images, (e) TEM and (f) HRTEM images for the as-prepared Fe3S4. | ||
To test the electrochemical performances of the as-prepared Fe3S4 particles, CR2016 coin-type cells were fabricated. Fig. 2a shows the galvanostatic discharge/charge processes for the as-prepared Fe3S4 electrode at a low current rate of 0.2 A g−1 in a range of 0.5–3.0 V. The Fe3S4 delivers a high initial discharge capacity of 571 mA h g−1, and the first charge capacity is 548 mA h g−1, which shows an impressive initial coulombic efficiency of 96%. After 100 cycles, it still delivers a reversible discharge capacity of 536 mA h g−1 (Fig. S1†). The cyclic voltammogram (CV) curves of the as-prepared Fe3S4 electrode at a scan rate of 0.2 mV s−1 show that the charge and discharge processes maintain stable curves after the initial cycle (Fig. S2a†). The long-term cycling is tested under a relatively high specific current (5 and 20 A g−1) as shown in Fig. 2b and S3.† After 50 cycles, the Fe3S4 anode delivers a stable discharge capacity of 435 mA h g−1 at 5 A g−1. After 1000 cycles, a capacity of 401 mA h g−1 is still obtained, showing an impressive cycling stability. Moreover, the shape of the capacity–voltage curves shows little change during cycling especially after 200 cycles, which confirms the stable and reversible discharge/charge processes (Fig. S2b†). The coulombic efficiency is kept at nearly 100% from beginning to end at such a high specific current. More attractively, a relatively high capacity of 275 mA h g−1 is also obtained after 3500 cycles even at a high specific current of 20 A g−1 (Fig. 2b). It should be pointed out that the superior cycling performance benefits from both the ether based electrolyte and the cut-off voltage, as shown in (Fig. S4†). Carbonate-based electrolytes (NaClO4/EC-DMC) suffer from rapid capacity fading (584 mA h g−1 at the first cycle and 15 mA h g−1 at the 200th cycle respectively, Fig. S4a and b†). Moreover, when we extend the operating voltage to 0.01–3 V, the capacity seriously fades from 748 to 132 mA h g−1 within 100 cycles at 2 A g−1 (Fig. S4c and d†).
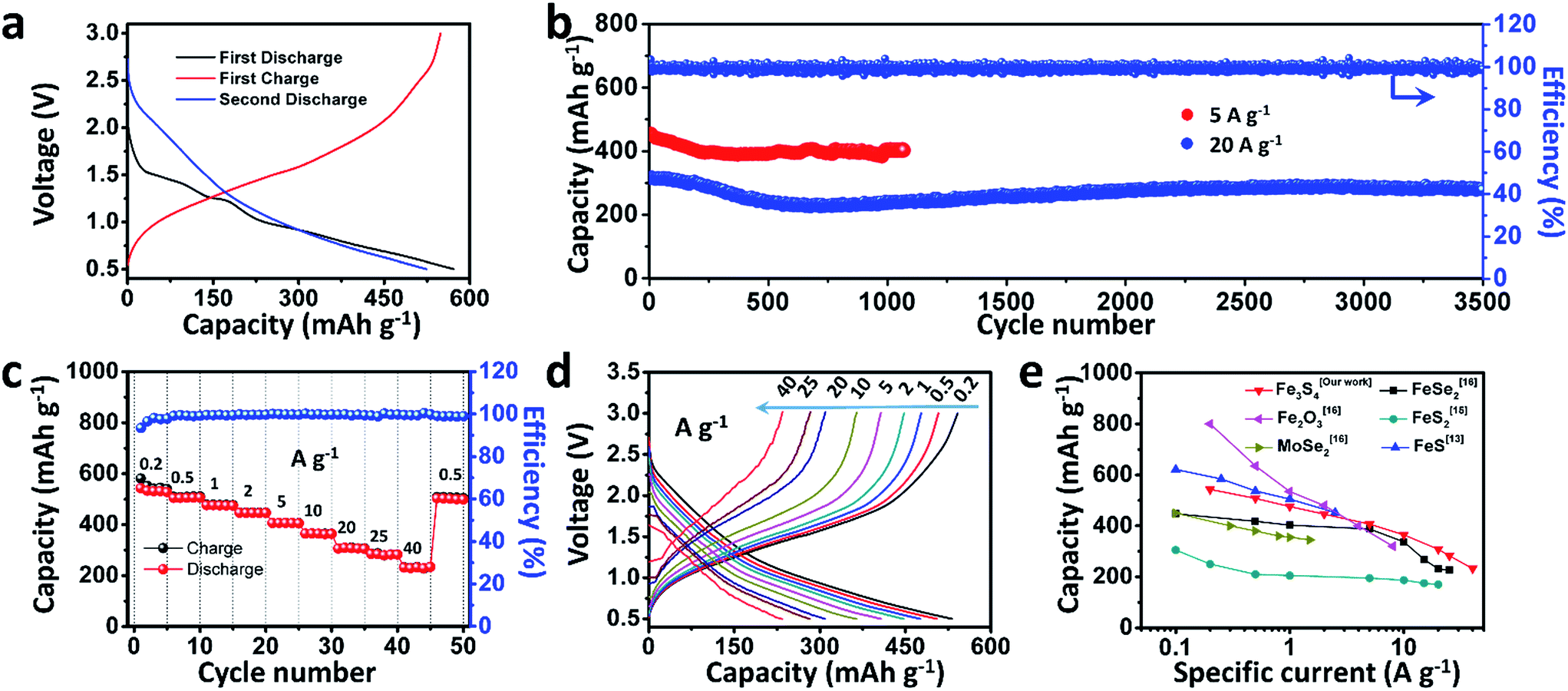 | ||
| Fig. 2 Electrochemical performance of the as-prepared Fe3S4. (a) Discharge–charge curves of the first two cycles at a specific current of 0.2 A g−1; (b) cycling performances at 5 and 20 A g−1; (c) rate capability and (d) discharge–charge curves at various current rates; (e) comparison of the as-prepared Fe3S4 with other conversion anode materials of SIBs. | ||
The rate capabilities of the as-prepared Fe3S4 electrode are further investigated at various specific currents ranging from 0.2 to 40 A g−1 (Fig. 2c). Fig. 2d exhibits the corresponding charge and discharge curves of the Fe3S4 electrode at different specific currents. The capacities show slight decline as the specific current gradually increases. When the specific currents reach 0.2, 0.5, 1, 2, 5, 10, 20, and 25 A g−1, the discharge capacities are 548, 508, 476, 446, 407, 365, 308, and 283 mA h g−1, respectively. It is noteworthy that even at an extremely high specific current of 40 A g−1, a capacity of 233 mA h g−1 is still achieved, corresponding to 43% capacity utilization within 21 s. Its corresponding areal current densities and areal capacities are shown in Table S1.† Comparing the excellent rate performance with the state of the art conversion type anode materials, the Fe3S4 anode has a distinct advantage at high specific currents (Fig. 2e).13,15,16
Sodium-storage mechanism
57Fe Mössbauer spectra and TEM were used to study intensively the sodium-storage mechanism of the as-prepared Fe3S4 particles (Fig. 3). Three states have been chosen to investigate the mechanism. The first charge and discharge curves (I (fresh state) → II (discharge to 0.5 V) → III (charge to 3 V)) of the Fe3S4-based battery at a specific current of 0.2 A g−1 are shown in Fig. 3a. The sodium-storage mechanism is first revealed via57Fe Mössbauer spectra. The representative 57Fe Mössbauer spectra recorded at room temperature for all samples are shown in Fig. 3c. The pristine Fe3S4 sample consists of two magnetic sextets and one central doublet, shown in Table S1.† Two sextets represent hyperfine interactions of Fe ions in octahedral and tetrahedral sites. The quadrupole doublet is probably associated with thermally relaxed fine particles present in the sample but already not visible by XRD.26 The obtained data is in good agreement with the previous findings for natural and synthetic greigite.27 The spectra of discharged and charged samples represent no magnetic sextets due to superparamagnetic behavior of small particles. The center shift (CS), quadrupole splitting (QS), and area ratio (A) obtained from analysis of the spectra are listed in Table S1.† For the discharged sample, the observed CS value of the bigger singlet (singlet 1) is −0.08 mm s−1, which clearly indicates that the iron is present in the nanostructured metallic state (α-Fe), the smaller singlet (singlet 2) can be attributed to nanoparticles of hexagonal FeS.28 Moreover, the HRTEM image is collected at the state II (Fig. 3c), which displays two sets of parallel fringes with the same d-spacing of 0.26 nm and an included angle of 60° between them, corresponding to the (200) and (020) planes of FeS (JCPDF no. 37-0477), which is consistent with the results obtained by Mössbauer techniques. To verify the state of sulphur, scanning TEM (STEM) and energy dispersive X-ray spectrometer (EDS) mappings were collected at state II (Fig. 3d) and III (Fig. 3e). When discharged to 0.5 V, the distribution of Na is well consistent with that of S, verifying the formation of Na2S. Fe shows a uniform distribution. Therefore, the initial discharge reaction can be expressed as eqn (S1).†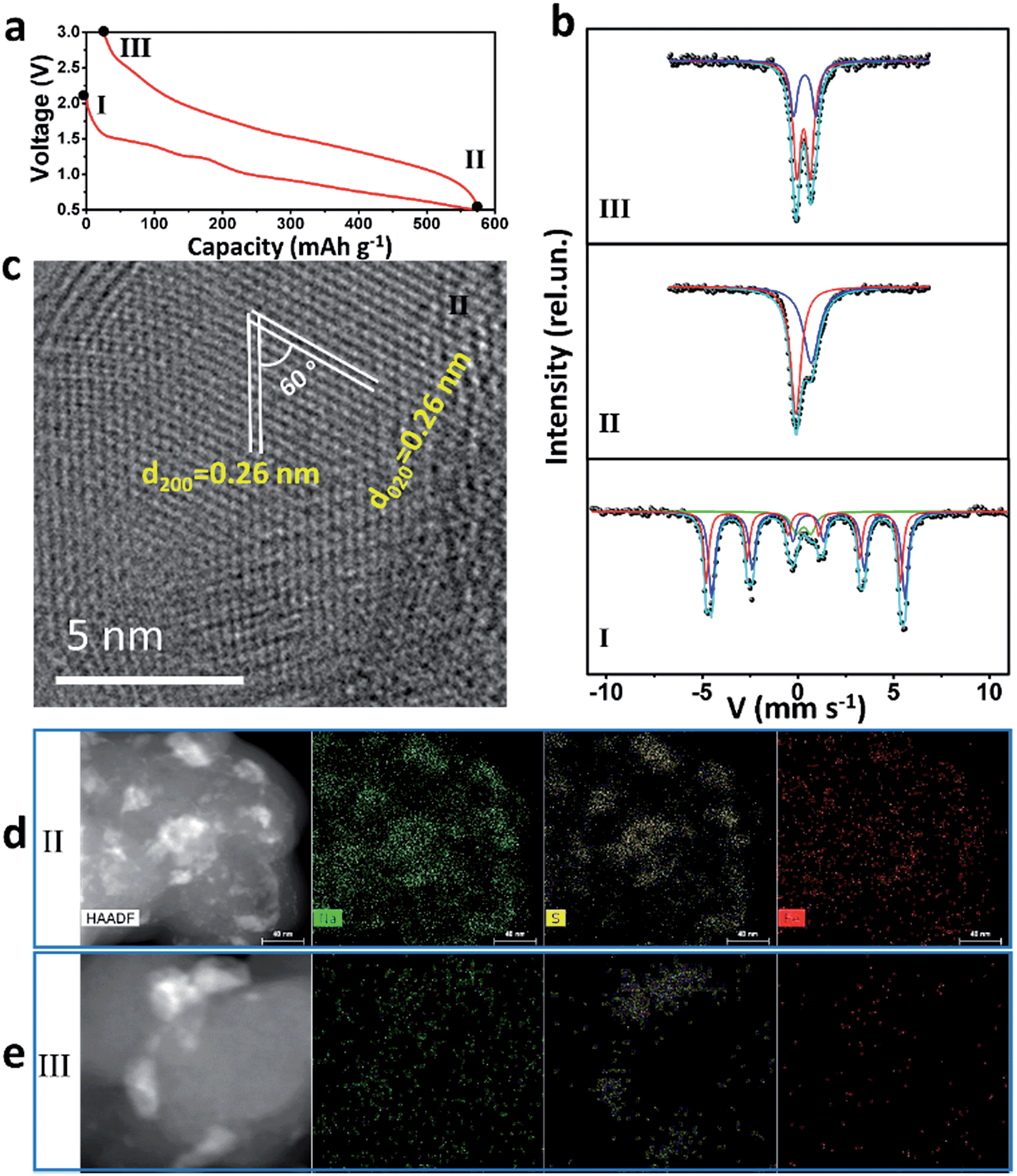 | ||
| Fig. 3 (a) Discharge–charge curves of the first cycle at the specific current of 0.2 A g−1; (b) 57Fe Mössbauer spectra, (c) TEM image, (d) STEM and (e) EDS mappings of the as-prepared Fe3S4 at different states. | ||
According to this equation, the theoretical capacity of Fe3S4 when discharging to 0.5 V is calculated to be 543 mA h g−1, which is very consistent with the reversible capacity of 548 mA h g−1 obtained at 0.2 A g−1. Furthermore, the Fe0 produced during the conversion reaction improves the conductivity of the electrode significantly, which is confirmed by the electrochemical impedance spectrum (EIS) (Fig. S5†). The EIS spectrum shows two compressed semicircles from the high to medium frequency range of each spectrum at the state II, for which the second semicircle describes the charge transfer resistance (Rct) of the electrode. After simulation, the values of Rct for the state I and II electrodes are calculated to be 98.8 and 10.7 Ω, respectively. The improved conductivity after the formation of Fe has great benefits for high-rate performance.29
For charging process, the Mössbauer spectra for the charged sample can be fitted with two doublets with similar CS values but different QS values of 0.69 and 1.15 mm s−1. The first more-intense doublet can be attributed to the ferrous low spin Fe2+ state, probably in tetrahedral FeSx.30 The nature of the second doublet is unclear, and the ferric ion Fe3+ state can be caused by some other iron sulfide species. These results indicate that, after first charging to 3 V, the active materials exist in the form of compounds, which can be defined as FeSx. The FeSx is also confirmed by the HRTEM image at state III (Fig. S6†), which is composed of FeS2, Fe3S4 and FeS. Notably, due to the conversion reaction, the FeSx compounds are pulverized to nanocrystals with the size of ∼1–10 nm (Fig. S6b†). This quantum size is of great significance for iron sulfide to achieve improved cycling and better rate capability because of the shorter diffusion lengths of Fe in iron sulfide (LD = 10−17 cm2 s−1 at 100 °C or ∼10−18 cm2 s−1 at room temperature in FeS2).14,31,32 The compound FeSx with quantum size, which is comparable or smaller than the Fe diffusion distance during cation exchange, overcomes the significant kinetic and thermodynamic constraints of chemical conversion to achieve an excellent cycling and rate capability.14 After charging back to 3 V, the elemental mapping images display the uniform distribution of Na and Fe, but the S is still concentrated in some areas (Fig. 3e). These results demonstrate that the S2− is at least in part transformed into S0. Therefore, the possible reaction during the reversible charging processes is summarized as eqn (S2).†
To confirm the active material in the subsequent cycles, TEM images of the electrode at full charge state after 200 cycles were collected (Fig. 4). The Selected Area Electron Diffraction (SAED) patterns confirm the coexistence of Fe3S4, FeS2 and FeS (Fig. 4b). The HRTEM image shows the nanocrystal of Fe3S4 and FeS2 (Fig. 4c). The whole sodium-storage mechanism is illustrated in Fig. 4d. In the sodiation process, Na+ exchanges with Fex+ to form Na2S, and the exchanged Fex+ obtain electrons to form Fe0. A portion of Fex+ still occupies octahedral sites to form FeS due to the controlled cut-off voltage. In the sodiation process, Fe0 exchanges with Na+ to form the Fe–S tetrahedron or octahedron, which further assembles to form the quantum-sized FeS2, Fe3S4 and FeS. The quantum-sized FeSx insure a synergistic and highly reversible conversion reaction which results in the superior cyclability and rate capability.14,33
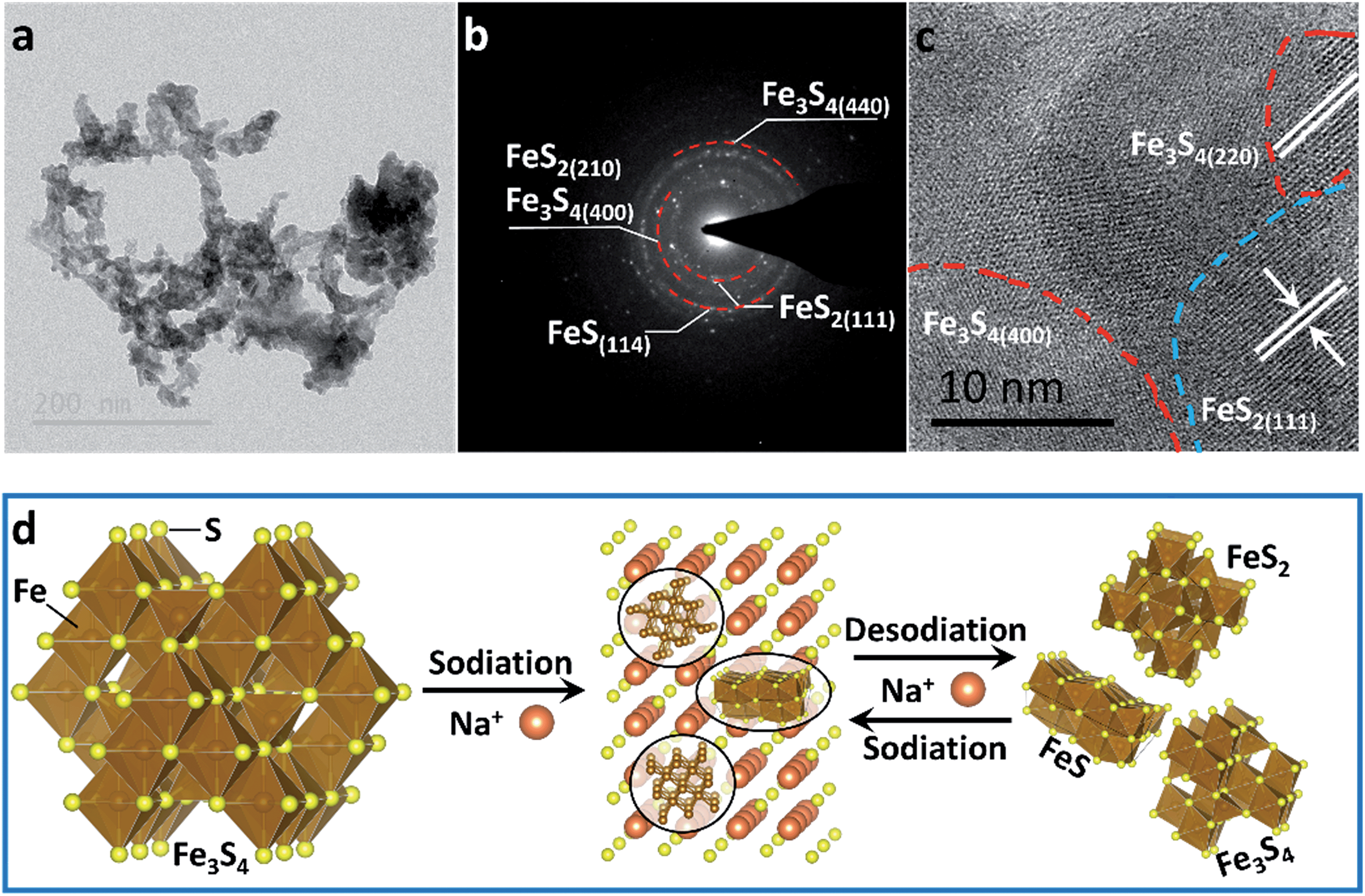 | ||
| Fig. 4 (a) TEM image, (b) SAED pattern and (c) HRTEM image of the as-prepared Fe3S4 at a full charge state after 200 cycles. (d) Schematic illustration of the sodium-storage mechanism in the Fe3S4 electrode. | ||
Conclusions
In summary, Fe3S4 particles have been prepared successfully and used as an anode material for SIBs for the first time. A conversion mechanism with 6 Na+ per formula has been proved between a wide operating window of 0.5–3 V. Due to the conversion reaction, Fe3S4 particles are pulverized to the quantum-sized compound FeSx which is composed of FeS, FeS2 and Fe3S4 quantum dots. The quantum-sized FeSx insure a synergistic and high reversible conversion reaction to provide the electrode with excellent cyclability and rate capability. As a result, Fe3S4 delivers a stable discharge capacity of 275 mA h g−1 after 3500 cycles at 20 A g−1. Even at 40 A g−1, a high discharge capacity of 233 mA h g−1 is obtained. This remarkable performance makes Fe3S4 a promising application candidate for the development of SIBs with high-rate capability and long-term cyclability. We believe that this high reversible conversion mechanism provides a new direction to improve the electrochemical performance of TMD materials for SIBs. Moreover, the involved electrochemical pulverization process provides a new route to synthesise quantum-sized materials.Acknowledgements
This work was supported by the National Basic Research Program of China (2013CB934103), the International Science & Technology Cooperation Program of China (2013DFA50840), the National Natural Science Foundation of China (51521001, 51272197, 51302203), the National Natural Science Fund for Distinguished Young Scholars (51425204), the Hubei Provincial Natural Science Fund for Distinguished Young Scholars (2014CFA035), and the Fundamental Research Funds for the Central Universities (WUT: 2016III001, 2016III002, 2016III003, 2016III004, 2016III006).Notes and references
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- L. Li, R. Jacobs, P. Gao, L. Gan, F. Wang, D. Morgan and S. Jin, J. Am. Chem. Soc., 2016, 138, 2838–2848 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- S. W. Kim, D. H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- M. S. Islam and C. A. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- Y. Sun, C. Liu, D. C. Grauer, J. Yano, J. R. Long, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699–17702 CrossRef CAS PubMed.
- N. Kornienko, J. Resasco, N. Becknell, C.-M. Jiang, Y.-S. Liu, K. Nie, X. Sun, J. Guo, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2015, 137, 7448–7455 CrossRef CAS PubMed.
- A. B. Wong, S. Brittman, Y. Yu, N. P. Dasgupta and P. Yang, Nano Lett., 2015, 15, 4096–4101 CrossRef CAS PubMed.
- X. Xu, Z. Fan, X. Yu, S. Ding, D. Yu and X. W. D. Lou, Adv. Energy Mater., 2014, 4, 1400902 CrossRef.
- X. Cao, C. Tan, X. Zhang, W. Zhao and H. Zhang, Adv. Mater., 2016, 28, 6167–6196 CrossRef CAS PubMed.
- Y.-X. Wang, J. Yang, S.-L. Chou, H. K. Liu, W.-x. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6, 8689 CrossRef CAS PubMed.
- A. Douglas, R. Carter, L. Oakes, K. Share, A. P. Cohn and C. L. Pint, ACS Nano, 2015, 9, 11156–11165 CrossRef CAS PubMed.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309–1316 CAS.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS PubMed.
- W. J. Li, C. Han, S. L. Chou, J. Z. Wang, Z. Li, Y. M. Kang, H. K. Liu and S. X. Dou, Chem.–Eur. J., 2016, 22, 590–597 CrossRef CAS PubMed.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- T. Kim, J. Choi, H. Ryu, G. Cho, K. Kim, J. Ahn, K. Cho and H. Ahn, J. Power Sources, 2007, 174, 1275–1278 CrossRef CAS.
- T. Kim, W. Jung, H. Ryu, K. Kim, J. Ahn, K. Cho, G. Cho, T. Nam, I. Ahn and H. Ahn, J. Alloys Compd., 2008, 449, 304–307 CrossRef CAS.
- R. Fong, J. Dahn and C. Jones, J. Electrochem. Soc., 1989, 136, 3206–3210 CrossRef CAS.
- D. Golodnitsky and E. Peled, Electrochim. Acta, 1999, 45, 335–350 CrossRef CAS.
- H. Gu, R. Zheng, X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 5664–5665 CrossRef CAS PubMed.
- Z. He, S. H. Yu, X. Zhou, X. Li and J. Qu, Adv. Funct. Mater., 2006, 16, 1105–1111 CrossRef CAS.
- I. Lyubutin, S. Starchikov, C.-R. Lin, S.-Z. Lu, M. O. Shaikh, K. Funtov, T. Dmitrieva, S. Ovchinnikov, I. Edelman and R. Ivantsov, J. Nanopart. Res., 2013, 15, 1–13 CrossRef.
- L. Chang, A. P. Roberts, Y. Tang, B. D. Rainford, A. R. Muxworthy and Q. Chen, J. Geophys. Res., 2008, 113, B06104 CrossRef.
- R. Vandenberghe, E. De Grave, P. De Bakker, M. Krs and J. Hus, Hyperfine Interact., 1992, 68, 319–322 CrossRef.
- W. Kim, I. J. Park and C. S. Kim, J. Appl. Phys., 2009, 105, 07D535 Search PubMed.
- Q. Xie, Y. Ma, X. Wang, D. Zeng, L. Wang, L. Mai and D.-L. Peng, ACS Nano, 2016, 10, 1283–1291 CrossRef CAS PubMed.
- I. Dékány, L. Turi, Z. Homonnay, A. Vértes and K. Burger, Colloids Surf., A, 1996, 119, 195–203 CrossRef.
- M. T. McDowell, Z. Lu, K. J. Koski, J. H. Yu, G. Zheng and Y. Cui, Nano Lett., 2015, 15, 1264–1271 CrossRef CAS PubMed.
- J. Chen and W. Harvey, Metall. Trans. B, 1975, 6, 331–339 CrossRef.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02716d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |