
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild and selective base-free C–H arylation of heteroarenes: experiment and computation†
Hannes P. L.
Gemoets
a,
Indrek
Kalvet
 b,
Alexander V.
Nyuchev
ac,
Nico
Erdmann
a,
Volker
Hessel
a,
Franziska
Schoenebeck
*b and
Timothy
Noël
*a
b,
Alexander V.
Nyuchev
ac,
Nico
Erdmann
a,
Volker
Hessel
a,
Franziska
Schoenebeck
*b and
Timothy
Noël
*a
aDepartment of Chemical Engineering and Chemistry, Micro Flow Chemistry & Process Technology, Eindhoven University of Technology, Den Dolech 2, 5612 AZ Eindhoven, The Netherlands. E-mail: t.noel@tue.nl
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: franziska.schoenebeck@rwth-aachen.de
cDepartment of Chemistry, N. I. Lobachevsky State University of Nizhny Novgorod, 23 Gagarin Avenue, 603950 Nizhny Novgorod, Russian Federation
First published on 5th September 2016
Abstract
A mild and selective C–H arylation strategy for indoles, benzofurans and benzothiophenes is described. The arylation method engages aryldiazonium salts as arylating reagents in equimolar amounts. The protocol is operationally simple, base free, moisture tolerant and air tolerant. It utilizes low palladium loadings (0.5 to 2.0 mol% Pd), short reaction times, green solvents (EtOAc/2-MeTHF or MeOH) and is carried out at room temperature, providing a broad substrate scope (47 examples) and excellent selectivity (C-2 arylation for indoles and benzofurans, C-3 arylation for benzothiophenes). Mechanistic experiments and DFT calculations support a Heck–Matsuda type coupling mechanism.
Introduction
The ubiquity of the heterobiaryl motif in pharmaceuticals, agrochemicals and materials illustrates its scientific and commercial value.1 Traditionally, these moieties have been prepared via cross-coupling strategies which require pre-functionalized substrates.2 However, over the last decade, transition metal-catalyzed C–H arylation protocols have been developed to enable the formation of C–C bonds.3 In contrast to classical cross-coupling chemistry, C–H arylation strategies enable direct functionalization of simple heteroarenes.The direct arylation of heteroarenes can be achieved via radical pathways, e.g., visible light photoredox catalysis4 and Meerwein arylation.5 However, these methods suffer from a number of disadvantages, including long reaction times, large excesses of substrates, selectivity issues and limited substrate scopes. Recently, there has been an increase in the number of new methods, particularly in the use of metal-catalyzed processes.6 In particular, the work by Gaunt,7 Sames,8 Sanford,9 DeBoef,10 Glorius,11 Ackermann,12 Fagnou13 and Larrosa14 has increased the number of useful C–H arylation transformations to enable heteroaryl-(hetero)aryl bond formation. Furthermore, these examples have deepened our fundamental understanding of the underlying challenges inherent in such processes. However, the state of the art is still far from competitive with classical cross coupling strategies, e.g. Suzuki–Miyaura cross coupling. Current hurdles include harsh reaction conditions (i.e. high temperature), the necessity of stoichiometric amounts of oxidants and/or additives, use of toxic solvent systems, limited selectivity and high catalyst loadings (typically 5 to 10 mol%). Consequently, the development of new, mild and broadly applicable C–H arylation strategies is still highly desirable.15 We anticipated that the design of a mild and selective C–H arylation protocol for heteroaromatics (i.e. indoles, benzofurans and benzothiophenes) could be of high interest for API synthesis (e.g. Bazedoxifene,16 Saprisartan17 and Raloxifene18). Recently, Correia et al. described a Pd-based arylation of heteroarenes using aryldiazonium salts.6i However, the protocol suffered from high catalyst loadings (10 to 20 mol% Pd), limited scope and impractical reaction conditions (e.g. biphasic reaction conditions, large excesses of reagents, and high reaction temperatures). Herein, we describe the development of a mild and selective palladium-based C–H arylation strategy (Scheme 1). Notable features of our open flask protocol are its operational simplicity in conjunction with low catalyst loadings, broad substrate scope, green solvent system, and short reaction times. No additional oxidants or additives are required. The strategy uses equimolar amounts or slight excesses of aryldiazonium salts as convenient arylating reagents.6l,19 Kinetic studies and DFT calculations suggest that a Heck–Matsuda-type mechanism occurs under our reaction conditions.
 | ||
| Scheme 1 Pd-catalyzed C-2 C–H arylation of indoles. | ||
Results and discussion
Optimization of reaction conditions
We commenced our optimization studies with the Pd-catalyzed C–H arylation of 1-methylindole (1a), which was reacted with 1.2 equivalents of benzenediazonium tetrafluoroborate (2a) in the presence of 10 mol% Pd(OAc)2 in DMF. A satisfying yield of 66% for 1-methyl-2-phenylindole (3a) was obtained within only 30 minutes of reaction time at room temperature (Table 1, Entry 1). The main byproducts were 3-(arylazo)-1-methylindole (1aa) and 2-aryl-3-(arylazo)-1-methylindole (3aa), due to the uncatalyzed electrophilic substitution reaction between the highly electrophilic nitrogen of the aryldiazonium salt and the C-3 position of 1-methylindole (Fig. 1b). Lowering the catalyst loading to 5 mol% Pd(OAc)2 in DMF resulted in a significant drop in yield (34%). An overnight control experiment showed that no product formation was observed in the absence of a palladium catalyst (Table 1, Entry 3). Using polar protic solvents (e.g. i-PrOH) resulted in generally high reactivity and moderate yields. A considerable amount of byproduct formation was consistently observed (see ESI†). It was generally found that carrying out the reaction in less polar (aprotic) solvents (e.g. DMF → THF → 1,4-dioxane) resulted in reduced byproduct formation. After solvent screening, THF was considered to be the best solvent (77%), combining both the desired reactivity and selectivity (Table 1, Entry 5). A control experiment using Schlenk techniques indicated that the catalyst was not affected by air and moisture (Table 1, Entry 5). Therefore, all the following experiments could be performed as open-flask reactions, making this procedure appealing for future scale-up. A catalyst survey demonstrated that only Pd(OAc)2, Pd(TFA)2 and, to a lesser extent, Pd2(dba)3 were active catalysts for this chemical transformation (Table 1, Entries 6–8). The use of Pd(OAc)2 was preferred over Pd(TFA)2 due to its cost efficiency and stability. Further optimization studies showed that it was possible to lower the catalyst loading further to 0.5 mol% in THF (Table 1, Entry 9). Even lower catalyst loading resulted in only trace amounts of product (Table 1, Entry 10). Finally, 2-MeTHF was evaluated. This solvent is recognized as a green solvent for synthetic organic chemistry because it can be readily produced from furfural, a common biomass material.20 Satisfyingly, 2-MeTHF showed even better selectivity for the desired product (87%), although an increased reaction time of 2 hours was required to obtain full conversion (Table 1, Entry 11). Continued optimization studies with green solvents revealed that the reaction time could be halved by using EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-MeTHF (1:1) as a solvent mixture (Table 1, Entry 12). Indeed, this solvent combination proved to be superior, as it enabled further lowering of the catalyst loading to 0.2 mol% Pd(OAc)2 (Table 1, Entry 10 vs. 13). However, in the case of 0.2 mol% Pd(OAc)2, significant increases of 1aa and 3aa were observed because the reactivity toward the desired arylation was diminished. Therefore, 0.5 mol% Pd(OAc)2 was considered to be optimal.
:2-MeTHF (1:1) as a solvent mixture (Table 1, Entry 12). Indeed, this solvent combination proved to be superior, as it enabled further lowering of the catalyst loading to 0.2 mol% Pd(OAc)2 (Table 1, Entry 10 vs. 13). However, in the case of 0.2 mol% Pd(OAc)2, significant increases of 1aa and 3aa were observed because the reactivity toward the desired arylation was diminished. Therefore, 0.5 mol% Pd(OAc)2 was considered to be optimal.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Reaction time | Yield GC-FID (%) |
| a Reaction conditions: catalyst, 0.5 mmol heteroarene and 1.2 equiv. benzenediazonium tetrafluoroborate in 2.5 mL solvent at rt, 0.1 equiv. decafluorobiphenyl as internal standard for GC-FID, open flask. b Solvent: H2O, AcOH, EtOAc, propylene carbonate, DMF, acetone, MeCN, Et2O, 1,4-dioxane, MeOH, EtOH, i-PrOH n-BuOH, DCM, DCE, CHCl3, toluene. c Schlenk line techniques used. d Catalyst: 10% Pd/C, PdCl2, Cu(OAc)2, Cu(OTf)2, Pd[P(C6H5)3]4, (MeCN)2Pd(II)Cl2 and (η3–C3H5)2Pd2Cl2, PEPPSI-SIPr. e 2 h premixing of Pd(OAc)2 with 1-methylindole, 1.0 equiv. of benzenediazonium tetrafluoroborate used. f isolated yield. | ||||
| 1 | Pd(OAc)2 (10.0) | DMF | 30 min | 66 |
| 2 | Pd(OAc)2 (5.0) | DMF | 30 min | 34 |
| 3 | — | DMF | 16 h | 0 |
| 4 | Pd(OAc)2 (5.0) | Solventb | 30 min | <72 |
| 5 | Pd(OAc)2 (5.0) | THF | 30 min | 77; 76c |
| 6 | Catalyst (5.0)d | THF | 2 h | 0 |
| 7 | Pd(TFA)2 (5.0) | THF | 30 min | 76 |
| 8 | Pd2(dba)3 (2.5) | THF | 30 min | 68 |
| 9 | Pd(OAc)2 (0.5) | THF | 1 h | 81 |
| 10 | Pd(OAc)2 (0.2) | THF | 1 h | Trace |
| 11 | Pd(OAc)2 (0.5) | 2-MeTHF | 2 h | 87 |
| 12 | Pd(OAc)2 (0.5) | EtOAc:2-MeTHF (1:1) |
1 h | 89 |
| 13 | Pd(OAc)2 (0.2) | EtOAc:2-MeTHF (1:1) |
1 h | 78 |
| 14e | Pd(OAc)2 (0.5) | EtOAc:2-MeTHF (1:1) |
30 min | 93; 90 |
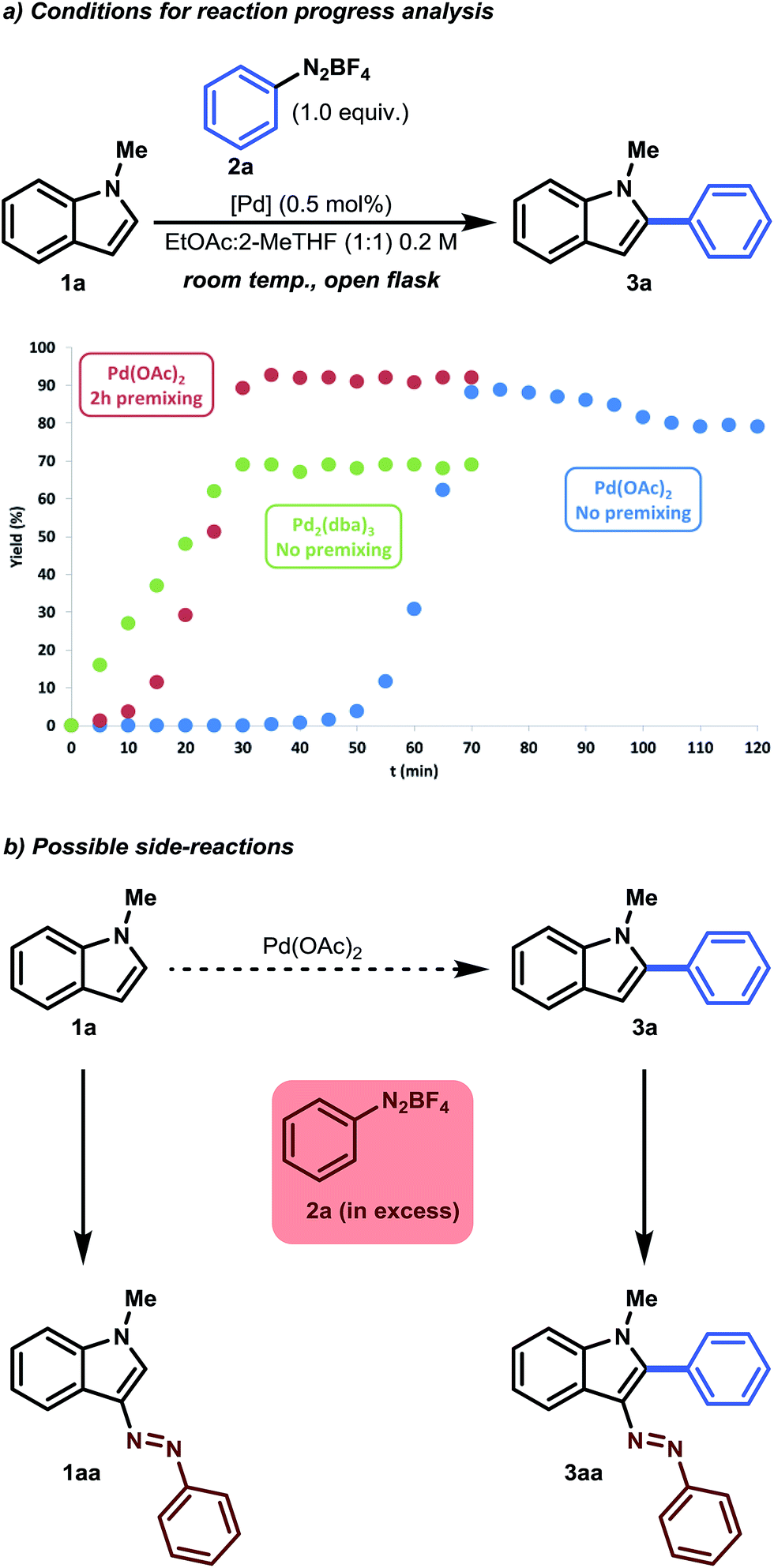 | ||
| Fig. 1 (a) Yield as a function of time. In the case of Pd(OAc)2 with no premixing (blue series), 1.1 equiv. of benzenediazonium tetrafluoroborate was used. (b) Observed side-reactions occurring in excess benzenediazonium tetrafluoroborate. | ||
In parallel with our optimization studies, a series of reaction progress kinetic experiments were performed to shed more light on the observed catalyst induction period. Unusual kinetics has often been reported in the field of C–H functionalization, but has seldom been investigated.21 Therefore, in order to obtain a more realistic view of this activation period, we monitored a series of reactions. As can be seen from Fig. 1a, an induction period of approximately 50 minutes was observed in the case of Pd(OAc)2 (Fig. 1a, blue series). As soon as the reaction began (>50 min), an initial acceleration occurred, resulting in S-curve behavior. It was postulated that a possible activation period could be necessary between the catalyst and the substrate. Therefore, premixing experiments were conducted. It was found that premixing 1-methylindole with Pd(OAc)2 (0.5 mol%) in EtOAc:2-MeTHF (1:1) for 2 hours could eliminate this observed induction period (Fig. 1a, red series). We surmised that Pd(II) is first reduced to a homogeneous Pd(0) complex and is stabilized by the π-donating character of 1-methylindole and/or by the ligand exchange of −OAc with 2-MeTHF.22 Indeed, a reaction performed with Pd2(dba)3 as a stable homogenous Pd(0) substitute showed that neither an induction period nor an initial acceleration occurred (Fig. 1a, green series). However, lower yields were obtained with Pd2(dba)3. This result gives us a first glimpse of the possible catalytic mechanism, indicating that palladium in its homogeneous zero state can act as an active catalyst.
As expected, the product 3a was even more prone to undergo a side reaction (i.e. an electrophilic substitution reaction) with benzenediazonium salt, as the inductive effect of the phenyl substituent makes the C-3 position more nucleophilic.23 This was especially noticeable when a slight excess of benzenediazonium tetrafluoroborate was used (Fig. 1a, blue series). A small yield of approximately 10% was observed after prolonged reaction time, which accounts for the 0.1 equivalent excess. To counteract this consecutive reaction, an equimolar amount (1.0 equiv. benzenediazonium tetrafluoroborate) was used. As a result, 90% of the desired product could be isolated (Table 1, Entry 14). Note that the reaction time could be halved again, to approximately 30 minutes, when using the premixing strategy. In addition, a slightly higher selectivity was obtained because side reactions were minimized. More information regarding reaction optimization and reaction progress analysis can be obtained from the ESI.†
Mechanistic studies: DFT calculations and experimental investigations
Since heteroarenes are good nucleophiles, it would be reasonable to assume a mechanism in which Pd(II) acts as an electrophile, consistent with numerous literature proposals in the context of C–H functionalization.8a,24 Similar to SEAr, these reactions are expected to be C-3 selective for indoles. However, our methodology yields C-2 arylated indoles selectively and thus requires a subsequent C-3/C-2 isomerization. In this context, Gaunt and co-workers showed that the presence of acid would facilitate a switch from C-3 to C-2 in the Pd-catalyzed C–H olefination of indoles,24 proposing that under acidic conditions, C-3 deprotonation of the indole moiety would be slowed. However, such a scenario appears unlikely in our case. For example, progressive 1H-NMR spectroscopy with equimolar quantities of Pd(OAc)2 and 1-methylindole in d8-THF showed that neither the HC-2 or the HC-3 peaks of 1-methylindole were affected (see ESI Section 3.1.1†).25Therefore, the employed Pd(OAc)2 likely serves as a pre-catalyst and is reduced to Pd(0) during the initiation period. Additionally, since we have shown that Pd(0) is catalytically active without any induction period (Table 1, Entry 8), it is reasonable to assume that the reaction proceeds via a Pd(0)/Pd(II) catalytic cycle.26 This cycle starts with an initial oxidative addition of the highly activated aryl diazonium salt to Pd(0) to yield a cationic Pd(II) complex which should subsequently serve as an electrophile in the reaction with the substrate (Scheme 2, I). The overall product selectivity would then again be determined by the C-3 to C-2 migration of Pd.24 However, our efforts to computationally locate the C-2 Pd complex yielded a structure that is 9.1 kcal mol−1 higher in energy than the preferred η2 π-complex Int1 (Fig. 2), suggesting that the migration is disfavored.27
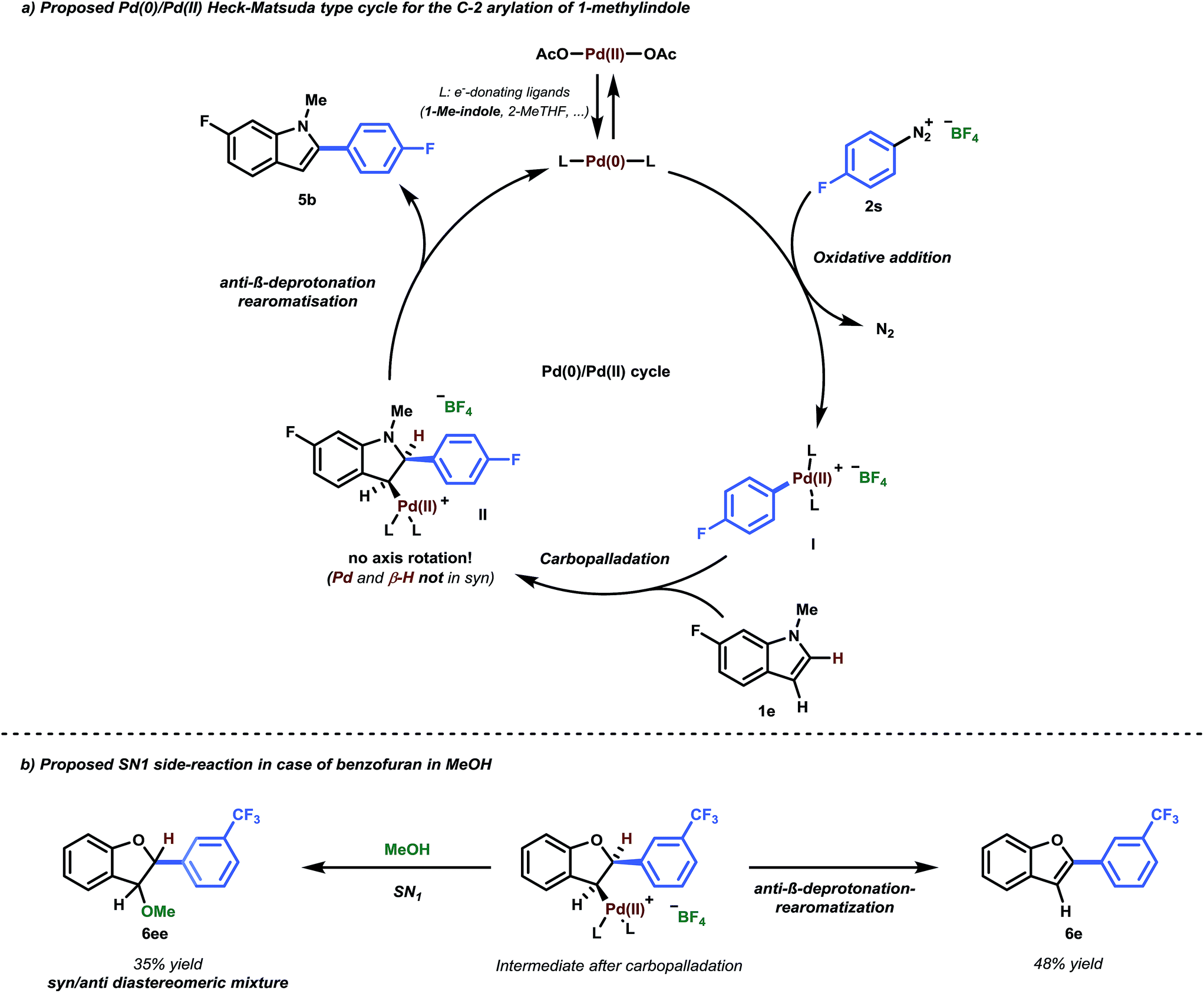 | ||
| Scheme 2 (a) Proposed Pd(0)/Pd(II) Heck–Matsuda-type cycle for the C-2 arylation of 1-methylindole. (b) Observed SN1 side-reaction in the case of benzofuran in MeOH. | ||
 | ||
| Fig. 2 Heck-type carbopalladation pathway and the prediction of selectivity via its transition states at the CPCM (THF) M06L/def2TZVP//wB97X-D/6-31G(d) SDD level of theory.31 Coordination by two THF molecules was found to be the preferred ligation state of Pd.29 Free energies are shown in kcal mol−1. | ||
Intermediate Int1 may alternatively undergo a Heck-type carbopalladation reaction.28 Our calculations suggest this process to be energetically feasible, being characterized by a relatively facile free energy barrier of 17.5 kcal mol−1 (Fig. 2). Thus, we subsequently calculated the expected selectivities (C-3 versus C-2) for C–H arylation for a carbopalladation mechanism. We considered several possible solvent coordinations to the cationic Pd; we determined that the coordination of two THF molecules is likely preferred.29 Our computed selectivities are in agreement with experiments. Complete C-2 selectivity was experimentally observed for 1-methylindole and benzofuran, consistent with our computational results (ΔΔG‡ = 2.4 kcal mol−1 and 0.7 kcal mol−1 in favor of C-2, respectively).30 By contrast, benzothiophene yielded the C-3 arylated product exclusively, which was also reproduced by computations (ΔΔG‡ = 1.9 kcal mol−1 in favor of C-3) (see Fig. 2).
The carbopalladation step in the traditional Heck-type reaction would be followed by syn-β-hydride elimination. Due to the rigidity of the ring system, however, there is no possibility of conventional syn-β-hydride elimination from the formed intermediate Int2. In contrast, it has been previously suggested that a base or solvent assisted anti-β-deprotonation rearomatisation could occur.14a,28b,32 While that step may also be involved in our case, due to the ionic and complex natures of the intermediates involved, an adequate computational description of the system would pose a number of difficulties.28b,33 However, in situ1H and 19F NMR analysis of the reaction have given us initial insights into the likely nature of the processes involved (see ESI Section 3.1.2 for a detailed description†). The data indicate that additional signals, assigned as BF3·2Me–THF and HF, appear in the 19F NMR spectrum at the same rate as the product 5b. Moreover, when using an alternative counterion for the aryldiazonium salt (e.g., 4-methoxybenzenediazonium mesylate), no product was observed (Table 2, 3h). It is therefore hypothesized that the BF4− counterion of the aryldiazonium salt plays a non-negligible role in the reaction mechanism, i.e. acting as a pseudo-base in the anti-β-deprotonation rearomatisation step. In addition, a crude 1H-NMR spectrum acquired from the reaction mixture (using THF-d8 as solvent) indicates that the lost proton appears quantitatively as a broad signal at 9.0 ppm (See ESI Section 2.3).
|
|
|---|
|
a Reaction conditions: 0.5 to 1.0 mol% Pd(OAc)2, 1.0 mmol heteroarene and 1.0 equiv. aryldiazonium salt in 5 mL EtOAc:2-MeTHF (1:1) at rt, open flask, 2 h premixing of Pd(OAc)2 with heteroarene.
b Pd2(dba)3 as catalyst, 1 h reaction.
c 1 mol% Pd(OAc)2, 1.2 equiv. aryldiazonium salt.
d 4-Methoxybenzenediazonium mesylate was used.
e Gram-scale experiment (10.0 mmol) yielded 2.47 g (83%), 4 h reaction time in 2-MeTHF as solvent.
f 1 mol% Pd(OAc)2.
g 2 mol% Pd(OAc)2.
h 2 mol% Pd(OAc)2, 1.2 equiv. aryldiazonium salt.
i 1.2 equiv. aryldiazonium salt at 40 °C; *no full conversion obtained.
j 0.01 M and 100 mol% Pd2(dba)3 was used.
|

|

Alternatively, a radical mechanism could be envisioned for this transformation. However, a large excess (5 to 100 equiv.) of the heteroarene substrate is generally required for satisfying results under such conditions. In our case, optimal results were achieved with equimolar quantities. In addition, test reactions via the radical pathway34 did not lead to the desired product. Moreover, radical scavenging tests failed to trap any radical intermediates (See ESI Section 3.2 for details). Finally, in radical chemistry, mixtures of C-2 and C-3 arylation are frequently observed,35 while our system displays complete selectivity.
Synthetic scope: heteroarenes and aryldiazonium salts
With the optimized conditions in hand, we next explored the scope of our developed methodology on indoles (Table 2). These substrates were reacted with equimolar amounts of aryldiazonium salts in the presence of 0.5 mol% Pd(OAc)2 in the case of 1-methylindoles and 1.0 mol% of Pd(OAc)2 for NH-indoles. A 1:1 mixture of EtOAc:2-MeTHF was used as the solvent. A broad set of substituted aryldiazonium substrates (3a–y) could be successfully coupled with 1-methylindole. Indole arylation with aryldiazonium salts bearing alkyl substituents (3c–g, 4a, b) proceeded well for both N-protected and free indoles, even in the presence of sterically demanding ortho-methyl substituents (3c, 3e). When using the more sterically hindered mesitylenediazonium tetrafluoroborate as the arylating agent, a mixture (C-2 and C-3 arylated product) was found for both the N-methylated and the free indoles (3f, 4c). Selectivity towards the C-3 arylated product was prevalent in 4c (C-2:C-3 1:3.3). For all other reactions, complete selectivity towards the C-2 arylated product was observed. Next, a scope of aryldiazonium salts, containing hydroxy-, phenoxy- and methoxy-substituents, was explored (3h–p). It was demonstrated that aryldiazonium salts bearing a free hydroxyl group showed some reactivity, although in lower yield (3p, 16%). A para-phenoxy group as an electron-donating substituent on the aryldiazonium salt resulted in good reactivity (3o, 79%).
Moreover, all methoxy-containing aryldiazonium salts (3h–n) showed good to excellent reactivity (69% to 93%), except for 3l, where no full conversion could be obtained. The yields obtained for compounds 3h–n showcase the applicability of our methodology for the C-2 arylation of indoles with arylating agents bearing methoxy-substituents, which are often reported to be cumbersome.7,8b,9b These substituents are functional handles which can be engaged in nickel-catalyzed cross-coupling chemistry via C–O activation.36 In addition, heterocyclic aryldiazonium salts were tolerated in this protocol: 3q was obtained in moderate yield (34%) overnight, while for 3r, a good yield (71%) was acquired within 1 hour reaction time. Notably, in the case of free NH-indoles (4a–d), an ortho-methyl substituent on the aryldiazonium salt proved necessary to avoid significant by-product formation (electrophilic substitution). However, it was found that by blocking the C-3 position of the NH-indole (i.e., via methylation), this side-reaction could be completely avoided (4eavs.4e).
Next, we explored a more challenging class of aryldiazonium salts bearing weakly (e.g., F) to highly electron-withdrawing (e.g., NO2) substituents (3s–y). Gratifyingly, 4-fluoro- and 3-iodobenzenediazonium tetrafluoroborate readily reacted with 1-methylindole (3s, 3w). The latter (3w) is particularly appealing, since it indicates that palladium undergoes oxidative addition at the electrophilic diazonium site (instead of breaking the C–I bond) at room temperature. In contrast, aryldiazonium salts bearing m-CF3 (3ta), p-NO2 (3ua), o-Cl (3va) and p-Br (3xa) as substituents did not deliver any arylated product when 1-methylindole was used as the substrate. It was observed that these aryldiazonium salts were too prone to electrophilic substitution reactions, resulting in the rapid formation of 3-(arylazo)-1-methylindoles (see Fig. 1b). However, as in the NH-indole case, this side reaction could be efficiently overcome by blocking the C-3 position. Consequently, the arylation scope could be expanded to electron-withdrawing substituents (3t, 3u, 3v, 3x) with high to excellent yields of the desired product (80% to 92%). This trend was also observed when aryldiazonium salts bearing an acyl moiety were used (3ya and 3y): 58% of the target product (3ya) was obtained for 1-methylindole, while an improved result was obtained for the C-3 methylated indole (80% yield, 3y).
Subsequently, several indole derivatives were subjected to the reaction conditions using benzenediazonium tetrafluoroborate as a benchmark coupling partner. For 5a and 5c, the reaction proceeded smoothly under equimolar conditions. 5d proved more challenging (22% yield) due to the electron-withdrawing nature of the methyl carboxylate substituent, which renders it a less nucleophilic substrate. Interestingly, an experiment with 1,2-dimethylindole and benzenediazonium salt showed that no C-3 arylated product could be formed over 5 hours. Instead, the substrate was fully converted to the electrophilic substituted product 1bb (93% yield). Moreover, during a control experiment with a stoichiometric amount of Pd2(dba)3, no 1bb was formed. This indicates that the benzenediazonium salt preferably underwent oxidative addition (see ESI Section 3.4†).
Next, a gram scale experiment was conducted to test the scalability of this mild procedure. The reaction was carried out with equimolar quantities of reactants (10 mmol) and 0.5 mol% Pd(OAc)2 in 2-MeTHF. With a slightly longer reaction time of 4 hours, a satisfying yield of 83% (2.47 g) of 3k was achieved under open flask conditions.
Having established a good coupling protocol for indoles, we subsequently examined the scope of benzofuran (1i) with various aryldiazonium salts (Table 3). Since benzofuran is not prone to electrophilic substitution, MeOH could be used as a more reactive solvent (see ESI Section 2.4†). These results are in agreement with the literature.19b Felpin et al. demonstrated with DFT and experimental results that the cationic palladium intermediates in the Heck cycle are exoergic with MeOH as the solvent.37 Moreover, it was observed that the addition of 1.0 equivalent of TFA resulted in an impressive rate acceleration (overnight to 30 minutes) while maintaining its selectivity (6e, 81%). The use of the protic solvent MeOH resulted in the formation of 2-aryl-3-methoxy-2,3-dihydrobenzofuran (6ee). It was speculated that 6ee was formed from the proposed carbopalladation intermediate II through a SN1 mechanism, resulting in the observed syn/anti diastereomeric mixture (Scheme 2b). However, a simple workup procedure consisting of 15 minutes of reflux under acidic conditions (i.e. acetyl chloride) was found to be sufficient to eliminate MeOH from the compound, affording the desired product in overall high yield. Benzofuran could be readily coupled with benzenediazonium tetrafluoroborate in good yield (6a, 70%).


Next, we carried out several reactions by coupling benzofuran with several halogenated aryldiazonium tetrafluoroborates (6b–h). Satisfyingly, all reactions proceeded smoothly in the presence of only 0.5 mol% Pd(OAc)2, thus showcasing the mild reaction conditions of this protocol.
Finally, we turned our attention towards a more challenging heteroarene, i.e. benzothiophene (1j) (Table 3). Because benzothiophene is the least nucleophilic heteroarene investigated herein, it was necessary to use slightly higher catalyst loadings (2.0 mol%) and 2.0 equivalents of aryldiazonium salt in order to achieve full conversion. Operating at 40 °C was decisive to obtain a good yield for both 7a (80%) and 7b (73%). In agreement with literature reports and DFT calculations (see above), we observed a complete shift in selectivity from C-2 to C-3 arylation. For compound 7a, a significant improvement in yield (80% vs. 69%) and a reduction in reaction time (16 h vs. 96 h) was observed, highlighting the relevance of our mild protocol.11b Taken together, this C–H activation protocol for the direct arylation of heteroarenes provides a convenient pathway towards a broad range of heteroaromatic arylated derivatives.
Application: synthesis of the drug precursor of Saprisartan
To further illustrate the efficacy of this mild strategy, we applied the C–H arylation process to the synthesis of methyl 2-(5-methylbenzofuran-2-yl)benzoate (8a) (Scheme 3). Compound 8a is a key intermediate in the total synthesis of Saprisartan, an approved drug belonging to the sartan family.17 Sartans act as AT1-antagonists and are among the most prescribed drugs for the treatment of hypertension and heart failure.38 Our mild procedure allowed us to successfully isolate the key intermediate 8a in 70% yield, which compares favorably to the patented process, which requires four consecutive steps for the same transformation with an overall low yield of 11%.17,39 | ||
| Scheme 3 Synthesis of the drug precursor 8a of Saprisartan. | ||
Conclusion
In summary, we have developed a mild and selective protocol for the C–H arylation of heteroarenes, including indoles, benzofurans and benzothiophenes, with aryldiazonium salts. The protocol is operationally simple and is insensitive to air and moisture. It utilizes low palladium loadings (0.5 to 2 mol% Pd), short reaction times, green solvents (EtOAc/2-MeTHF or MeOH) and is carried out at room temperature. Notably, no oxidants or other additives are required. The substrate scope is broad and displays excellent selectivity (C-2 arylation for indoles and benzofurans, C-3 arylation for benzothiophenes). To illustrate the efficacy of this procedure, a key intermediate (8a) for the drug Saprisartan was synthesized, comparing favorably to the patented process (70% vs. 11%). Mechanistic experiments and DFT calculations support a Heck–Matsuda-type coupling mechanism. Moreover, the first experimental results indicate that BF4− anions could be involved in the anti-β-deprotonation rearomatisation step of the catalytic cycle. We expect this protocol will find widespread application due to its mild character and excellent selectivity.Acknowledgements
Financial support is provided by the Dutch Science Foundation (NWO) via an ECHO grant (Grant No. 713.013.001) and a VIDI grant for T.N. (Grant No. 14150). We also acknowledge the European Union for a Marie Curie CIG grant for T.N. (Grant No. 333659) and an ERC Advanced Grant for V.H. (Grant No. 267443). IK and FS gratefully acknowledge the computing time granted by the JARA-HPC “Vergabegremium” on the RWTHBull Cluster in Aachen (Grant JARA0091). HG and TN thank Cecilia Bottecchia (TU/e) for helping with NMR analysis and Bart Hendricks (TU/e) for helping with the synthesis of aryldiazonium salts. We would like to thank referee 2 for stimulating discussions during the revision stage of this manuscript.Notes and references
- (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- (a) C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef PubMed; (b) A. Meijere and F. diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) T. Colacot, New Trends in Cross-Coupling: Theory and Applications, RSC, Cambridge, 2015 Search PubMed; (d) T. Noel and S. L. Buchwald, Chem. Soc. Rev., 2011, 40, 5010 RSC; (e) T. Noël and A. J. Musacchio, Org. Lett., 2011, 13, 5180 CrossRef PubMed.
- (a) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed; (b) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) K. Shin, S. W. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 8584 CrossRef CAS PubMed; (e) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863 CrossRef CAS PubMed; (f) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS PubMed.
- (a) D. P. Hari and B. Konig, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (b) D. P. Hari, P. Schroll and B. Konig, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed.
- (a) S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902 RSC; (b) H. Bonin, M. Sauthier and F. X. Felpin, Adv. Synth. Catal., 2014, 356, 645 CrossRef CAS; (c) F. P. Crisostomo, T. Martin and R. Carrillo, Angew. Chem., Int. Ed., 2014, 53, 2181 CrossRef CAS PubMed; (d) A. Honraedt, M. A. Raux, E. Le Grognec, D. Jacquemin and F. X. Felpin, Chem. Commun., 2014, 50, 5236 Search PubMed; (e) S. Gowrisankar and J. Seayad, Chem.–Eur. J., 2014, 20, 12754 CrossRef CAS PubMed.
- (a) L. Duan, R. Fu, B. Zhang, W. Shi, S. Chen and Y. Wan, ACS Catal., 2016, 6, 1062 CrossRef CAS; (b) Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495 CrossRef CAS PubMed; (c) L. J. Durak, J. T. Payne and J. C. Lewis, ACS Catal., 2016, 6, 1451 CrossRef CAS PubMed; (d) H.-C. Wu, J.-H. Chu, C.-W. Li, L.-C. Hwang and M.-J. Wu, Organometallics, 2016, 35, 288 CrossRef CAS; (e) S. G. Modha and M. F. Greaney, J. Am. Chem. Soc., 2015, 137, 1416 CrossRef CAS PubMed; (f) J. Malmgren, A. Nagendiran, C. W. Tai, J. E. Backvall and B. Olofsson, Chem.–Eur. J., 2014, 20, 13531 CrossRef CAS PubMed; (g) K. Ueda, K. Amaike, R. M. Maceiczyk, K. Itami and J. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 13226 CrossRef CAS PubMed; (h) H. P. L. Gemoets, V. Hessel and T. Noël, Org. Lett., 2014, 16, 5800 CrossRef CAS PubMed; (i) A. F. P. Biajoli, E. T. da Penha and C. R. D. Correia, RSC Adv., 2012, 2, 11930 RSC; (j) J. G. Taylor, A. V. Moro and C. R. D. Correia, Eur. J. Org. Chem., 2011, 2011, 1403 CrossRef; (k) L. Wang, W.-B. Yi and C. Cai, Chem. Commun., 2011, 47, 806 RSC; (l) C. Colas and M. Goeldner, Eur. J. Org. Chem., 1999, 1999, 1357 CrossRef.
- R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed.
- (a) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050 CrossRef CAS PubMed; (b) B. S. Lane and D. Sames, Org. Lett., 2004, 6, 2897 CrossRef CAS PubMed.
- (a) A. M. Wagner and M. S. Sanford, Org. Lett., 2011, 13, 288 CrossRef CAS PubMed; (b) N. R. Deprez, D. Kalyani, A. Krause and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 4972 CrossRef CAS PubMed.
- (a) S. Potavathri, K. C. Pereira, S. I. Gorelsky, A. Pike, A. P. LeBris and B. DeBoef, J. Am. Chem. Soc., 2010, 132, 14676 CrossRef CAS PubMed; (b) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137 CrossRef CAS PubMed.
- (a) D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809 CrossRef CAS PubMed; (b) D. T. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450 CrossRef CAS PubMed.
- (a) Y. Zhu, M. Bauer, J. Ploog and L. Ackermann, Chem.–Eur. J., 2014, 20, 13099 CrossRef CAS PubMed; (b) L. Ackermann, M. Dell'Acqua, S. Fenner, R. N. Vicente and R. Sandmann, Org. Lett., 2011, 13, 2358 CrossRef CAS PubMed.
- D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172 CrossRef CAS PubMed.
- (a) C. Colletto, S. Islam, F. Julia-Hernandez and I. Larrosa, J. Am. Chem. Soc., 2016, 138, 1677 CrossRef CAS PubMed; (b) N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926 CrossRef CAS PubMed.
- (a) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (b) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC.
- (a) H. Li, H. Xiao, L. Lin, D. Jou, V. Kumari, J. Lin and C. Li, J. Med. Chem., 2014, 57, 632 CrossRef CAS PubMed; (b) Ž. Hodnik, L. Peterlin Mašič, T. Tomašić, D. Smodiš, C. D'Amore, S. Fiorucci and D. Kikelj, J. Med. Chem., 2014, 57, 4819 CrossRef PubMed.
- D. B. Judd, M. D. Dowle, D. Middlemiss, D. I. Scopes, B. C. Ross, T. I. Jack, M. Pass, E. Tranquillini, J. E. Hobson and T. A. Panchal, et al. , J. Med. Chem., 1994, 37, 3108 CrossRef CAS PubMed.
- S. Dadiboyena, Eur. J. Med. Chem., 2012, 51, 17 CrossRef CAS PubMed.
- (a) M. N. Hopkinson, B. Sahoo, J. L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (b) F. X. Felpin, L. Nassar-Hardy, F. Le Callonnec and E. Fouquet, Tetrahedron, 2011, 67, 2815 CrossRef CAS; (c) A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed.
- (a) V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369 CrossRef CAS PubMed; (b) R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC; (c) D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 CrossRef CAS.
- (a) R. B. Bedford, S. J. Durrant and M. Montgomery, Angew. Chem., Int. Ed., 2015, 54, 8787 CrossRef CAS PubMed; (b) Y. Tan, F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 3683 CrossRef CAS PubMed; (c) R. D. Baxter, D. Sale, K. M. Engle, J. Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 4600 CrossRef CAS PubMed; (d) D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302 CrossRef CAS PubMed.
- (a) A. H. L. Machado, H. M. S. Milagre, L. S. Eberlin, A. A. Sabino, C. R. D. Correia and M. N. Eberlin, Org. Biomol. Chem., 2013, 11, 3277 RSC; (b) A. A. Sabino, A. H. L. Machado, C. R. D. Correia and M. N. Eberlin, Angew. Chem., Int. Ed., 2004, 43, 2514 CrossRef CAS PubMed; (c) J. M. Brown and K. K. Hii, Angew. Chem., Int. Ed., 1996, 35, 657 CrossRef CAS.
- S. Daly, K. Hayden, I. Malik, N. Porch, H. Tang, S. Rogelj, L. V. Frolova, K. Lepthien, A. Kornienko and I. V. Magedov, Bioorg. Med. Chem. Lett., 2011, 21, 4720 CrossRef CAS PubMed.
- N. P. Grimster, C. Gauntlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125 CrossRef CAS PubMed.
- Even if C–H activation was to occur under these conditions, then the introduction of the aryl group would require a subsequent oxidative addition of the diazonium reagent to a Pd(II) complex, leading to a Pd(II)/Pd(IV) catalytic cycle. Calculations, however, are suggesting a prohibitively high barrier of around 56 kcal mol−1 for such transformation.
- (a) R. Yamashita, K. Kikukawa, F. Wada and T. Matsuda, J. Organomet. Chem., 1980, 201, 463 CrossRef CAS; (b) K. Kikukawa and T. Matsuda, Chem. Lett., 1977, 159 CrossRef CAS.
- The C-2 Pd complex was located only by locking the Pd(II) center to C-2. However, upon unfreezing this interaction the structure converged back to the η2 π-complex. For similar difficulties in locating the migration of Pd from one carbon to another in the context of C–H activation of thiophenes see ref. 28a.
- (a) S. Y. Tang, Q. X. Guo and Y. Fu, Chem.–Eur. J., 2011, 17, 13866 CrossRef CAS PubMed; (b) M. Steinmetz, K. Ueda, S. Grimme, J. Yamaguchi, S. Kirchberg, K. Itami and A. Studer, Chem.–Asian J., 2012, 7, 1256 CrossRef CAS PubMed; (c) B. Glover, K. A. Harvey, B. Liu, M. J. Sharp and M. F. Tymoschenko, Org. Lett., 2003, 5, 301 CrossRef CAS PubMed; (d) K. Maeda, E. J. Farrington, E. Galardon, B. D. John and J. M. Brown, Adv. Synth. Catal., 2002, 344, 104 CrossRef CAS.
- Various ligation states of Pd were considered: THF molecules, Indole substrate and the combination of both, with the THF molecules being the preferred one.
- Benzofuran was reacted in MeOH, which offers various different likely coordination states. Thus, it is challenging to describe this system adequately with computations. For the coordination state considered, we predict selectivities in line with experiments. See ESI for additional information†.
- (a) M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed; (b) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532 CrossRef CAS PubMed.
- M. Ikeda, S. A. A. El Bialy and T. Yakura, Heterocycles, 1999, 51, 1957 CrossRef CAS.
- (a) T. Sperger, H. C. Fisher and F. Schoenebeck, WIREs Comput. Mol. Sci., 2016, 6, 226 CrossRef CAS; (b) K. J. Bonney and F. Schoenebeck, Chem. Soc. Rev., 2014, 43, 6609 RSC; (c) T. Sperger, I. A. Sanhueza and F. Schoenebeck, Acc. Chem. Res., 2016, 49, 1311 CrossRef CAS PubMed.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS PubMed.
- D. P. Hari, T. Hering and B. König, Org. Lett., 2012, 14, 5334 CrossRef CAS PubMed.
- (a) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed; (b) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC; (c) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (d) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 1728 CrossRef CAS PubMed.
- F.-X. Felpin, K. Miqueu, J.-M. Sotiropoulos, E. Fouquet, O. Ibarguren and J. Laudien, Chem.–Eur. J., 2010, 16, 5191 CrossRef CAS PubMed.
- P. A. James, S. Oparil and B. L. Carter, et al. , JAMA, J. Am. Med. Assoc., 2014, 311, 507 CrossRef CAS PubMed.
- M. D. Dowle and D. B. Judd, USA Pat., US5332831 A, 1994.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02595a |
| This journal is © The Royal Society of Chemistry 2017 |