
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantiodivergent Steglich rearrangement of O-carboxylazlactones catalyzed by a chirality switchable helicene containing a 4-aminopyridine unit†
Chien-Tien
Chen
*,
Cheng-Che
Tsai
,
Pei-Kang
Tsou
,
Gou-Tao
Huang
and
Chin-Hui
Yu
*
Department of Chemistry, National Tsing Hua University, No. 101, Section 2, Kuang-Fu Road, Hsinchu 30013, Taiwan. E-mail: ctchen@mx.nthu.edu.tw; chyu@mx.nthu.edu.tw
First published on 25th August 2016
Abstract
A pseudo-enantiomeric pair of optically switchable helicenes containing a catalytic 4-N-methylaminopyridine (MAP) bottom unit and a C2-symmetric, (10R,11R)-dimethoxymethyl-dibenzosuberane top template was synthesized. They underwent complementary photoswitching at 290 nm (P/M′, <1/>99) and 340 nm (P/M′, 91/9) and unidirectional thermo-rotation at 130 °C (P/M′, >99/<1). They were utilized to catalyze enantiodivergent Steglich rearrangement of O- to C-carboxylazlactones, with formation of either enantiomer with up to 91% ee (R) and 94% ee (S), respectively.
The development of enantiodivergent catalysts has garnered significant interest in recent years, because it allows for the production of either enantiomerically enriched compound based on a single chiral catalyst.1 To date, several enantiodivergent catalytic systems under various conditions have been reported, in which the chiral environment of the complementary transition states can be tuned. For instance, reaction temperatures,2 solvent effects,3 achiral co-catalysts,4 structural modifications of functional substrates,5 and different coordinating metal ions6 were found to facilitate enantiodivergent catalyses with good to high enantioselectivities (89–97% ee). Nevertheless, such specific conditions may not be suitable for all desired transformations and substrate scopes. In marked contrast, enantiodivergent catalysis using pseudo-enantiomers that can be interconverted by an external stimulus constitutes a more practical approach. For example, the enantiodivergent hydrosilylation of styrenes was first accomplished by Suginome and co-workers by using a polymer-based chiral catalyst, whose helical chirality depended on the solvent’s polarity and attributes.7 Moreover, Canary and co-workers demonstrated an enantiodivergent, conjugate addition of diethyl malonates to nitrostyrene [up to 70% ee (R) and 72% ee (S)] by using redox-reconfigurable copper(I/II) complexes.8 In addition, Feringa and co-workers described a temperature-modulated, light-triggered enantiodivergent Michael addition [up to 54% ee (R) and 50% ee (S)], Henry reaction [up to 72% ee (R) and 42% ee (S)], and Pd-catalyzed allylic desymmetrization [up to 88% ee (S,R) and 86% ee (R,S)], utilizing a thiourea/DMAP hybrid organocatalyst or biphosphine ligand employing a helically chiral molecular motor scaffold.9 Photoswitchable catalysts that are capable of modulating reaction enantioselectivity [from 50% ee (S,S) to 5% ee (S,S)] have been established.10 However, as compared to enantiodivergent catalyses, the catalytic processes based on light- or heat-modulated pseudo-enantiomeric catalysts remain challenging. Towards this end, herein we report a highly selective, enantiodivergent Steglich rearrangement of O-carboxylazlactones by using a nucleophilic helical catalyst with an embedded MAP unit, whose chirality can be switched.
Asymmetric Steglich rearrangement, an enantioselective O- to C-carboxyl group transfer reaction, is a reliable method for the construction of a quaternary stereogenic center. Until now, a variety of chiral nucleophilic catalysts for asymmetric Steglich rearrangements have been developed with remarkable success. For example, planar- and central-chiral derivatives of 4-dimethylaminopyridine (i.e. DMAP) were first introduced by Fu,11 Richards,12 and Vedejs.13 Recently, easily accessible C-3 functionalized DAMPs were described by Poisson,14 Mandai and Suga.15 In addition, chiral tetrahydropyrimidine-based isothioureas and chiral N-heterocyclic carbenes (i.e. NHC) utilized by Smith,16 chiral ammonium betaines employed by Ooi,17 and chiral bicyclic imidazoles designed by Zhang18 were developed with better enantiocontrol or broader substrate scope. In these distinguished works, however, only one of two possible enantiomers in the products could be obtained, except in Smith's work using pseudo-enantiomeric isothioureas prepared from different chiral sources. With respect to the requirements of pharmacological studies, a feasible access to both enantiomeric C-carboxyazlactones should be developed. In this study, we describe an enantiodivergent approach to obtain both enantiomeric C-carboxylazlactones by using a chirality switchable nucleophilic helicene containing a 4-aminopyridine unit.
As part of our on-going research on the C2-symmetric dibenzosuberane (DBS)-based helicenes as conformationally flippable, chirochromic optical switches in liquid crystal materials and supramolecular organogels,19 we evaluated the feasibility of their use in enantiodivergent catalysis. In this study, we synthesized a new photo-switchable and unidirectional thermo-rotatable helicene 1, whose catalytic activity was originated from 4-N-methylaminopyridine (i.e., MAP).20,21 Notably, the proximity of the catalytic site (i.e., N7′) to the upper chiral DBS template facilitates the asymmetric discrimination in the pyridine-centered activation event (Scheme 1).
 | ||
| Scheme 1 Molecular design of chirality-switchable, helicene-based, 4-N-methylaminopyridine (MAP) as an organocatalyst. | ||
The key synthetic steps towards the pyridine-incorporated helicene (10R,11R,P)-1 [i.e., (P)-1)] are shown in Scheme 2 (see ESI† for details). Hydrazone 5, prepared from t-butyl-1,2,3,4-tetrahydro-1,6-naphthyridine-1-carboxylate in three steps, was converted to the corresponding diazo compound by treatment with PhI(OCOCH3)2 at −20 °C in a 1/1 mixture of DMF/CH2Cl2. Subsequent reaction of the diazo compound with freshly prepared thioketone 4 afforded (10R,11R,1′S)-MOM-DBS-based episulfide 3 in 48% isolated yield as a single diastereomer. Support for the (S)-absolute configuration at C1′ in 3 was obtained using circular dichroism (CD) analysis and comparing its sign of specific optical rotation and exciton chirality with those of analogous episulfides previously reported by us.19a,c Stereospecific, reductive desulfurization of episulfide 3 with HMPT at 0 °C gave helicene (P)-2 in 76% isolated yield. Finally, reductive methylation of N-Boc (P)-2 with DIBAL in THF at 50 °C furnished the desired N-methyl (P)-1 in 80% yield without epimerization of the helicene, as evidenced by HPLC analysis. The (P)-form helical chirality of 1 was confirmed by observing a negative exciton chirality at 221 nm using CD analysis (see Fig. 1c).22
 | ||
| Scheme 2 Synthesis of (10R,11R,P)-helicene, (P)-1, bearing catalytic 4-N-methylaminopyridine (MAP) lower template. HMPT stands for hexamethylphosphorous triamide. DIBAL stands for diisobutylaluminum hydride. | ||
 | ||
| Fig. 1 Control and monitoring of helical chirality. (a) Complementary photoisomerization of helicenes (P)-1 and (M)-1′ in degassed CH2Cl2 (1 × 10−4 M) and unidirectional thermorotation of (M)-1′ in xylene at 130 °C. (b) UV-Vis and difference spectra of (P)-1 and (M)-1′ in degassed CH2Cl2 (1 × 10−4 M). (c) Dynamic trace experiments with CD stacked plots before and after irradiation of (P)-1 at 290 nm in degassed CH2Cl2 (1.0 × 10−4 M). | ||
In order to identify two distinctive irradiation wavelengths to induce photochemical switching between the two pseudo-enantiomeric helicenes [i.e., (P)-1 and (M′)-1], their individual UV/Vis and difference spectra with complementary changes in the extinction coefficient (Δε) at two given wavelengths should be identified. This information provides their relative abundance and thus the composition of the photostationary state (i.e., pss) at a given irradiation wavelength can be assessed. The diastereomeric (i.e., pseudo-enantiomeric) excess of the pss from irradiation at a given wavelength (i.e., [de]pss)23 can often be directly determined by the extinction coefficient difference under conditions where the photoisomerization quantum yields (ΦM′→P and ΦP→M′) for both processes are similar. Therefore, to induce efficient switching in a highly diastereoselective (pseudo-enantiomeric in this system) manner, irradiation should be targeted at regions with significant and complementary differences in the extinction coefficients.
The photochemical switching experiments were carried out by irradiating individual (P)-1 and (M)-1′ (ref. 24) in degassed CH2Cl2 under a monochromator light source (Fig. 1a). The irradiation wavelengths were set at the wavelengths of 290 nm and 340 nm with the largest and complementary differences in the extinction coefficients which were determined by using the UV-Vis difference spectrum between (P)-1 and (M)-1′ (Fig. 1b). Photochemical switching of (P)-1 was first performed at 290 nm, which led to the exclusive formation of (M)-1′ [(P)-1/(M)-1′, <1/>99]. The relative composition of the reaction mixture was monitored using HPLC on a Chiralcel AD column until the pss was reached (see Fig. S1–S3 in ESI†) with the detection wavelength set at the isosbestic point (i.e., 310 nm) of (P)-1 and (M)-1′. Photoisomerization of pure (M)-1′ was then performed at 340 nm, which led to the predominant enrichment of (P)-1 [(P)-1/(M)-1′, 91/9]. The diastereomerically pure (P)-1 can be regenerated either by unidirectional thermo-rotation of the 91/9 [(P)-1/(M)-1′] mixture or by unidirectional thermo-rotation of pure (M)-1′ in p-xylene at 130 °C for two hours (Fig. 1a and S4 in ESI†). A stacked plot of the dynamic circular dichroism (CD) traces of the photoisomerization experiments of (P)-1 is shown in Fig. 1c. The observed positive exciton chirality at 221 nm clearly indicated the reversal of helical chirality upon irradiation of (P)-1 at 290 nm.
Having established the complementary photo-switching profiles of pseudo-enantiomeric helicenes (P)-1 and (M)-1′, we investigated their individual performance as chiral organocatalysts in the Steglich rearrangement of O-carboxylazlactone (Table 1). Both (P)-1 and (M)-1′ in the catalytic reaction are used in diastereomerically pure form. In an initial study, O-1,1,1-trichloro-tert-butoxycarbonyl (Cl3-t-Boc) derivative 6a was treated with 5 mol% of (P)-1 in CH2Cl2 at ambient temperature, resulting in C-carboxylated product 7a in a modest yield of 57% with only 10% ee (entry 1). Replacement of the migrating Cl3-t-Boc group with benzoxycarbonyl [BnOC(O)] and phenoxycarbonyl [PhOC(O)] led to a significant improvement in the enantioselectivities (41% and 63% ee; entries 2 and 3).25
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Solvent | Catalyst | Time (h) | Yieldb (%) | eec (%) (config)d |
| a The reactions were carried out in the presence of 5 mol% of (P)-1 at −40 °C under nitrogen atmosphere. b Isolated yield. c The enantiomeric purity of 7 was determined by using a chiral column (DAICEL Chiralcel OD-H) with hexane/propan-2-ol as eluents. d The absolute configuration of 7a–c were determined by comparison of the HPLC retention time with those reported in ref. 11a, 12 and 13. e The reaction was carried out at ambient temperature. | ||||||
| 1e | CMe2CCl3 (6a) | CH2Cl2 | (P)-1 | 48 | 57 | 10 (R) |
| 2 | Bn (6b) | CH2Cl2 | (P)-1 | 6 | 85 | 41 (R) |
| 3 | Ph (6c) | CH2Cl2 | (P)-1 | 6 | 90 | 63 (R) |
| 4 | Ph (6c) | Toluene | (P)-1 | 6 | 67 | 80 (R) |
| 5 | Ph (6c) | Et2O | (P)-1 | 48 | 78 | 79 (R) |
| 6 | Ph (6c) | THF | (P)-1 | 12 | 80 | 72 (R) |
| 7 | Ph (6c) | DME | (P)-1 | 6 | 82 | 80 (R) |
| 8 | Ph (6c) | DME/tAA (1/1) | (P)-1 | 24 | 84 | 87 (R) |
| 9 | Ph (6c) | DME/tAA (1/1) | (M)-1′ | 24 | 81 | 91 (S) |

Further improvement in the enantioselectivity to around 80% ee was achieved by changing the solvent to toluene, ether, or 1,2-dimethoxyethane (entries 4–7), suggesting that ionic pair intermediates may be solvated in polar solvents or stabilized by aromatic solvents through cation-π interactions.26 Final optimization by using a 1/1 mixture of 1,2-dimethoxyethane (DME)/t-amyl alcohol (tAA) led to the best results, giving (R)-7c in 84% yield with 87% ee (entry 8).27 Conversely, the reaction of O-carboxylazlactone 6c promoted by the pseudo-enantiomeric catalyst (M)-1′ gave enantiomeric (S)-7c in 81% yield with 91% ee (entry 9). The switch of enantiocontrol indicated a complementary helical asymmetric environment in the pseudo-enantiomeric catalyst (M)-1′.
With the optimal reaction conditions in hand, the enantiodivergent Steglich rearrangements of various O-carboxylazlactones, 6d–6k, with pseudo-enantiomeric catalysts (P)-1 and (M)-1′ were further examined (Table 2). It was found that rearrangements of 4-ethyl- and 4-i-butyl-1,3-oxazolyl phenyl carbonates (R′ = Et and i-Bu) catalyzed by either (P)-1 or (M)-1′ proceeded with excellent and complementary enantioselectivities (87–88% ee and 90–91% ee, respectively). Both products 7d, e were delivered in 80–85% yields (entries 1–4). Reaction of substrate 6f bearing the 4-i-propyl-substituent with (P)-1 or (M)-1′ gave the corresponding products in 71 and 70% yield with complementary enantiomeric excess of 69 and 72% (entries 5 and 6). These poorer yields were due to partial hydrolysis of the carbonate group in the resulting products, consistent with the works of Smith16a,c and Zhang.18 Substrates 6f–k bearing 4-allyl (entries 7 and 8), 4-(2-methylthio)ethyl (entries 9 and 10), 4-benzyl (entries 11 and 12), 4-(4-benzyloxy)benzyl (entries 13 and 14), and 4-(4-phenoxycarbonyloxy)benzyl (entries 15 and 16) groups were efficiently transformed into the corresponding products 7f–k in 81–86% yields with similarly excellent and complementary enantioselectivities (87–91% ee and 91–94% ee, respectively).
|
|
||||
|---|---|---|---|---|
| Entry | R′ | Catalyst | Yieldb (%) | eec (%) (config)d |
a The reaction was carried out in the presence of 5 mol% of (P)-1 or (M)-1′ in 1,2-dimethoxyethane/tert-amyl alcohol (v/v 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at −40 °C under nitrogen atmosphere.
b Isolated yields.
c The enantiomeric purity of 7 was determined by using a chiral column (DAICEL Chiralcel OD-H or AD-H) with hexane/propan-2-ol as eluents.
d The absolute configuration of 7d and 7f–h was determined by comparison of the HPLC retention times with those reported in ref. 16a. :1) at −40 °C under nitrogen atmosphere.
b Isolated yields.
c The enantiomeric purity of 7 was determined by using a chiral column (DAICEL Chiralcel OD-H or AD-H) with hexane/propan-2-ol as eluents.
d The absolute configuration of 7d and 7f–h was determined by comparison of the HPLC retention times with those reported in ref. 16a.
|
||||
| 1 | Et (6d) | (P)-1 | 85 | 88 (R) |
| 2 | Et (6d) | (M)-1′ | 83 | 91 (S) |
| 3 | i-Bu (6e) | (P)-1 | 84 | 87 (R) |
| 4 | i-Bu (6e) | (M)-1′ | 80 | 90 (S) |
| 5 | i-Pr (6f) | (P)-1 | 71 | 69 (R) |
| 6 | i-Pr (6f) | (M)-1′ | 70 | 72 (S) |
| 7 | Allyl (6g) | (P)-1 | 86 | 88 (R) |
| 8 | Allyl (6g) | (M)-1′ | 82 | 91 (S) |
| 9 | CH2CH2SMe (6h) | (P)-1 | 84 | 87 (R) |
| 10 | CH2CH2SMe (6h) | (M)-1′ | 81 | 91 (S) |
| 11 | Bn (6i) | (P)-1 | 85 | 90 (R) |
| 12 | Bn (6i) | (M)-1′ | 82 | 94 (S) |
| 13 | 4-BnOC6H4CH2 (6j) | (P)-1 | 83 | 91 (R) |
| 14 | 4-BnOC6H4CH2 (6j) | (M)-1′ | 81 | 93 (S) |
| 15 | 4-PhO2COC6H4CH2 (6k) | (P)-1 | 85 | 87 (R) |
| 16 | 4-PhO2COC6H4CH2 (6k) | (M)-1′ | 82 | 90 (S) |

In order to gain insight into the mechanism of Steglich rearrangement catalyzed by (P)-1 or (M)-1′, the reversibility of the O- to C-carboxyl group transfer process was investigated through some control experiments (Scheme 3). An initial crossover study was carried out by treatment of a 50:50 mixture of O-carboxylazlactones 6c and 6l with 5 mol% of catalyst (P)-1 at −40 °C in DME/tAA (1/1) mixed solvents. After 24 hours and reaction completion, a 22:27:23:28 mixture of four C-carboxylated products 7c, 7l, 7h, and 7i was observed (see Fig. S11 and 12 in ESI†). A further control experiment was performed by reaction of a mixture of C-carboxyl 7c and O-carboxyl 6l under the same reaction conditions, giving C-carboxyl 7c and 7l, exclusively (see Fig. S13 in ESI†). These results indicated that C-carboxylated products are configurationally stable under these reaction conditions and that crossover only occurs in the O-carboxylation stage, which is consistent with those reported by Fu using planar chiral DAMPs11a and by Smith using chiral isothioureas or chiral NHCs.16b,c Moreover, the complete crossover caused by rapid transcarboxylation between two different O-carboxyl azlactones indicated that ion pair intermediates are fully stabilized in the DME/tAA (1/1) mixed solvents.
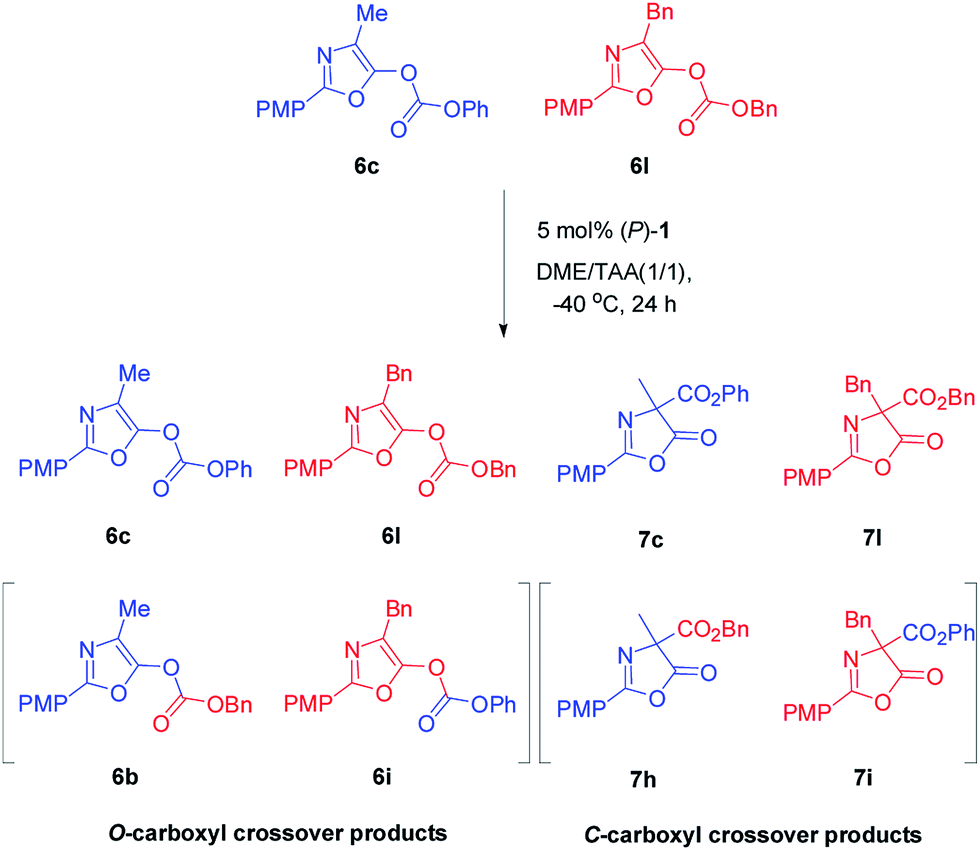 | ||
| Scheme 3 Crossover experiments of O-carboxylazlactones 6c and 6l under optimal reaction conditions. | ||
Based on the control experiments, it was proposed that the catalytic process may proceed through an initial and reversible nucleophilic carboxyl substitution of the substrate carbonate moiety by catalyst (P)-1, resulting in the formation of a stabilized ion pair between enolate-anion I and pyridinium-cation II (Scheme 4).11b The carbonyl group in the phenoxycarbonyl moiety in II is anti with respect to the 3-chloro appendage in the top template, thus avoiding stereoelectronic repulsion between the lone pairs of the chlorine and the carbonyl oxygen. Subsequently, an irreversible C-carboxylation of enolate-anion I takes place with pyridinium-cation II in high enantiocontrol, giving the C-carboxylazlactone in R configuration.
 | ||
| Scheme 4 Proposed mechanism for the Steglich rearrangement of O-carboxylazlactone catalyzed by (P)-1. | ||
To gain further insight into the origin of enantiocontrol in the asymmetric catalytic process, molecular simulations of the transition state assemblies in the ion pair were performed by DFT calculations.28 The preferred transition state assembly, A-syn, with Re-face attack leading to (R)-7 is shown in Fig. 2a.
 | ||
| Fig. 2 Plausible transition state assemblies of Steglich rearrangement by using (P)-1. (a) A-syn and A-anti transition state assemblies using GaussView (side view) and ChemDraw (side and bottom views) presentations. (b) B-anti transition state assembly using ChemDraw (bottom view) presentation. | ||
Presumably, the synclinal approach in A-syn assembly is more favored than the corresponding antiperiplanar approach in A-anti assembly in view of the advantageous donor–acceptor type, π–π interaction in A-syn and the unfavorable steric repulsion between the 4-methoxyphenyl group and the C11-methoxymethyl group in A-anti.
In transition state assembly B-anti with an antiperiplanar approach and Si-face attack leading to (S)-7 (Fig. 2b), calculations show that the barrier for the Si-face attack is 2.35 kcal mol−1 higher than that for the Re-face attack (see Fig. S8–S10 in ESI†). A greater overlap between HOMO and LUMO is observed for the Re-face attack based on the frontier molecular orbital analysis; therefore, the preference for the Re-face attack is rationalized. In addition, the stereoelectronic repulsion between the enolate oxygen lone pairs and the C11-methoxymethyl group as well as between the 4-methoxyphenyl and the N-methyl groups in the bottom template also contribute to the higher Gibbs free energy mentioned above.
Conclusions
In summary, we have documented a pseudo-enantiomeric pair of optically switchable helicene catalysts (10R,11R,P)-1 and (10R,11R,M)-1′, which contain a 4-N-methylaminopyridine (MAP) moiety. The helicene pair underwent complementary photo-switching profiles at 290 nm (1/1′, <1/>99) and 340 nm (1/1′, 91/9) and unidirectional thermo-rotation (1/1′, >99/<1), as evidenced by UV-Vis difference, CD, and HPLC analyses. They can efficiently catalyze enantiodivergent Steglich rearrangements of O- to C-carboxyazlactones at −40 °C in DME/tAA mixed solvents, resulting in the formation of either enantiomeric product of bio-medicinal importance29 in up to 91% (R) and 94% (S) ee, respectively. Control experiments were performed to understand that the catalytic process may proceed through a reversible O-carboxylation and a subsequent irreversible C-carboxylation; molecular simulations of the transition state assemblies of the incipient ion pairs indicate that synclinal Re-face attack is favored in catalyst (P)-1 due to greater HOMO–LUMO interactions with minimal stereoelectronic repulsion. To our knowledge, this system provides the best complementary enantioselectivities among several light- or heat-controlled enantiodivergent catalytic reactions. Investigations toward examining other substrate classes, as well as applications to other organo-catalytic systems are under way.Acknowledgements
Financial support from the Ministry of Science of Technology of Taiwan (MOST 104-2113-M-007-002-MY3 and 104-2113-M-007-014) was greatly acknowledged.Notes and references
- For reviews of enantioselectivity switches using a single chiral source, see: (a) G. Zanoni, F. Castronovo, M. Franzini, G. Vidari and E. Giannini, Chem. Soc. Rev., 2003, 32, 115–129 RSC; (b) T. Tanaka and M. Hayashi, Synthesis, 2008, 3361–3376 CAS; (c) M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef PubMed; (d) J. Escorihuela, M. I. Burguete and S. V. Luis, Chem. Soc. Rev., 2013, 42, 5595–5617 RSC.
- G. Storch and O. Trapp, Angew. Chem., Int. Ed., 2015, 54, 3580–3586 CrossRef CAS PubMed.
- Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 9254–9257 CrossRef CAS PubMed.
- S. A. Moteki, J. Han, S. Arimitsu, M. Akakura, K. Nakayama and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1187–1190 CrossRef CAS PubMed.
- J. Wang, J. Chen, C. W. Kee and C.-H. Tan, Angew. Chem., Int. Ed., 2012, 51, 2382–2386 CrossRef CAS PubMed.
- Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928–4932 CrossRef CAS PubMed.
- (a) T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS PubMed; (b) Y. Akai, T. Yamamoto, Y. Nagata, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 11092–11095 CrossRef CAS PubMed.
- S. Mortezaei, N. R. Catarineu and J. W. Canary, J. Am. Chem. Soc., 2012, 134, 8054–8057 CrossRef CAS PubMed.
- (a) J. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed; (b) M. Vlatković, L. Bernard, E. Otten and B. L. Feringa, Chem. Commun., 2014, 50, 7773–7775 RSC; (c) D. Zhao, T. M. Neubauer and B. L. Feringa, Nat. Commun., 2015, 6, 6652 CrossRef CAS PubMed.
- For reviews of photoswitchable catalysis, see: (a) B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS; (b) R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC. For a representative example of photochemically modulated enantioselectivity in an asymmetric reaction, see: (c) D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019–2021 CrossRef CAS PubMed.
- (a) J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532–11533 CrossRef CAS; (b) I. D. Hills and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 3921–3924 CrossRef CAS PubMed.
- H. V. Nguyen, D. C. D. Butler and C. J. Richards, Org. Lett., 2006, 8, 769–772 CrossRef CAS PubMed.
- S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368–13369 CrossRef CAS PubMed.
- T. Poisson, S. Oudeyer and V. Levacher, Tetrahedron Lett., 2012, 53, 3284–3287 CrossRef CAS.
- H. Mandai, T. Fujiwara, K. Noda, K. Fujii, K. Mitsudo, T. Korenaga and S. Suga, Org. Lett., 2015, 17, 4436–4439 CrossRef CAS PubMed.
- (a) C. Joannesse, C. P. Johnston, C. Concellon, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed; (b) C. D. Campbell, C. Concellon and A. D. Smith, Tetrahedron: Asymmetry, 2011, 22, 797–811 CrossRef CAS; (c) C. Joannesse, C. P. Johnston, L. C. Morrill, P. A. Woods, M. Kieffer, T. A. Nigst, H. Mayr, T. Lebl, D. Philp, R. A. Bragg and A. D. Smith, Chem.–Eur. J., 2012, 18, 2398–2408 CrossRef CAS PubMed.
- D. Uraguchi, K. Koshimoto, S. Miyake and T. Ooi, Angew. Chem., Int. Ed., 2010, 49, 5567–5569 CrossRef CAS PubMed.
- Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 CrossRef CAS PubMed.
- (a) C.-T. Chen and Y.-C. Chou, J. Am. Chem. Soc., 2000, 122, 7662–7672 CrossRef CAS; (b) W.-C. Chen, Y.-W. Lee and C.-T. Chen, Org. Lett., 2010, 12, 1472–1475 CrossRef CAS PubMed; (c) W.-C. Chen, P.-C. Lin, C.-H. Chen and C.-T. Chen, Chem.–Eur. J., 2010, 16, 12822–12830 CrossRef CAS PubMed; (d) C.-T. Chen, C.-H. Chen and T.-G. Ong, J. Am. Chem. Soc., 2013, 135, 5294–5297 CrossRef CAS PubMed.
- For reviews of chiral dialkylaminopyridine catalysts in asymmetric synthesis, see: (a) R. P. Wurz, Chem. Rev., 2007, 107, 5570–5595 CrossRef CAS PubMed; (b) T. Furuta and T. Kawabata, in Science of Synthesis: Asymmetric Organocatalysis, ed. B. List and K. Maruoka, Thieme, 2012, pp. 497–546 Search PubMed.
- M. R. Heinrich, H. S. Klisa, H. Mayr, W. Steglich and H. Zipse, Angew. Chem., Int. Ed., 2003, 42, 4826–4828 CrossRef CAS PubMed.
- The negative exciton chirality is attributed to the electric transition dipole moment change from the bottom template to the top right.
- The diastereomeric excess (de) of the pss at a given irradiation wavelength is given by [de]pss = (P − M′)/(P + M′) = [(εM′ΦM′→P − εPΦP→M′)/[(εM′ΦM′→P+ εPΦP→M′)].
- (M)-1′ is unavailable by chemical synthesis but subsequently obtained by photoisomerization of (P)-1 at 290 nm in CH2Cl2 for 1 hour.
- The ee values of the resulting products were improved by about 10%, when the reaction temperature was lowered from 0 to −40 °C.
- The calculated energy differences between the transition states in various solvents are less than 0.5 kcal mol−1; therefore, the similar enantioselectivities in toluene, Et2O, and DME are rationalized.
- The control experiments in this work indicated the formation of a solvent-separated ion pair in the DME/tAA (1/1) mixed solvents, suggesting that the enolate anion in the ion-pair intermediate may be further stabilized by the additional tAA through partial hydrogen-bonding interaction.
- Four transition states derived from (P)-1 or (M)-1′ along with two possibilities of facial selectivity of enolate anion-I are shown in Gaussview presentation (Fig. S5) as well as in Chem 3D and Chem Draw presentations (see Fig. S6 and 7 in ESI†).
- For a review of the significance of α,α-disubstituted azlactones, see: (a) R. A. Mosey, J. S. Fisk and J. J. Tepe, Tetrahedron: Asymmetry, 2008, 19, 2755–2762 CrossRef CAS. For a representative example of the bio-medicinal importance of α,α-disubstituted azlactones, see: (b) T. Zhou, R. C. Hider, P. Jenner, B. Campbell, C. J. Hobbs, S. Rose, M. Jairaj, K. A. Tayayrani-Binazir and A. Syme, Bioorg. Med. Chem. Lett., 2013, 23, 5279–5282 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02646j |
| This journal is © The Royal Society of Chemistry 2017 |