
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetalloporphyrin-modified semiconductors for solar fuel production†
D.
Khusnutdinova
,
A. M.
Beiler
,
B. L.
Wadsworth
,
S. I.
Jacob
and
G. F.
Moore
*
School of Molecular Sciences and the Biodesign Institute Center for Applied Structural Discovery (CASD), Arizona State University, Tempe, AZ 85287-1604, USA. E-mail: gfmoore@asu.edu
First published on 5th August 2016
Abstract
We report a direct one-step method to chemically graft metalloporphyrins to a visible-light-absorbing gallium phosphide semiconductor with the aim of constructing an integrated photocathode for light activating chemical transformations that include capturing, converting, and storing solar energy as fuels. Structural characterization of the hybrid assemblies is achieved using surface-sensitive spectroscopic methods, and functional performance for photoinduced hydrogen production is demonstrated via three-electrode electrochemical testing combined with photoproduct analysis using gas chromatography. Measurements of the total per geometric area porphyrin surface loadings using a cobalt-porphyrin based assembly indicate a turnover frequency ≥3.9 H2 molecules per site per second, representing the highest reported to date for a molecular-catalyst-modified semiconductor photoelectrode operating at the H+/H2 equilibrium potential under 1-sun illumination.
Introduction
Energy and environmental issues will likely dominate science and society for the next several decades as climate change threatens the wellbeing of the planet.1 In this scenario, the development of advanced materials and techniques for controlling matter and energy at the nanoscale is receiving increased global attention2 as a technological path to restoring a safe operating space for humanity.3 Artificial photosynthesis, which uses concepts inspired by its biological counterpart to produce fuels, is an attractive approach to storing solar energy.4 To this end, the immobilization of molecules on semiconductor materials is gaining interest.5 Although some recent progress has been made in development of such assemblies,6 finding new and more effective ways to interface catalysts to semiconductor surfaces remains a major challenge.7Metalloporphyrins serve important roles in biology and as components in emerging molecular-based materials.8 As electrocatalysts, they are capable of chemically transforming protons into hydrogen as well as converting carbon dioxide into carbon monoxide when electrochemically activated in solution or immobilized at a conductive substrate polarized at an appropriate potential. Herein, we report a one-step method to chemically graft metalloporphyrin complexes onto p-type GaP(100), a midsize optical band gap semiconductor that has shown promise in light-emitting-diode technologies and in applications for solar energy transduction as light capture and conversion components.9 The cobalt and iron porphyrin analogs used in this report are prepared via a novel synthetic strategy to yield a macrocycle with a pendent 4-vinylphenyl surface attachment group at the β-position of the porphyrin ring structure. This modification allows use of the UV-induced immobilization chemistry of olefins10 to attach intact metalloporphyrin complexes to the semiconductor surface. While the mechanistic details of the vinyl group attachment chemistry are not settled, molecular binding appears to occur over bridging oxygen atoms on GaP surfaces.6b,i,10a
Results and discussion
Materials preparation
Synthesis of the 4-vinylphenyl functionalized metalloporphyrins is described in detail as ESI.† Preparation of the GaP substrates for subsequent photochemical functionalization using the structurally modified porphyrins begins with buffered hydrofluoric acid treatment to remove the bulk surface oxide layers. The freshly etched wafers are placed into a sealed quartz flask containing an argon-sparged solution of the appropriate porphyrin and illuminated with shortwave UV light (254 nm) for 2 h. The porphyrin-functionalized wafers are then removed from the flask, ultrasonically cleaned, and dried under nitrogen (see Experimental section for details).Structural characterization
Grazing angle total reflectance Fourier transform infrared (GATR-FTIR) spectra of unmodified GaP(100) substrates following acid treatment are characterized by significant residual surface oxygen coverage, and static water contact angles of <10° indicate a dominant coverage by hydrophilic hydroxyl groups (Fig. S13†). However, GATR-FTIR absorbance spectra collected using samples following cobalt or iron porphyrin functionalization, yielding CoP–GaP or FeP–GaP (Fig. 1a), are characterized by unique vibrational features corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond ring modes of the porphyrin, appearing at 1607 cm−1, as well as transitions that are assigned to the Cβ–H, Cα–N, and Cβ–Cβ vibrations of the macrocycle (Fig. S14 and S15†). FTIR spectra of the cobalt and iron porphyrins prior to surface immobilization show similar CC bond ring modes centered at 1607 cm−1, but also include an additional pronounced peak centered at 1626 cm−1 associated with the vinyl CC bond (Fig. S16†). The lack of this pronounced feature at 1626 cm−1 in spectra of the metalloporphyrin-modified GaP samples indicates undetectable to no vinyl functionality on the surface, consistent with the proposed mechanism of the vinyl group grafting chemistry on hydroxyl and oxygen-terminated surfaces.6b,i,10a–c Further, the Co–N and Fe–N vibrations observed on the surfaces of the CoP–GaP or FeP–GaP (1001 cm−1 and 997 cm−1, respectively) provide compelling evidence that the porphyrin metal centers remain intact following the grafting procedure (Fig. 1b and c). In contrast, the N–H vibration of analogous free-base porphyrins occurs at 966 cm−1 (Fig. S7, S8, & S17†). The similarity in positions of the nitrogen–metal vibrations observed on the metalloporphyrin-functionalized GaP surfaces with those observed in spectra of analogous non-surface-attached metalloporphyrins indicates the porphyrin metal centers maintain a similar vibrational environment following immobilization. Lastly, spectra of control samples, in which metalloporphyrins without the vinyl group functionality (CoTTP or FeTTP) are used during the photochemical grafting step, show no evidence of porphyrin complexes at the GaP surface.
C bond ring modes of the porphyrin, appearing at 1607 cm−1, as well as transitions that are assigned to the Cβ–H, Cα–N, and Cβ–Cβ vibrations of the macrocycle (Fig. S14 and S15†). FTIR spectra of the cobalt and iron porphyrins prior to surface immobilization show similar CC bond ring modes centered at 1607 cm−1, but also include an additional pronounced peak centered at 1626 cm−1 associated with the vinyl CC bond (Fig. S16†). The lack of this pronounced feature at 1626 cm−1 in spectra of the metalloporphyrin-modified GaP samples indicates undetectable to no vinyl functionality on the surface, consistent with the proposed mechanism of the vinyl group grafting chemistry on hydroxyl and oxygen-terminated surfaces.6b,i,10a–c Further, the Co–N and Fe–N vibrations observed on the surfaces of the CoP–GaP or FeP–GaP (1001 cm−1 and 997 cm−1, respectively) provide compelling evidence that the porphyrin metal centers remain intact following the grafting procedure (Fig. 1b and c). In contrast, the N–H vibration of analogous free-base porphyrins occurs at 966 cm−1 (Fig. S7, S8, & S17†). The similarity in positions of the nitrogen–metal vibrations observed on the metalloporphyrin-functionalized GaP surfaces with those observed in spectra of analogous non-surface-attached metalloporphyrins indicates the porphyrin metal centers maintain a similar vibrational environment following immobilization. Lastly, spectra of control samples, in which metalloporphyrins without the vinyl group functionality (CoTTP or FeTTP) are used during the photochemical grafting step, show no evidence of porphyrin complexes at the GaP surface.
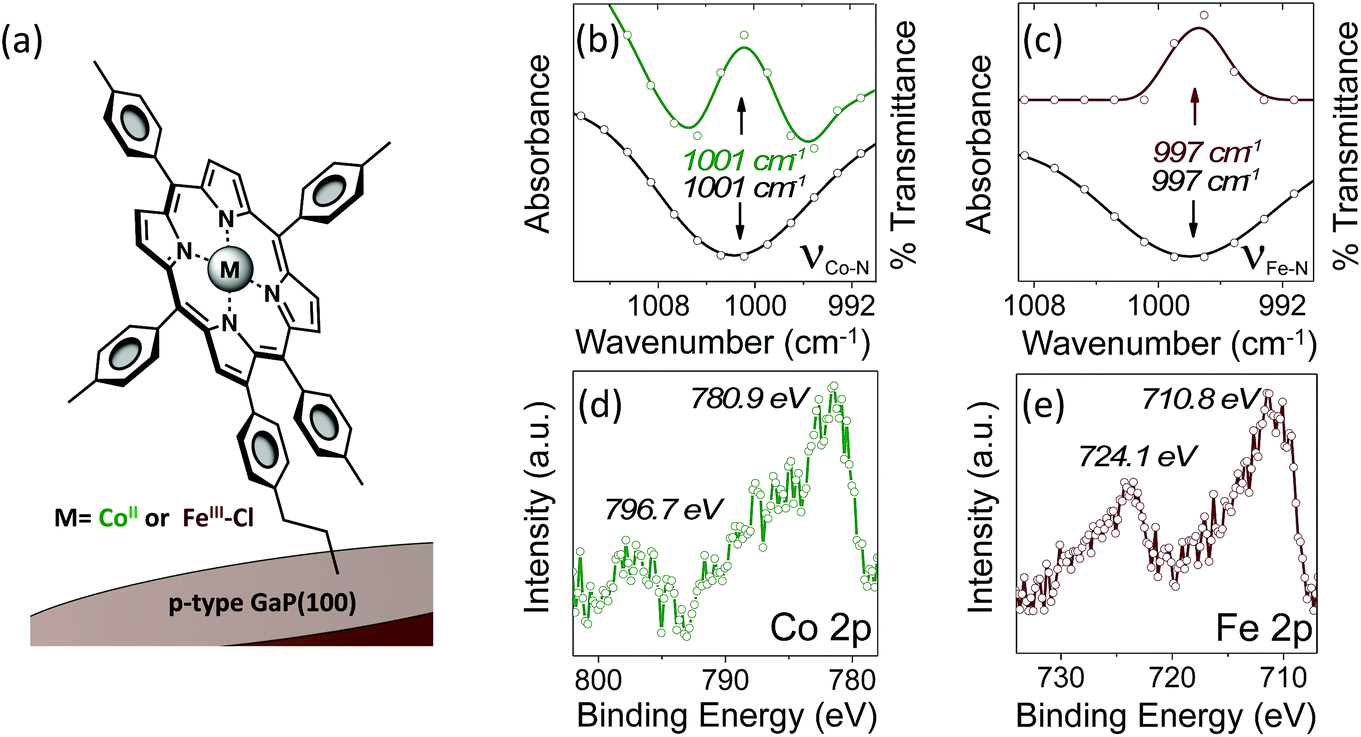 | ||
| Fig. 1 (a) Schematic representation of the CoP–GaP and FeP–GaP constructs. (b) GATR-FTIR absorbance spectra showing the porphyrin pyrollic nitrogen–cobalt vibration, νCo–N, at the surface of CoP–GaP (green) and FTIR transmission spectra showing the νCo–N of the non-surface-attached cobalt porphyrin (black). (c) GATR-FTIR absorbance spectra showing the porphyrin pyrollic nitrogen–iron vibration, νFe–N, at the surface of FeP–GaP (dark red) and FTIR transmission spectra showing the νFe–N of the non-surface-attached iron porphyrin (black). (d) Co 2p core level XP spectra of CoP–GaP. (e) Fe 2p core level XP spectra of FeP–GaP. | ||
X-ray photoelectron (XP) spectroscopy provides additional characterization and evidence of successful functionalization. As compared to spectra obtained using unmodified GaP samples, survey XP spectra of CoP–GaP surfaces show the presence of additional N, Co, and C elements associated with attached cobalt porphyrins, and spectra of FeP–GaP surfaces show the presence of additional N, Fe, and C elements associated with attached iron porphyrins (Fig. S20 & S22†). In addition, high-energy resolution Co 2p core level spectra of the CoP–GaP samples show peaks centered at 780.9 eV (2p3/2) and 796.7 eV (2p1/2) with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 branching ratio (Fig. 1d). The Co 2p3/2 signal indicates a complex multiplet structure, consistent with the oxidation state +2 and the open-shell (d7) character of the Co ion (Fig. S21†). For the FeP–GaP substrates, Fe 2p core level spectra contain features characteristic of FeIII porphyrins, including peaks centered at 710.8 eV (2p3/2) and 724.1 eV (2p1/2) (Fig. 1e). For both constructs, analysis of the metal 2p and nitrogen 1s spectral intensity ratios yields metal:nitrogen ratios of 1:4, indicating no detectable loss of metal from the attached porphyrin units following UV-induced grafting.
:1 branching ratio (Fig. 1d). The Co 2p3/2 signal indicates a complex multiplet structure, consistent with the oxidation state +2 and the open-shell (d7) character of the Co ion (Fig. S21†). For the FeP–GaP substrates, Fe 2p core level spectra contain features characteristic of FeIII porphyrins, including peaks centered at 710.8 eV (2p3/2) and 724.1 eV (2p1/2) (Fig. 1e). For both constructs, analysis of the metal 2p and nitrogen 1s spectral intensity ratios yields metal:nitrogen ratios of 1:4, indicating no detectable loss of metal from the attached porphyrin units following UV-induced grafting.
Photoelectrochemical measurements
Illumination of CoP–GaP electrodes polarized at 0 V vs. RHE in pH neutral aqueous solutions results in hydrogen generation at an initial rate of ∼10 μL min−1 cm−2 (Fig. 2a–d and Table 1). This rate of hydrogen evolution exhibits less than 10% loss of activity over 4 h of photoelectrochemical (PEC) testing (Fig. S27†). By contrast, the iron-based constructs show significant diminution of performance during PEC testing, including a rapid loss in current density following illumination during bulk-electrolysis measurements (Fig. 2a). Further, the relatively stable photocurrent densities that are measured after the drop off are similar in value to those initially achieved using unmodified GaP electrodes polarized at the same potential (0 V vs. RHE). Thus, there is a nearly complete loss of the photocurrent gains afforded by FeP functionalization. Although iron porphyrins are notorious for their relative instability, including a propensity to form μ-oxo dimers and undergo auto-reduction reactions,8q,11 a detailed analysis of the photocurrent degradation pathways regarding the FeP–GaP constructs is currently unavailable. These results do, however, illustrate the synthetic versatility of the porphyrin architecture, including selection of the catalytic metal site for controlling activity, and presence of ligand auxiliaries for tailoring their molecular structure as well as associated electronic properties.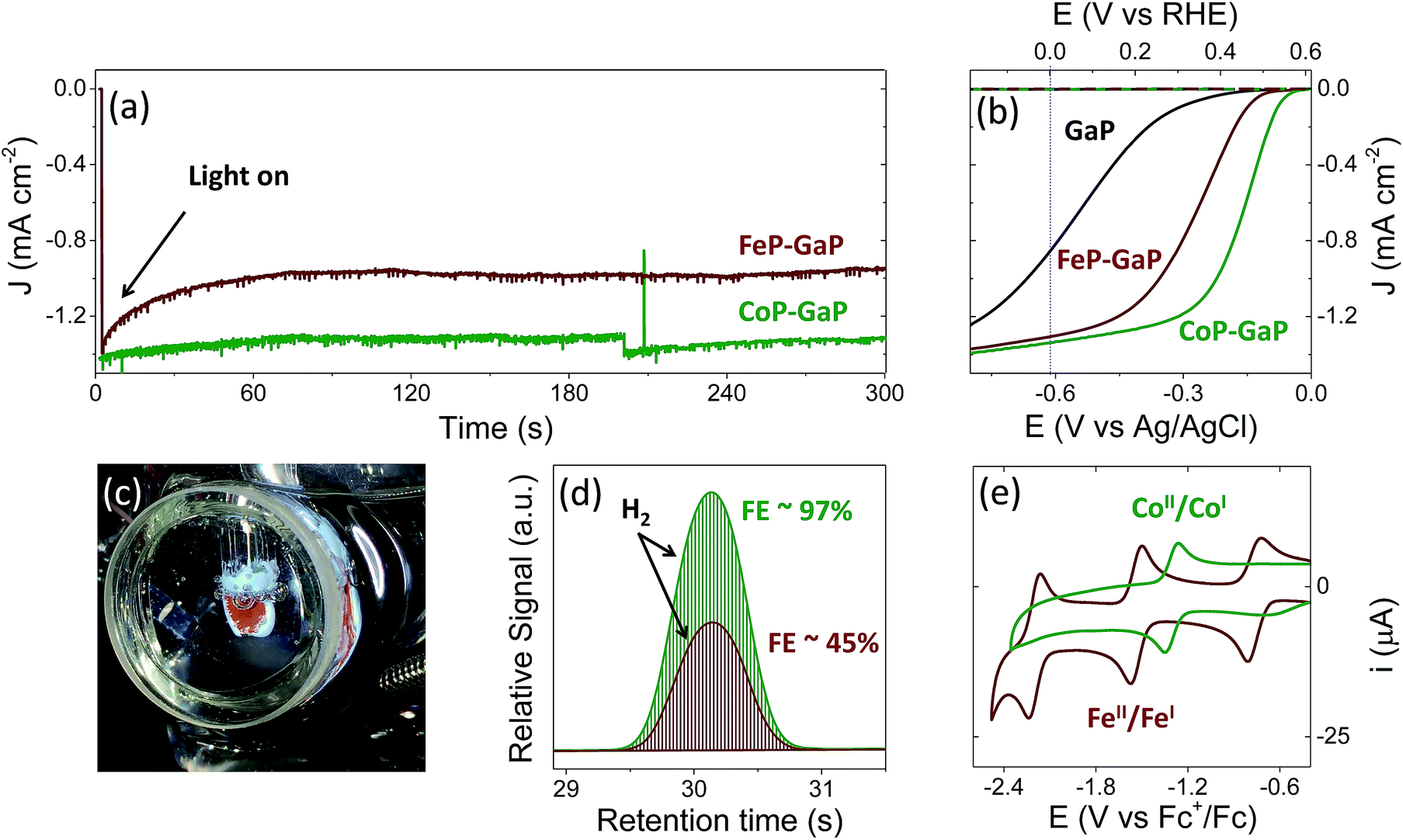 | ||
| Fig. 2 Three-electrode electrochemical data collected using unmodified GaP(100) (black), CoP–GaP (green), or FeP–GaP (dark red) working electrodes in phosphate buffer (pH 7) including (a) chronoamperograms using working electrodes polarized at a constant potential of 0 V vs. RHE and under 1-sun illumination (100 mW cm−2), (b) linear sweep voltammograms recorded in the dark (dashed) or under 1-sun illumination (solid), (c) an image of a CoP–GaP photocathode under photoelectrochemical testing, and (d) gas chromatograms obtained using samples of headspace gas collected from sealed photoelectrochemical cells containing working electrodes polarized at a constant potential of 0 V vs. RHE and under 1-sun illumination. The amount of hydrogen produced in these experiments corresponds to a faradaic efficiency (FE) of 97% following 30 min of illumination using CoP–GaP and 45% following 6 min of illumination using FeP–GaP. (e) Cyclic voltammetry data recorded using butyronitrile solutions of model cobalt (green) or iron (dark red) porphyrin compounds are included for comparison. | ||
| Construct | V oc (V vs. RHE) | E at −1 mA cm−2 (V vs. RHE) | J at 0 V vs. RHE (mA cm−2) |
|---|---|---|---|
| GaP | 0.57 ± 0.03 | −0.04 ± 0.06 | −0.86 ± 0.21 |
| CoP–GaP | 0.61 ± 0.01 | 0.35 ± 0.03 | −1.31 ± 0.03 |
| FeP–GaP | 0.61 ± 0.01 | 0.23 ± 0.07 | −1.29 ± 0.04 |
During PEC testing, the formation of gas bubbles at the surface of the porphyrin-modified electrode are transiently observed in linear sweep voltammetry experiments, when the electrodes are polarized at potentials generating cathodic currents, and continuously observed during bulk photoelectrolysis experiments (Fig. 2c). Gas chromatography analysis of the photoproducts confirms the production of hydrogen with near-unity faradaic efficiency (measured at ∼97% following 30 min of illumination) when using CoP–GaP working electrodes (Fig. 2d). These results confirm that no measurable hydrogen is present prior to illumination of the electrode surface (Fig. S24†) and the rate of hydrogen production is directly correlated with the current produced by the cell during illumination. Measurements performed using FeP–GaP working electrodes polarized at 0 V vs. RHE also confirm the photoproduction of hydrogen. However, the faradaic efficiency is ∼45% following 6 min of illumination.
To facilitate comparisons with data obtained using the metalloporphyrin-modified GaP constructs in aqueous conditions, cyclic voltammograms of CoTTP and FeTTP recorded in organic solvents with a supporting electrolyte (0.1 M tetrabutylammonium hexafluorophosphate in butyronitrile) are included in this report (Fig. 2e). Under these conditions, the difference in potential between the midpoints of the CoII/CoI and FeII/FeI couples is 230 mV, with the cobalt relay occurring at less negative potentials (Table S1†). For the metalloporphyrin-modified GaP surfaces, a difference in potential to access the catalytically active cobalt or iron redox state in aqueous conditions may contribute to the 120 mV offset required to achieve a −1 mA cm−2 current density using the CoP–GaP versus FeP–GaP photocathodes (Table 1). However, other factors, including differences in hydricity of the metal centers12 and possible changes in electronic structure of the underlying semiconductors upon functionalization13 may contribute to this divergence. Nonetheless, the saturating current densities, measured at 0 V vs. RHE using CoP–GaP working electrodes, do increase approximately linearly with illumination intensity (Fig. 3), indicating that photocarrier transport to the interface in part limits the performance and that improvement in the spectral coverage and photophysical properties of the underpinning semiconductor could yield additional efficiency gains.
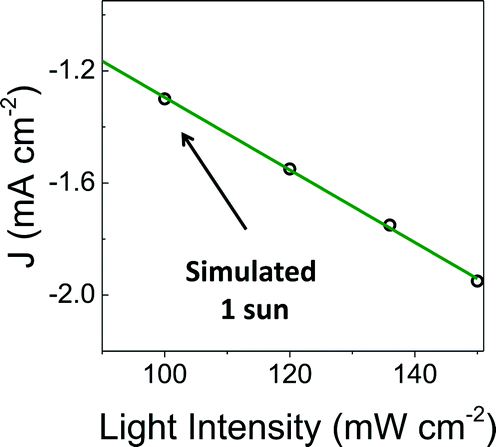 | ||
| Fig. 3 Photocurrent density recorded at increasing illumination intensity using a CoP–GaP working electrode polarized at 0 V vs. RHE. | ||
A comparison of the photon flux striking the CoP–GaP surface at simulated 1-sun intensity (Fig. 3 & S29†) with the electron flux measured during PEC testing allows an analysis of external quantum efficiency (EQE). Considering only photons in the GaP actinic range (Fig. S11 & S12†), i.e. those with energies higher than the 2.26 eV GaP band gap, the EQE = 19% for CoP–GaP electrodes polarized at 0 V vs. RHE. A similar analysis of the optical to chemical power conversion efficiency (η)14a is achieved by comparing the spectral irradiance at this wavelength range with the output chemical power represented by the rate of hydrogen production. Using the enthalpy of H2 combustion (286 kJ mol−1) or change in Gibb's free energy (237 kJ mol−1), η = 11% or 9%, respectively. We emphasize that these measurements are performed using a three-electrode configuration14b and thus represent energetics and efficiencies associated with a photocathode component, not a device.
Total cobalt loadings on the CoP–GaP surface were obtained using inductively coupled plasma mass spectroscopy (ICP-MS) (see Experimental section for details), yielding a cobalt porphyrin surface concentration of 0.59 ± 0.03 nmol cm−2. The loadings obtained from this analysis combined with the current densities measured in polarization experiments using CoP–GaP working electrodes yield information on the activity of the electrodes per number of porphyrins assembled on the surface and thus an estimate of the immobilized porphyrin turnover frequency (TOF). Using only the increase in current density obtained for a GaP electrode polarized at 0 V vs. RHE following cobalt porphyrin surface functionalization, this equates to a TOF ≥3.9 H2 molecules site−1 s−1, representing the highest reported to date for a molecular-catalyst-modified semiconductor photoelectrode operating at the H+/H2 equilibrium potential under 1-sun illumination. In future work, implementation of porphyrins with improved catalytic features and the development of synthetic methodologies to achieve higher porphyrin surface loading as well as improved interfacial dynamics may lead to further performance gains.
Conclusions
We describe a one-step method to chemically graft metalloporphyrin catalysts onto p-type gallium phosphide (100). The porphyrin complexes are structurally modified with a 4-vinylphenyl group essential to successful semiconductor attachment using the UV-induced grafting method. Structural analysis of the constructs using surface-sensitive characterization techniques, including XP and GATR-FTIR spectroscopy, provides evidence of successful grafting. The resulting hybrid material can be used as a photocathode for driving the hydrogen evolution half-reaction and shows significantly improved photoelectrochemical performance over unmodified electrodes. When using GaP(100) with identical doping conditions (i.e. cut from the same ingot), the PEC results using CoP–GaP show an enhanced rate and stability of photoinduced hydrogen production over the analogous FeP–GaP assemblies as well as those previously reported6j using cobaloxime-polymer-modified GaP electrodes prepared using a two-step attachment chemistry (Fig. S28†). Unlike the cobaloximes,15 the Co and Fe porphyrins permit access to metalI/metal0 redox couples and are known catalysts for the electrochemical reduction of carbon dioxide.8 Thus, methods to covalently graft metalloporphyrins to semiconductor substrates could lead to new perspectives and approaches of photoelectrochemically activating carbon dioxide. In addition, the porphyrins are synthetically versatile, allowing tailoring of their molecular structure and electronic properties as new discoveries and material developments emerge. Key features of the constructs reported here include use of metalloporphyrins with built-in chemical sites for direct grafting to a GaP semiconductor, creating hybrid assemblies capable of converting photonic energy to fuel.Experimental
Materials and synthesis
All compounds were synthesized from commercially available starting materials (see ESI,† Molecular synthesis and characterization). All reagents were purchased from Aldrich. Solvents were obtained from Aldrich or Mallinckrodt. Dichloromethane, hexanes, toluene and p-tolyl aldehyde were freshly distilled before use. Milli-Q water (18.2 MΩ cm) was used to prepare all aqueous solutions.Single crystalline p-type gallium phosphide wafers were purchased from University Wafers. The material is single side polished to an epi-ready finish. The p-type Zn-doped GaP(100) wafers have a resistivity of 0.2 Ω cm, a mobility of 66 cm2 V−1 s−1, and a carrier concentration of 4.7 × 1017 cm−3, with an etch pit density of less than 8 × 104 cm−2.
Wafer cleaning procedure
Diced semiconductor samples were degreased by wiping the surface with an acetone soaked cotton swab and ultrasonically cleaning in acetone and isopropanol for 5 min each, followed by drying under nitrogen. Samples were then exposed to an air-generated oxygen plasma (Harrick Plasma, U.S.) at 30 W for 2 min. Surface oxide layers were then removed by immersion of the plasma-treated samples in buffered hydrofluoric acid (6:1 HF/NH4F in H2O) for 5 min, followed by rinsing with Milli-Q water.
Wafer functionalization
Freshly etched wafers were put into an argon-sparged solution of the appropriate porphyrin (1 mM) in toluene and exposed to 254 nm UV light for 2 h. After thoroughly rinsing with toluene the wafers were dried under nitrogen and stored under vacuum.Electrode fabrication
GaP working electrodes were fabricated by applying an indium–gallium eutectic (Aldrich) to the backside of a wafer, then fixing a copper wire to the back of the wafer using a conductive silver epoxy (Circuit Works). The copper wire was passed through a glass tube, and the wafer was insulated and attached to the glass tube with Loctite 615 Hysol Epoxi-patch adhesive. The epoxy was allowed to fully cure before testing the electrodes.Instrument descriptions and experimental details
Acknowledgements
This work was supported by The College of Liberal Arts and Sciences at Arizona State University, the Biodesign Institute Center for Applied Structural Discovery (CASD) and LightWorks. We thank Gwyneth Gordon for assistance with ICP-MS measurements and Timothy Karcher for assistance with XP data collection. A. M. B. and B. L. W gratefully acknowledge IGERT-SUN fellowships funded by the National Science Foundation (Award 1144616). NMR studies were performed using the Magnetic Resonance Research Center at Arizona State University.Notes and references
- IPCC 5th Assessment Report, Geneva, Switzerland, 2014 Search PubMed.
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi and K. B. Yoon, Energy Environ. Sci., 2013, 6, 695 Search PubMed.
- (a) J. Rockström, W. Steffen, K. Noone, Å. Persson, F. S. Chapin, E. F. Lambin, T. M. Lenton, M. Scheffer, C. Folke, H. J. Schellnhuber and B. Nykvist, Nature, 2009, 461, 472 CrossRef PubMed; (b) W. Steffen, K. Richardson, J. Rockström, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpetner, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sörlin, Science, 2015, 347, 736 CrossRef CAS PubMed.
- (a) A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141 CrossRef CAS; (b) G. F. Moore and G. W. Brudvig, Annu. Rev. Condens. Matter Phys., 2011, 2, 303 CrossRef CAS; (c) R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis and T. A. Moore, Science, 2011, 332, 805 CrossRef CAS PubMed; (d) P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902 RSC; (e) J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357 RSC; (f) D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- (a) N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90 CrossRef CAS; (b) A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861 CrossRef CAS PubMed; (c) A. Krawicz, D. Cedeno and G. F. Moore, Phys. Chem. Chem. Phys., 2014, 16, 15818 RSC; (d) D. Cedeno, A. Krawicz, P. Doak, M. Yu, J. B. Neaton and G. F. Moore, J. Phys. Chem. Lett., 2014, 5, 3222 CrossRef CAS PubMed; (e) C. A. Downes and S. C. Marinescu, J. Am. Chem. Soc., 2015, 137, 13740 CrossRef CAS PubMed; (f) H. J. Kim, J. Seo and M. J. Rose, ACS Appl. Mater. Interfaces, 2016, 8, 1061 CrossRef CAS PubMed; (g) J. Gu, Y. Yan, J. L. Young, K. X. Steirer, N. R. Neale and J. A. Turner, Nat. Mater., 2015, 15, 456 CrossRef PubMed; (h) M. Schreier, J. Luo, P. Gao, T. Moehl, T. M. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 1938 CrossRef CAS PubMed; (i) A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, ACS Appl. Mater. Interfaces, 2016, 8, 10038 CrossRef CAS PubMed; (j) A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, Ind. Eng. Chem. Res., 2016, 55, 5306 CrossRef CAS.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- (a) W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105 CrossRef PubMed; (b) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983 CrossRef CAS PubMed; (c) A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138, 2492 CrossRef CAS PubMed; (d) S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed; (e) M. L. Rigsby, D. J. Wasylenko, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2015, 137, 4296 CrossRef CAS PubMed; (f) J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V. Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681 CrossRef CAS PubMed; (g) C. Costentin, M. Robert and J.-M. Savéant, Acc. Chem. Res., 2015, 48, 2996 CrossRef CAS PubMed; (h) S. R. Ahrenholtz, C. C. Epley and A. J. Morris, J. Am. Chem. Soc., 2014, 136, 2464 CrossRef CAS PubMed; (i) S. A. Yao, R. E. Ruther, L. Zhang, R. A. Franking, R. J. Hamers and J. F. Berry, J. Am. Chem. Soc., 2012, 134, 15632 CrossRef CAS PubMed; (j) D. J. Sommer, M. D. Vaughn and G. Ghirlanda, Chem. Commun., 2014, 50, 15852 RSC; (k) G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389 RSC; (l) G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466 CrossRef CAS PubMed; (m) J. S. Lindsey and D. F. Bocian, Acc. Chem. Res., 2011, 44, 638 CrossRef CAS PubMed; (n) T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and F. Etsuko, J. Phys. Chem. A, 1999, 103, 7742 CrossRef CAS; (o) J.-M. Savéant, Chem. Rev., 2008, 108, 2348 CrossRef PubMed; (p) D. Lexa, J. Mispelter and J.-M. Savéant, J. Am. Chem. Soc., 1981, 103, 6806 CrossRef CAS; (q) A. R. Oveisi, K. Zhang, A. Khorramabadi-zad, O. K. Farha and J. T. Hupp, Sci. Rep., 2015, 5, 10621 CrossRef CAS PubMed; (r) Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed; (s) M. R. Civic and P. H. Dinolfo, ACS Appl. Mater. Interfaces, 2016, 8, 20465 CrossRef CAS PubMed; (t) I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302 CrossRef CAS; (u) B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541 CrossRef CAS PubMed; (v) S. Ardo, D. Achey, A. J. Morris, M. Abrahamsson and G. J. Meyer, J. Am. Chem. Soc., 2011, 133, 16572 CrossRef CAS PubMed.
- (a) M. Halmann, Nature, 1978, 275, 115 CrossRef CAS; (b) M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed; (c) C. Liu, N. P. Dasgupta and P. Yang, Chem. Mater., 2014, 26, 415 CrossRef CAS; (d) B. Kaiser, D. Fertig, J. Ziegler, J. Klett, S. Hoch and W. Jaegermann, ChemPhysChem, 2012, 13, 3053 CrossRef CAS PubMed; (e) E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342 CrossRef CAS PubMed; (f) M. J. Price and S. Maldonado, J. Phys. Chem. C, 2009, 113, 11988 CrossRef CAS; (g) G. Zeng, J. Qiu, Z. Li, P. Pavaskar and S. B. Cronin, ACS Catal., 2014, 4, 3512 CrossRef CAS; (h) A. Standing, S. Assali, L. Gao, M. A. Verheijen, D. van Dam, Y. Cui, P. H. L. Notten, J. E. M. Haverkort and E. P. A. M. Bakkers, Nat. Commun., 2015, 6, 7824 CrossRef CAS PubMed; (i) J. Sun, C. Liu and P. Yang, J. Am. Chem. Soc., 2011, 133, 19306 CrossRef CAS PubMed; (j) C. Liu, J. Sun, J. Tang and P. Yang, Nano Lett., 2012, 12, 5407 CrossRef CAS PubMed.
- (a) G. F. Moore and I. D. Sharp, J. Phys. Chem. Lett., 2013, 4, 568 CrossRef CAS PubMed; (b) R. Franking, E. C. Landis, H. Kim and R. J. Hamers, ACS Appl. Mater. Interfaces, 2009, 1, 1013 CrossRef PubMed; (c) D. Richards, D. Zemlyanov and A. Ivanisevic, Langmuir, 2010, 18, 10676 Search PubMed; (d) M. Seifert, A. H. R. Koch, F. Deubel, T. Simmet, L. A. Hess, M. Stutzmann, R. Jordan, J. A. Garrido and I. D. Sharp, Chem. Mater., 2013, 25, 466 CrossRef CAS; (e) M. Steenackers, A. M. Gigler, N. Zhang, F. Deubel, M. Seifert, L. H. Hess, C. H. Lim, K. P. Loh, J. A. Garrido, R. Jordan, M. Stutzmann and I. D. Sharp, J. Am. Chem. Soc., 2011, 133, 10490 CrossRef CAS PubMed; (f) R. L. Cicero, M. R. Linford and C. E. D. Chidsey, Langmuir, 2000, 16, 5688 CrossRef CAS.
- (a) K. Shin, S. K. Kramer and H. M. Goff, Inorg. Chem., 1987, 26, 4103 CrossRef CAS; (b) S. Modi, V. P. Shedbalkar and D. V. Behere, Inorg. Chim. Acta, 1990, 173, 9 CrossRef CAS; (c) A. L. Balch, B. C. Noll, M. M. Olmstead and S. L. Phillips, Inorg. Chem., 1996, 35, 6495 Search PubMed; (d) T. N. St. Claire and A. L. Balch, Inorg. Chem., 1999, 38, 684 CrossRef CAS PubMed.
- (a) D. L. DuBois and D. E. Berning, Appl. Organomet. Chem., 2000, 14, 860 CrossRef CAS; (b) C. Creutz and M. H. Chou, J. Am. Chem. Soc., 2009, 131, 2794 CrossRef CAS PubMed; (c) S. J. Connelly, E. S. Wiedner and A. M. Appel, Dalton Trans., 2015, 44, 5933 RSC.
- (a) M. Barroso, A. J. Cowan, S. R. Pendlebury, M. Gratzel, D. R. Klug and J. R. Durrant, J. Am. Chem. Soc., 2011, 133, 14868 CrossRef CAS PubMed; (b) B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294 CrossRef CAS PubMed; (c) F. Lin and S. W. Boettcher, Nat. Mater., 2013, 13, 81 CrossRef PubMed; (d) M. M. Waegele, X. Chen, D. M. Herlihy and T. Cuk, J. Am. Chem. Soc., 2014, 136, 10632 CrossRef CAS PubMed; (e) J. E. Thorne, S. Li, C. Du, G. Qin and D. Wang, J. Phys. Chem. Lett., 2015, 6, 4083 CrossRef CAS PubMed.
- (a) A. Nozik, Nature, 1975, 257, 383–386 CrossRef CAS; (b) Z. Chen, T. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 3 CrossRef CAS.
- (a) S. R. Soltau, J. Niklas, P. D. Dahlberg, O. G. Poluektov, D. M. Tiede, K. L. Mulfort and L. M. Utschig, Chem. Commun., 2015, 51, 10628 RSC; (b) D. W. Wakerley and E. Reisner, Phys. Chem. Chem. Phys., 2014, 16, 5739 RSC; (c) B. S. Veldkamp, W.-S. Han, S. M. Dyar, S. W. Eaton, M. A. Ratner and M. R. Wasielewski, Energy Environ. Sci., 2013, 6, 1917 RSC; (d) E. S. Andreiadis, P.-A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2012, 5, 48 CrossRef PubMed; (e) S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127 CrossRef CAS PubMed; (f) L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988 RSC; (g) C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164 CrossRef CAS PubMed; (h) F. Lakadamyali, A. Reynal, M. Kato, J. Durrant and E. Reisner, Chem.–Eur. J, 2012, 18, 15464 CrossRef CAS PubMed; (i) J. T. Muckerman and E. Fujita, Chem. Commun., 2011, 47, 12456 RSC; (j) B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2011, 50, 11252 CrossRef CAS PubMed; (k) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed; (l) P. Du, K. Knowles and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 12576 CrossRef CAS PubMed; (m) C. Baffert, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817 CrossRef CAS PubMed; (n) X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723 RSC; (o) M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786 CrossRef CAS PubMed; (p) X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988 CrossRef CAS PubMed; (q) P. Connelly and J. H. Espenson, Inorg. Chem., 1986, 25, 2684 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular synthesis and characterization, surface characterization, photoelectrochemical data. See DOI: 10.1039/c6sc02664h |
| This journal is © The Royal Society of Chemistry 2017 |